

Summary
Fungal infections are major causes of morbidity and mortality, especially in immunocompromised individuals. The innate immune system senses fungal pathogens through Syk-coupled C-type lectin receptors (CLRs), which signal through the conserved immune adaptor Card9. Although Card9 is essential for antifungal defense, the mechanisms that couple CLR-proximal events to Card9 control are not well defined. Here, we identify Vav proteins as key activators of the Card9 pathway. Vav1, Vav2, and Vav3 cooperate downstream of Dectin-1, Dectin-2, and Mincle to engage Card9 for NF-κB control and proinflammatory gene transcription. Although Vav family members show functional redundancy, Vav1/2/3−/− mice phenocopy Card9−/− animals with extreme susceptibility to fungi. In this context, Vav3 is the single most important Vav in mice, and a polymorphism in human VAV3 is associated with susceptibility to candidemia in patients. Our results reveal a molecular mechanism for CLR-mediated Card9 regulation that controls innate immunity to fungal infections.
Graphical Abstract
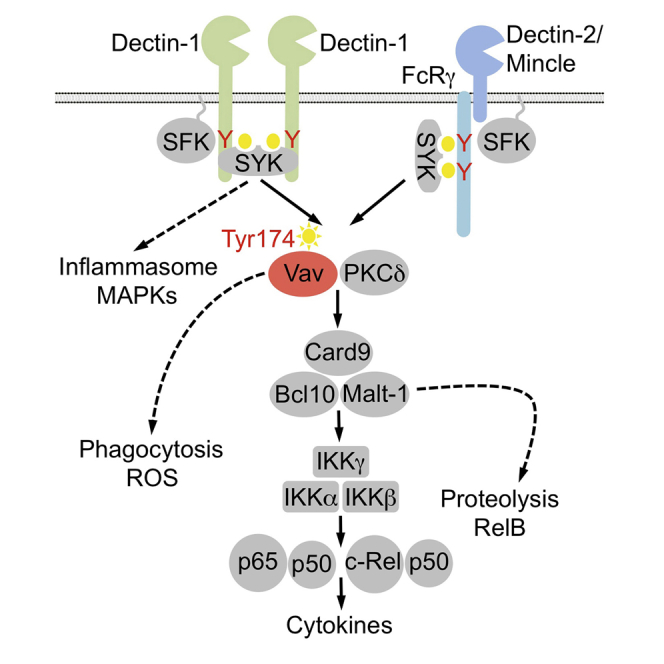
Highlights
-
•
Vav proteins control CLR-mediated inflammatory responses
-
•
CLR-induced NF-κB activation is regulated by Vav proteins
-
•
Vav/Card9 signaling is critical for antifungal host defense
CLR/Card9 signaling is essential for antifungal immunity. Roth et al. have identified the Vav family of proteins as key activators of the Card9/NF-kB pathway, essential for CLR-induced inflammatory responses and host defense against fungi.
Introduction
Cells of the innate immune system sense microbial components or cell damage-associated structures via germline-encoded pattern recognition receptors (PRRs) that subsequently signal for host defense and tissue homeostasis (Takeuchi and Akira, 2010). One important family of signaling PRRs on myeloid cells are the Syk-coupled C-type lectin receptors (Kerrigan and Brown, 2011, Sancho and Reis e Sousa, 2012). These PRRs play a broad role in innate immunity and are particularly important for host defense against fungal infections (Ferwerda et al., 2009, Robinson et al., 2009, Saijo et al., 2007, Saijo et al., 2010, Sato et al., 2006, Taylor et al., 2007, Wells et al., 2008), which constitute an increasing health threat because of growing numbers of patients at risk mainly because of immunosuppressive medical interventions and AIDS. In the context of antifungal immunity, the C-type lectin receptor (CLR) family member Dectin-1 senses β-glucans in fungal cell walls (Brown and Gordon, 2001), whereas Mincle and Dectin-2 detect α-mannose, glycolipids, and α-mannans, respectively (Sancho and Reis e Sousa, 2012). Agonist binding by Dectin-1 leads to the phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM)-like motifs in its cytoplasmic tail by Src family kinases, resulting in activation of the tyrosine kinase Syk. Likewise, Dectin-2 and Mincle also activate Syk, indicating that these CLR signals engage common effector mechanisms (Mócsai et al., 2010, Sancho and Reis e Sousa, 2012). The innate immune adaptor protein Card9 is critical for CLR signaling. It assembles signaling complexes that also contain Bcl10 and Malt1 (Card9-BCl10-Malt1 [CBM] complexes) and that serve as scaffolds for activation of the canonical nuclear factor κB (NF-κB) pathway (Roth and Ruland, 2013). This mechanism activates the inhibitor of kappa B (IκB) kinase (IKK) complex, which phosphorylates inhibitory IκBs, leading to their proteasomal degradation and the release of NF-κB dimers to the nucleus to activate gene transcription (Vallabhapurapu and Karin, 2009). Malt1 can also function as a protease upon CBM complex assembly that cleaves a set of NF-κB regulators, including RelB, to fine-tune immune gene expression (Hailfinger et al., 2011, Jaworski et al., 2014). These Card9 signaling complexes operate downstream of all tested Syk-coupled CLRs (Roth and Ruland, 2013) and are essential for innate antifungal immunity. Indeed, Card9-deficient mice are highly susceptible to infection with Candida (Gross et al., 2006, Jia et al., 2014), Aspergillus (Jhingran et al., 2012), and Cryptococcus (Yamamoto et al., 2014) species. Moreover, loss-of-function mutations in human CARD9 have been identified as causes of mucocutaneous and invasive fungal infections (Glocker et al., 2009, Pérez de Diego et al., 2015). Nevertheless, despite the critical role for CLR-triggered Card9 signaling in innate immunity and mammalian host defense, the molecular mechanisms that link CLR ligation to Card9-dependent effector mechanisms are not well understood.
Here, we used a mass spectrometry-based proteomic approach and identified Vav proteins as regulators of Card9 signaling. Vav1, Vav2, and Vav3 cooperate downstream of Dectin-1, Dectin-2, and Mincle to engage Card9 complexes for NF-κB control and proinflammatory gene transcription. Like Card9-deficient mice, Vav1/2/3 triple-deficient mice are severely impaired in inflammatory responses to Candida albicans infection and host defense against the fungus. Moreover, we report a human polymorphism in VAV3 that is associated with susceptibility to candidemia. Thus, our results establish Vav proteins as essential regulators of CLR-mediated Card9 control in innate antifungal immunity.
Results
Fungal Infection Induces Tyrosine Phosphorylation of Vav in Myeloid Cells
To investigate the mechanisms of Syk-coupled CLR signaling, we stimulated wild-type murine bone marrow-derived dendritic cells (BMDCs), comprising conventional DCs and monocyte-derived macrophages (Helft et al., 2015), for 10 min with zymosan, a yeast cell wall preparation that is highly enriched in Dectin-1 and Dectin-2 agonists, and subsequently affinity-purified tyrosine-phosphorylated proteins for mass spectrometric analysis (Strasser et al., 2012). Under these conditions, we observed signal-induced tyrosine phosphorylation of Vav1 and Vav3, which are cytosolic signaling scaffolds and guanine nucleotide exchange factors that can play context-specific roles in immune receptor pathways (Bustelo, 2014). To validate these findings, we stimulated BMDCs with C. albicans hyphae and specifically analyzed Vav1 phosphorylation by western blot analysis. Indeed, Vav1 was tyrosine-phosphorylated after C. albicans infection (Figure 1A). These data are in line with previously published results that demonstrated that stimulation with β-glucan or zymosan triggers tyrosine phosphorylation of Vav1 also in microglial cells or neutrophils (Li et al., 2011, Shah et al., 2009). Further analysis with a phosphospecific antibody raised against the regulatory Tyr174 residue of Vav1 additionally revealed that this specific residue is phosphorylated upon C. albicans detection, indicating Vav1 activation (Aghazadeh et al., 2000), but not upon LPS stimulation (Figure 1B).
Figure 1.

Vav1 Is Tyrosine-Phosphorylated in Response to Candida
(A) BMDCs were untreated (−) or stimulated with C. albicans hyphae (MOI 5) for 15 min. Proteins from cell lysates were immunopurified with anti-phospho-tyrosine antibodies and analyzed by immunoblot with a Vav1 antibody (left). Also shown is quantification of Vav1 by densitometry (right).
(B) Immunoblot analysis of BMDCs unstimulated (−), treated with C. albicans hyphae (MOI 0.5) for the indicated times, or stimulated with LPS (100 ng/ml) for 45 min and probed with antibodies against phospho-Vav1 (Tyr174) or Vav1 (left). Also shown is densitometrical quantification of phospho-Vav1 relative to total Vav1, normalized to BMDCs treated with C. albicans for 45 min (right).
(C) BMDCs were untreated (−) or preincubated with the Syk inhibitor R406 (0.5, 1, and 2 μM) or the Src-kinase inhibitor PP2 (1.5, 3, and 6 μM) or its inactive analog PP3 (1.5, 3, and 6 μM) and then stimulated with C. albicans hyphae (MOI 1) for 30 min. Lysates were immunoblotted with anti-phospho-Vav1 (Tyr174) or anti-Vav1 antibodies (left). Also shown is densitometrical quantification of phospho-Vav1 relative to total Vav1, normalized to BMDCs treated with C. albicans without any inhibitors (right). Data of at least three independent experiments are shown as mean ± SEM.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; Student’s t test (A) and one-way ANOVA and post hoc Tukey-Kramer test (C). n.s., not significant.
Recent work has demonstrated that Vav phosphorylation downstream of different ITAM-containing receptors is mediated by Src and Syk kinases (Bustelo, 2014). To determine the importance of the CLR-proximal Src and Syk kinases in Vav phosphorylation, we pretreated BMDCs with the Src inhibitor PP2 or the Syk inhibitor R406 prior to the addition of C. albicans. Both Src and Syk inhibition blocked Vav1-Tyr174 phosphorylation in a dose-dependent manner, whereas pretreatment with PP3, an inactive analog of PP2, did not affect C. albicans-induced phosphorylation of Vav1 at Tyr174 (Figure 1C). Together, these results indicate that Vav1 is activated in myeloid antigen-presenting cells specifically after fungal sensing via an Src and Syk kinase-dependent mechanism.
CLR-Mediated Inflammatory Responses Are Critically Dependent on Vav Proteins
Next, we studied the functional relevance of Vav proteins in C. albicans-induced inflammatory responses. Because dendritic cells express Vav2 (Spurrell et al., 2009) in addition to Vav1 and Vav3, which have partially redundant functions in immune cells (Fujikawa et al., 2003, Tybulewicz, 2005), we investigated BMDCs from mice that lack each individual Vav isoform, two Vav isoforms, or all three Vav family members. Notably, the lack of any one or all Vav molecules did not impair BMDC differentiation (data not shown; Graham et al., 2007, Spurrell et al., 2009). However, BMDCs from Vav1/2/3 triple-deficient mice were severely defective in C. albicans-induced TNF production, whereas tumor necrosis factor (TNF) secretion in response to Toll-like receptor (TLR) stimulation with LPS or Pam3CSK4 was unaffected in these cells (Figure 2A). Moreover, the production of interleukin-2 (IL-2) and IL-10 was also defective in BMDCs from Vav1/2/3−/− mice (Figure 2A). The finding that Vav1, Vav2, or Vav3 single-deficient or Vav2/3−/− myeloid antigen-presenting cells are only partially impaired in C. albicans-induced TNF, IL-2, and IL-10 generation (Figure 2B) indicates redundancy for Vav proteins in fungus-induced cytokine responses.
Figure 2.

Vav Proteins Are Essential for Syk-Coupled CLR-Induced Proinflammatory Responses
(A and B) TNF, IL-2, and IL-10 levels in the supernatants of wild-type (WT) and Vav1/2/3-/- BMDCs (A), or Vav single- or double mutant BMDCs of the indicated genotypes (B) that were untreated (−) or stimulated with C. albicans hyphae (MOI 0.3), LPS (500 ng/ml), or Pam3CSK4 (50 ng/ml) for 16 hr were analyzed by ELISA.
(C) IL-1β in the supernatants of WT and Vav1/2/3−/− BMDCs left untreated (−) or stimulated with C. albicans hyphae (MOI 1) for 6 hr.
(D) Quantitative real-time PCR analysis of IL-1β transcripts in WT and Vav1/2/3−/− BMDCs left untreated or stimulated with C. albicans hyphae (MOI 1) (left) or LPS (100 ng/ml) (right) for the indicated times; results are relative to those of β-actin mRNA.
(E) Immunoblot analysis of mature Caspase-1 (p10) in cell culture supernatants of WT and Vav1/2/3−/− (−/−) BMDCs left untreated (−), stimulated with C. albicans hyphae (MOI 1) for 4 hr (C.a.), or pre-stimulated with LPS (50 ng/ml) for 6 hr prior to the addition of ATP (5 mM) for 45 min (A+L). Quantification of mature Caspase-1 p10 by densitometry is shown to the right of the western blot.
(F and G) BMDCs from WT and Vav1/2/3-/- mice (F), or Vav single- or double mutant mice of the indicated genotypes (G) were untreated (−), stimulated with the Dectin-1 ligand curdlan (10 μg/ml), plate-bound anti-Dectin-2 (12.5 μg/ml) or isotype control antibodies (Isotype), or the Mincle ligand TDB (100 μg/ml). TNF levels in the cell culture supernatants were quantified by ELISA.
(H) WT and Vav1/2/3−/− mice were injected intravenously with TDM containing oil-in-water emulsions or with the vehicle control (−). 24 hr post-injection, TNF, IL-1β, and IL-10 levels in sera were determined by CBA.
(A, B, F, and G) Data are expressed as percent of WT ± SEM of three independent experiments.
(C–E and H) Data of at least three independent experiments are presented as mean ± SEM.
∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001, Student’s t test (A, C, F, and H), and one-way ANOVA and post hoc Tukey-Kramer test (B and G).
One important inflammatory cytokine for antifungal immunity is IL-1β (Vonk et al., 2006), the generation of which is controlled by NF-κB-dependent pro-IL-1β gene transcription followed by NLRP3 inflammasome-mediated Caspase-1-dependent pro-IL-1β processing (Gross et al., 2009, Hise et al., 2009). Stimulation of wild-type BMDCs with C. albicans induced the robust secretion of mature IL-1β into the culture supernatant (Figure 2C). In contrast, Vav1/2/3-deficient cells were almost completely defective in Candida-induced IL-1β production (Figure 2C). This defect was caused by the selective impairment of pro-IL-1β gene transcription in Vav1/2/3−/− cells (Figure 2D), whereas the deficiency of Vav proteins did not affect NLRP3 inflammasome-dependent Caspase-1 activation (Figure 2E). Together, these experiments demonstrate that Vav proteins are critical for cytokine production by myeloid cells following the detection of whole fungal cells.
Next, we studied the specific requirement of Vav proteins for Dectin-1-, Dectin-2-, and Mincle-induced cytokine responses using selective agonists for each individual receptor. Stimulation of Dectin-1 with curdlan, a purified particulate β-glucan (LeibundGut-Landmann et al., 2007), induced the robust production of TNF in wild-type BMDCs, whereas Vav1/2/3-deficient cells failed to produce TNF in response to Dectin-1 triggering (Figure 2F). Likewise, stimulation of BMDCs with agonistic antibodies against Dectin-2 or with trehalose-6,6-dibehenate (TDB), a synthetic analog of the mycobacterial cord factor trehalose 6,6′-dimycolate (TDM), which specifically activates Mincle (Lobato-Pascual et al., 2013, Miyake et al., 2013, Schoenen et al., 2010), induced TNF in wild-type cells, but this response was severely impaired in Vav1/2/3-deficient BMDCs (Figure 2F). Therefore, although Vav single- or double-mutant cells exhibited a partially impaired cytokine response upon selective Dectin-1, Dectin-2, or Mincle stimulation (Figure 2G), the Vav family as a whole is essential for cytokine production after Syk-coupled CLR ligation in vitro.
Vav Proteins Regulate CLR-Induced Inflammatory Responses In Vivo and Are Essential for Antifungal Defense
To investigate the function of Vav proteins in CLR-mediated inflammatory responses in vivo, we intravenously injected the Mincle ligand TDM (Ishikawa et al., 2009, Miyake et al., 2013) into wild-type and Vav1/2/3−/− mice. Consistent with previously published results (Ishikawa et al., 2009), TDM injection resulted in the strong systemic production of TNF, IL-1β, and IL-10 in wild-type mice (Figure 2H). However, these responses were defective in Vav1/2/3 triple-deficient animals (Figure 2H). Next, we studied the role of Vav proteins in inflammatory cytokine responses after fungal infection in vivo. To this end, we intravenously injected wild-type or Vav1/2/3 triple knockout mice with live C. albicans and measured serum TNF and IL-6 levels after 6 hr. Notably, the production of both inflammatory cytokines was almost completely abolished in Vav1/2/3−/− mice (Figure 3A). Subsequently, we studied the importance of individual Vav proteins in host protection against fungi by infecting wild-type or Vav single-, double-, or triple-deficient mice with 1 × 105 colony-forming units (CFUs) of C. albicans. Four days later, we assessed intravital fungal growth in the kidneys, the main target organ of the fungus (Brieland et al., 2001). Although wild-type mice cleared the fungus readily, the kidneys of all investigated Vav1/2/3−/− mice were enlarged and infiltrated with macroscopically visible fungal colonies (Figure 3B). Quantitatively, we detected more than 100-fold higher titers of C. albicans in the kidneys of Vav1/2/3 triple-deficient animals compared with the wild-type (Figure 3C), and histopathology confirmed intravital fungal growth (Figure 3D). The fungal burdens in Vav3 single knockout and Vav2/3 double knockout mice were increased by trend compared with the wild-type, but a selective loss of Vav1 or Vav2 did not result in higher fungal titers compared with wild-type mice (Figure 3C). Finally, we studied the role of Vav signaling in the survival of mice after fungal infection. In line with the incapacity to control fungal invasion of the organs, Vav1/2/3-deficient mice died rapidly after injection of 1 × 105 CFU of C. albicans, whereas the wild-type control animals survived this challenge (Figure 3E). These experiments reveal that Vav proteins play essential roles in fungus-induced cytokine production and antifungal immunity in vivo, with Vav3 being the single most important Vav family member for protection against C. albicans infection.
Figure 3.

Vav Proteins Are Required for Systemic Antifungal Host Defense
(A–E) Mice of the indicated genotypes were intravenously infected with 1 × 105 CFUs of C. albicans.
(A) Serum IL-6 and TNF levels in WT and Vav1/2/3−/− mice were determined by CBA 6 hr after infection.
(B and C) After 4 days, kidneys were macroscopically examined (B; scale bar, 10 mm), and C. albicans titers were determined in the kidneys (C).
(D) Kidney sections from C. albicans-infected WT and Vav1/2/3−/− mice were stained with H&E or periodic acid-Schiff (PAS; scale bars, 100 μm).
(E) WT (n = 5) and Vav1/2/3−/− (n = 4) mice were monitored daily for health and survival. Statistical survival analyses were performed using the log-rank test (p < 0.005).
(A and C) Each symbol represents an individual mouse; small horizontal lines indicate the mean; error bars indicate the SEM. Data of one experiment in each case are presented. ∗p < 0.05, Student’s t test (A and C, left) and one-way ANOVA (C, right, p = 0.06).
Vav Proteins Are Regulators of CLR-Induced NF-κB Activation
The susceptibility of Vav1/2/3 triple-deficient animals and the failure to induce CLR-mediated cytokine responses are reminiscent of the severe phenotype observed in Card9-deficient mice, suggesting that Vav proteins and Card9 may operate in a common signaling cascade. Because of the partially overlapping functions of the three Vav isoforms, we focused subsequent biochemical studies on Vav1/2/3 triple knockout BMDCs. Upon C. albicans stimulation, phosphorylation of the tyrosine kinase Syk and of the Dectin-1 signal transducer PLCγ2 (Xu et al., 2009) did not differ between wild-type and Vav1/2/3−/− BMDCs (Figure 4A), indicating that Vav proteins are not required for the immediate receptor-proximal events. Moreover, because Erk1/2, p38, and Jun N-terminal kinase (JNK) were similarly phosphorylated in C. albicans-stimulated wild-type and Vav1/2/3−/− BMDCs (Figure 4B), we conclude that Vavs are not essential for the regulation of mitogen-activated protein kinase (MAPK) signaling after fungal infection. However, activation of protein kinase C δ (PKCδ), which controls Card9 engagement (Strasser et al., 2012), was partially impaired in BMDCs lacking all three Vav isoforms (Figure 4C; Figure S1A). Likewise, PKCδ deficiency partially compromised Vav1 phosphorylation in response to C. albicans or curdlan stimulation (Figure S1B). Moreover, although C. albicans or curdlan stimulation of wild-type BMDCs induces the rapid activation of the IKK complex, as determined by measuring IKKα/β phosphorylation (Figure 4C; Figures S1A and S1B), this response is almost completely abolished in the absence of Vav proteins (Figure 4C; Figure S1A) or PKCδ (Figure S1B; Strasser et al., 2012). Consistent with these findings, C. albicans-induced nuclear translocation of the transcriptional active NF-κB subunits p65, c-Rel, and RelB were also severely defective in Vav1/2/3−/− BMDCs, as they are in Card9-deficient cells (Figures 4D and 4E; Figures S1C and S1D), demonstrating a critical role for Vav proteins in CLR-dependent NF-κB control.
Figure 4.

Vav Proteins Control CLR-Triggered NF-κB Activation
(A–E) BMDCs from the indicated genotypes were stimulated with C. albicans hyphae (MOI 0.5) for various times or with LPS (100 ng/ml) for 45 min.
(A) Syk and PLCγ2 phosphorylation was determined in cell lysates by immunoblot with anti-phospho-Syk and anti-phospho-PLCγ2 antibodies.
(B) Activation of the MAP kinases Erk1/2, p38, and JNK was analyzed with anti-phospho-Erk1/2, anti-phospho-p38, and anti-phospho-JNK antibodies.
(C) Cell lysates were analyzed by immunoblot with anti-phospho-PKCδ and anti-phospho-IKKα/β antibodies. β-actin served as a loading control.
(D and E) Nuclear extracts from WT and Vav1/2/3-/- (D), or Card9-/- BMDCs (E) were analyzed by immunoblot with antibodies against the NF-κB subunits p65, c-Rel, and RelB; anti-lamin B antibodies indicate equal protein loading.
(F and G) BMDCs from mice of the indicated genotypes were pretreated with the proteasome inhibitor MG132 for 30 min and then stimulated with curdlan (0.5 mg/ml) (F) or C. albicans hyphae (MOI 1) (G) for 2 or 4 hr. Cell lysates were analyzed by immunoblot using antibodies against RelB and β-actin.
(H) WT and Vav1/2/3−/− BMDCs were stimulated with curdlan (0.2 mg/ml) for 3 hr. Shown is the GSEA enrichment score of NF-κB target genes differentially expressed in curdlan-stimulated wild-type cells compared with Vav1/2/3-deficient BMDCs.
Data are representative of at least three independent experiments. See also Figure S1.
As indicated above, the activation of the Card9/Bcl10/Malt1 complex not only regulates IKK activation but also induces Malt1 proteolytic activity (Jaworski et al., 2014). Consistently, stimulation of wild-type BMDCs with CLR ligands results in the rapid proteolytic processing of the Malt1 substrate RelB (Figures 4F and 4G; Figures S1E and S1F; Gewies et al., 2014, Hailfinger et al., 2011, Jaworski et al., 2014). Curdlan-induced RelB cleavage was completely defective in BMDCs from Malt1−/− mice and from mice with gene-targeted inactivation of the Malt1 paracaspase function (Malt1PM/−) (Gewies et al., 2014; Figure 4F; Figure S1E) and, therefore, can be used as a marker for CBM complex activation. Likewise, Malt1-mediated RelB cleavage upon C. albicans stimulation was also defective in Vav1/2/3 triple knockout cells (Figure 4G; Figure S1F), which, together with the defective IKK activation in Vav1/2/3−/− BMDCs, indicates that Vav proteins operate upstream of the Card9/Bcl10/Malt1 signalosome.
Finally, we studied the role of Vav proteins in CLR-induced gene expression in a global manner. To this end, we performed high-throughput cDNA sequencing (RNA sequencing [RNA-seq]) followed by gene set enrichment analysis (GSEA) of curdlan-stimulated and untreated BMDCs. In Dectin-1-stimulated wild-type BMDCs, we detected a significant enrichment of upregulated NF-κB-dependent transcripts compared with Vav1/2/3−/− cells (Figure 4H), which is consistent with the defective IKK activation described above. These results further validate Vav proteins as essential regulators of CLR-mediated NF-κB control.
A Human Vav3 Polymorphism Is Associated with Susceptibility to Candidemia
After identifying Vav proteins as integral regulators of the Dectin-1/Card9/NF-κB signaling cascade in murine cells, we were interested in the potential roles for Vav molecules in human antifungal immunity. Therefore, we investigated whether genetic variations linked to any of the VAV genes correlate with susceptibility to candidemia in patients by studying a previously described cohort of candidemia patients and an appropriate control group (Jaeger et al., 2015). Interestingly, the analysis of SNPs associated with VAV3 revealed a significant association with candidemia, with the significant SNPs distributed across the linkage disequilibrium (LD) region of the gene. The association does not extend beyond the LD region, and the strongest association was with the SNP rs4914950 (Figure 5A). The risk genotype of the SNP rs4914950 TT is increased from 10% (control population) to 18% (candidemia patients) (Figure 5B).
Figure 5.

A VAV3 Polymorphism Is Associated with Increased Susceptibility to Candidemia
(A) Polymorphisms associated with candidemia were investigated in a group of 227 patients with Candida-positive blood cultures and compared with 176 patients from the same clinical wards without infection. SNPs associated with the VAV3 locus revealed a significant association with candidemia, with the significant SNPs distributed across the LD region of the gene. The VAV3 gene region on chromosome 1 is shown. Using the SNAP server, the SNP of interest (rs4914950) (biggest orange diamond) was plotted with all annotated polymorphisms in this gene (other diamonds). R2 and recombination rate are indicated on the left and right y axis, respectively. Diamonds on the same horizontal line as the SNP of interest (biggest orange diamond) are in complete linkage disequilibrium (R2 = 1) with this SNP and have the same disease association values. Diamonds below this line have a lower LD value with the SNP of interest and, therefore, differ in the association with the disease. The table below shows the top 15 polymorphisms that are linked to rs4914950.
(B) Genotype frequencies for the rs4914950 SNP that showed the strongest association with candidemia. The table below shows the total numbers and percentages (in parentheses) of genotypes in Candida-infected (candidemia patients) and non-infected (controls) individuals. Statistical comparisons of genotype frequencies between the two groups were made using χ2 test.
Discussion
Using a complementary approach of immunology, molecular biology, and genetic studies, the results presented in this manuscript define an essential role for Vav proteins in CLR-mediated inflammatory responses and establish these molecules as essential signaling platforms that relay proximal events from Syk-coupled CLRs to the Card9 signaling complex for NF-κB control and antifungal defense (Dambuza and Brown, 2015, Osorio and Reis e Sousa, 2011, Roth and Ruland, 2013). Although individual Vav isoforms can compensate for each other, their combined absence results in a blockade of CLR-dependent NF-κB activation similar to Card9 deficiency, with Vav3 being the most important family member in this pathway in mice and, presumably, in humans.
When engaged by fungal particles, Syk-coupled CLRs activate various intracellular signaling pathways that regulate phagocytosis, microbicidal activities, and gene transcription. Using C. albicans as a clinically relevant model organism that is sensed by multiple CLRs and induces CLR signaling (Robinson et al., 2009, Taylor et al., 2007, Wells et al., 2008), we observed Vav activation downstream of Syk. In addition, using defined agonists of Dectin-1, Dectin-2, and Mincle, we established a general role for Vav proteins in CLR signaling in myeloid antigen-presenting cells. However, our results that CLR-mediated MAPK activation and inflammasome signaling are not impaired in Vav1/2/3 triple-deficient BMDCs reveal that Vav proteins only mediate specific CLR effector mechanisms. Recent studies have indicated the involvement of Vav1 and Vav3 in Dectin-1-mediated zymosan phagocytosis and reactive oxygen species (ROS) production in neutrophils and the recruitment of Vav1 to phagocytic cups for C. albicans engulfment by macrophages (Li et al., 2011, Strijbis et al., 2013). Although these effector mechanisms are important during innate immune responses, they are not regulated by the Card9 pathway (Drewniak et al., 2013, Goodridge et al., 2009, Gross et al., 2006). However, the Card9 signaling pathway appears to be the central mammalian host defense mechanism against fungi, given that multiple genetic studies in humans have recently identified that several Card9 loss-of-function defects are causative for several fungal diseases, including various types of mucocutaneous candidiasis; superficial, extensive, and deep dermatophytosis with Trichophyton spp.; subcutaneous phaeohyphomycosis; invasive Candida infections of the digestive tract and central nervous system; Candida endophthalmitis and osteomyelitis; and disseminated Exophiala disease of the liver, brain, and lung (Grumach et al., 2015, Pérez de Diego et al., 2015).
We assessed enzymatic Malt1 paracaspase activity as a direct functional marker for Card9 complex activity in Vav-deficient mice. Given that this activity was severely impaired in Vav1/2/3 triple-deficient BMDCs, our results establish Vav molecules as regulators of the Card9/Bcl10/Malt1 complex. Consistent with the essential function of a Vav/Card9-mediated mechanism for CLR-induced NF-κB activation, the NF-κB response is blocked in Vav1/2/3-deficient mice in response to C. albicans infection or selective CLR triggering, and Vav1/2/3-deficient animals exhibit dramatically impaired proinflammatory cytokine production in response to C. albicans infection and CLR stimulation in vitro and in vivo. Therefore, we propose a mechanistic model in which Syk is engaged by CLRs upon fungal sensing, thereby triggering the phosphorylation and activation of Vav proteins, which subsequently activate the Card9 complex for NF-κB control and antifungal gene transcription.
How Vav proteins are mechanistically coupled to Card9 control remains to be determined. We previously demonstrated that Card9 is phosphorylated by PKCδ at Thr231 to induce Card9 effector function. However, the activation of PKCδ is only partially impaired in Vav-deficient BMDCs, whereas IKK activation is almost completely defective. Similarly, in PKCδ-deficient cells, IKK phosphorylation is abolished, whereas phosphorylation of Vav1 is only slightly compromised, suggesting, together, that Vav and PKCδ operate at the same level upstream of Card9. Because Vav proteins can control cytoskeletal reorganization and serve as scaffolding platforms in other signaling systems (Acton et al., 2012, Bustelo, 2014, Spurrell et al., 2009, Tybulewicz, 2005), we speculate that Vav family members could bring Card9 into the vicinity of key upstream regulators such as PKCδ and potential, still unknown factors to allow Card9 activation. Moreover, because Vav1/2/3 do not only regulate CLR-induced Card9-NF-κB signaling but also control other cellular responses, such as phagocytosis and ROS production (Li et al., 2011, Strijbis et al., 2013), the precise molecular interdependence of these mechanisms should be defined in the future.
In accordance with our molecular model, similar to Card9 deficiency, a complete deficiency in all Vav isoforms results in a massive susceptibility to C. albicans infection. Likewise, a conditional deficiency of Syk only in dendritic cells also abolishes innate resistance to acute systemic C. albicans infection (Whitney et al., 2014), and Whitney et al. (2014) elegantly demonstrated that the early innate cytokine response is required to orchestrate innate anti-fungal functions, including neutrophil anti-fungal activity. Nevertheless, in our infection models presented here, it must be noted that the Vav mutant mice analyzed were germline Vav-deficient animals and that Vav proteins are not selectively expressed in Syk and Card9-containing innate immune cells, including dendritic cells, macrophages, and neutrophils, but are also expressed in lymphocytes, which signal through Card9-independent mechanisms (Gross et al., 2006, Hara et al., 2007). Still, the rapid death of Vav1/2/3 triple-deficient mice upon fungal infection indicates that an innate immune defect is indeed responsible for the observed phenotype. This hypothesis is also in line with the established roles of Vav1 and Vav3 in mediating phagocytosis and ROS production in macrophages and neutrophils upon fungal sensing (Li et al., 2011, Strijbis et al., 2013). Future studies with conditional Vav mutant mice will further dissect the individual roles of Vav signaling in dendritic cells, macrophages, and neutrophils as well as in adaptive immune subsets during complex fungal infection scenarios in vivo. In addition, in vivo results in individual Vav1-, 2-, and 3-deficient animals also revealed that, although there is functional redundancy among the three Vav family members in infection control, Vav3 is the single most important Vav family member for antifungal defense in mice. This finding is consistent with our results in patients who link a polymorphism in the VAV3 gene to susceptibility to Candida infection, suggesting that the function of Vav molecules is similar to Card9 function conserved between mice and men.
In addition to fungal components, CLRs also recognize structures present on mycobacteria (Ishikawa et al., 2009, Lobato-Pascual et al., 2013, Miyake et al., 2013, Schoenen et al., 2010), viruses such as dengue virus (Chen et al., 2008), and parasites such as Schistosoma mansoni (Ritter et al., 2010), as well as endogenous ligands under sterile inflammatory conditions (Roth and Ruland, 2013). We therefore believe that our findings have implications beyond antifungal immunity. Consistent with this hypothesis, Vav1/2/3−/− mice were almost completely impaired in systemic inflammatory responses upon innate immune stimulation with the mycobacterial pathogen-associated molecular patterns (PAMP) analog TDM, implying that, like Card9 (Dorhoi et al., 2010), Vav proteins might also be critical for innate anti-mycobacterial immune responses. In addition, accumulating evidence indicates important roles for CLR/Card9 signaling in inflammatory disorders. Several independent studies have shown an association between Card9 polymorphisms and ulcerative colitis and Crohn’s disease (Roth and Ruland, 2013), which are supported by a recent study in mice demonstrating a protective role for Card9 in intestinal inflammation (Sokol et al., 2013). Deep re-sequencing of inflammatory bowel disease genome-wide association study (GWAS) loci confirmed an association between Card9 and ulcerative colitis and Crohn’s disease (Beaudoin et al., 2013, Hong et al., 2016). Moreover, additional genome-wide association studies in humans have identified significant associations between CARD9 and VAV3 gene loci and immunoglobulin A (IgA) nephropathy (Kiryluk et al., 2014). In conjunction with the results presented here, these data suggest that the described Vav/Card9-dependent signaling mechanism could also play a role in sterile inflammatory diseases.
Experimental Procedures
Mice
Vav1−/− (Turner et al., 1997), Vav2−/− (Doody et al., 2001), Vav3−/− (Sauzeau et al., 2006), Card9−/− (Gross et al., 2006), Prkcd−/− (Leitges et al., 2001), Malt1−/− (Ruland et al., 2003), and Malt1PM/- (Gewies et al., 2014) mice were described previously. Vav double-deficient (Vav2/3−/−) and triple-deficient (Vav1/2/3−/−) mice were generated by intercrossings of Vav single knockout animals and were provided by X.R.B. and V.L.T.. All animal work was conducted in accordance with German federal animal protection laws and approved by the Institutional Animal Care and Use Committee at the Technical University of Munich. Animals were used at 8–16 weeks of age.
Cell Culture and Stimulation
BMDCs were derived from bone marrow as described previously (Gross et al., 2006) and stimulated with curdlan (Wako Pure Chemicals Industries), LPS (ultrapure, from Escherichia coli strain K12, InvivoGen), Pam3CSK4 (InvivoGen), Dectin-2 monoclonal antibody (AbD Serotec), Rat IgG2a isotype control (R&D Systems), TDB (Avanti Polar Lipids), or C. albicans strain SC5314 as indicated. For tyrosine kinase or proteasome inhibition, the Syk inhibitor R406 (Rigel), the Src kinase inhibitor PP2 or its inactive analog PP3 (both from Calbiochem), or MG132 (Sigma) were used. Cell culture reagents were purchased from Invitrogen, and fetal calf serum (FCS) was purchased from HyClone. Mouse recombinant granulocyte-macrophage colony-stimulating factor was purchased from PeproTech.
Cytokine Measurement
Cytokine levels were measured by ELISA (BD Biosciences and eBioscience) or by cytometric bead array (CBA; BD Biosciences) according to the manufacturer’s instructions.
Immunoprecipitation and Immunoblot Analysis
Cell lysates or cell supernatants were subjected to standard immunoblot analysis techniques as described previously (Strasser et al., 2012). Cytosolic and nuclear extracts were prepared as described previously (Ferch et al., 2007). For immunoprecipitation experiments, BMDCs were left unstimulated or stimulated with C. albicans as indicated and lysed in an NP-40-containing buffer. Lysates were pre-cleared by incubation with protein G Sepharose (GE Healthcare 4 Fast Flow) and then incubated with anti-phospho-tyrosine antibodies. Immune complexes were precipitated with protein G Sepharose and subjected to protein immunoblotting as indicated. Quantification of immunoblots was performed by densitometry using ImageJ software. Densitometry data were expressed relative to levels of appropriate loading controls and normalized to specific treatment conditions.
Statistical Analysis
Data were analyzed and graphed using Excel (Microsoft Office) and Prism (GraphPad). For comparison between two groups, two-tailed Student’s t test was used. Analysis across more than two groups on a single dataset was performed using one-way ANOVA and post hoc Tukey-Kramer test. Survival data were analyzed using the log-rank test. Genotype frequencies between controls and candidemia patients were compared using the χ2 test. The level of significance was defined as ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Author Contributions
S.R. and J.R. designed the study. S.R., H.B., M.J., K.N., and P.A.K. performed experiments. S.R., H.B., M.J., K.N., P.A.K., M.N., and J.R. analyzed the results. M.J., V.K., M.J., and M.N. conducted patient sample collection and clinical data analyses. A.Y. and B.H. performed bioinformatic analyses. S.R., M.J., and A.Y. generated the figures. C.P.C., L.V., M.M.M., V.L.T., and X.R.B. provided critical reagents. S.R. and J.R. wrote the paper.
Acknowledgments
We thank Mathias Heikenwälder for excellent help with histological analysis, Michael Leitges for providing Prkcd−/− mice, and Manuel Ritter and Nathalie Knies for assistance with in vivo experiments. This work was supported by research grants from the Helmholtz Alliance Preclinical Comprehensive Cancer Center, the DFG (SFB 1054 and RU 695/6-1), and an ERC Advanced Grant (FP7, grant agreement 322865) (to J.R.). The work of X.R.B. has been supported by grants from the Spanish Ministry of Economy and Competitiveness (RD12/0036/0002 and SAF2015-64556-R), Worldwide Cancer Research (14-1248), and the Ramón Areces Foundation. M.G.N. was supported by an ERC Consolidator Grant (310372). V.L.T. was supported by the Francis Crick Institute which receives its core funding from the MRC (FC001194), Cancer Research UK (FC001194) and the Wellcome Trust (FC001194).
Published: December 6, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and one figure and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.11.018.
Accession Numbers
The accession number for the gene expression profiling raw data reported in this paper is NCBI GEO: GSE83736.
Supplemental Information
References
- Acton S.E., Astarita J.L., Malhotra D., Lukacs-Kornek V., Franz B., Hess P.R., Jakus Z., Kuligowski M., Fletcher A.L., Elpek K.G. Podoplanin-rich stromal networks induce dendritic cell motility via activation of the C-type lectin receptor CLEC-2. Immunity. 2012;37:276–289. doi: 10.1016/j.immuni.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghazadeh B., Lowry W.E., Huang X.Y., Rosen M.K. Structural basis for relief of autoinhibition of the Dbl homology domain of proto-oncogene Vav by tyrosine phosphorylation. Cell. 2000;102:625–633. doi: 10.1016/s0092-8674(00)00085-4. [DOI] [PubMed] [Google Scholar]
- Beaudoin M., Goyette P., Boucher G., Lo K.S., Rivas M.A., Stevens C., Alikashani A., Ladouceur M., Ellinghaus D., Törkvist L., Quebec IBD Genetics Consortium. NIDDK IBD Genetics Consortium. International IBD Genetics Consortium Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet. 2013;9:e1003723. doi: 10.1371/journal.pgen.1003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieland J., Essig D., Jackson C., Frank D., Loebenberg D., Menzel F., Arnold B., DiDomenico B., Hare R. Comparison of pathogenesis and host immune responses to Candida glabrata and Candida albicans in systemically infected immunocompetent mice. Infect. Immun. 2001;69:5046–5055. doi: 10.1128/IAI.69.8.5046-5055.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G.D., Gordon S. Immune recognition. A new receptor for beta-glucans. Nature. 2001;413:36–37. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]
- Bustelo X.R. Vav family exchange factors: an integrated regulatory and functional view. Small GTPases. 2014;5:9. doi: 10.4161/21541248.2014.973757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.T., Lin Y.L., Huang M.T., Wu M.F., Cheng S.C., Lei H.Y., Lee C.K., Chiou T.W., Wong C.H., Hsieh S.L. CLEC5A is critical for dengue-virus-induced lethal disease. Nature. 2008;453:672–676. doi: 10.1038/nature07013. [DOI] [PubMed] [Google Scholar]
- Dambuza I.M., Brown G.D. C-type lectins in immunity: recent developments. Curr. Opin. Immunol. 2015;32:21–27. doi: 10.1016/j.coi.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doody G.M., Bell S.E., Vigorito E., Clayton E., McAdam S., Tooze R., Fernandez C., Lee I.J., Turner M. Signal transduction through Vav-2 participates in humoral immune responses and B cell maturation. Nat. Immunol. 2001;2:542–547. doi: 10.1038/88748. [DOI] [PubMed] [Google Scholar]
- Dorhoi A., Desel C., Yeremeev V., Pradl L., Brinkmann V., Mollenkopf H.J., Hanke K., Gross O., Ruland J., Kaufmann S.H. The adaptor molecule CARD9 is essential for tuberculosis control. J. Exp. Med. 2010;207:777–792. doi: 10.1084/jem.20090067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewniak A., Gazendam R.P., Tool A.T., van Houdt M., Jansen M.H., van Hamme J.L., van Leeuwen E.M., Roos D., Scalais E., de Beaufort C. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood. 2013;121:2385–2392. doi: 10.1182/blood-2012-08-450551. [DOI] [PubMed] [Google Scholar]
- Ferch U., zum Büschenfelde C.M., Gewies A., Wegener E., Rauser S., Peschel C., Krappmann D., Ruland J. MALT1 directs B cell receptor-induced canonical nuclear factor-kappaB signaling selectively to the c-Rel subunit. Nat. Immunol. 2007;8:984–991. doi: 10.1038/ni1493. [DOI] [PubMed] [Google Scholar]
- Ferwerda B., Ferwerda G., Plantinga T.S., Willment J.A., van Spriel A.B., Venselaar H., Elbers C.C., Johnson M.D., Cambi A., Huysamen C. Human dectin-1 deficiency and mucocutaneous fungal infections. N. Engl. J. Med. 2009;361:1760–1767. doi: 10.1056/NEJMoa0901053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa K., Miletic A.V., Alt F.W., Faccio R., Brown T., Hoog J., Fredericks J., Nishi S., Mildiner S., Moores S.L. Vav1/2/3-null mice define an essential role for Vav family proteins in lymphocyte development and activation but a differential requirement in MAPK signaling in T and B cells. J. Exp. Med. 2003;198:1595–1608. doi: 10.1084/jem.20030874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewies A., Gorka O., Bergmann H., Pechloff K., Petermann F., Jeltsch K.M., Rudelius M., Kriegsmann M., Weichert W., Horsch M. Uncoupling Malt1 threshold function from paracaspase activity results in destructive autoimmune inflammation. Cell Rep. 2014;9:1292–1305. doi: 10.1016/j.celrep.2014.10.044. [DOI] [PubMed] [Google Scholar]
- Glocker E.O., Hennigs A., Nabavi M., Schäffer A.A., Woellner C., Salzer U., Pfeifer D., Veelken H., Warnatz K., Tahami F. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med. 2009;361:1727–1735. doi: 10.1056/NEJMoa0810719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodridge H.S., Shimada T., Wolf A.J., Hsu Y.M., Becker C.A., Lin X., Underhill D.M. Differential use of CARD9 by dectin-1 in macrophages and dendritic cells. J. Immunol. 2009;182:1146–1154. doi: 10.4049/jimmunol.182.2.1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham D.B., Stephenson L.M., Lam S.K., Brim K., Lee H.M., Bautista J., Gilfillan S., Akilesh S., Fujikawa K., Swat W. An ITAM-signaling pathway controls cross-presentation of particulate but not soluble antigens in dendritic cells. J. Exp. Med. 2007;204:2889–2897. doi: 10.1084/jem.20071283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross O., Gewies A., Finger K., Schäfer M., Sparwasser T., Peschel C., Förster I., Ruland J. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature. 2006;442:651–656. doi: 10.1038/nature04926. [DOI] [PubMed] [Google Scholar]
- Gross O., Poeck H., Bscheider M., Dostert C., Hannesschläger N., Endres S., Hartmann G., Tardivel A., Schweighoffer E., Tybulewicz V. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459:433–436. doi: 10.1038/nature07965. [DOI] [PubMed] [Google Scholar]
- Grumach A.S., de Queiroz-Telles F., Migaud M., Lanternier F., Filho N.R., Palma S.M., Constantino-Silva R.N., Casanova J.L., Puel A. A homozygous CARD9 mutation in a Brazilian patient with deep dermatophytosis. J. Clin. Immunol. 2015;35:486–490. doi: 10.1007/s10875-015-0170-4. [DOI] [PubMed] [Google Scholar]
- Hailfinger S., Nogai H., Pelzer C., Jaworski M., Cabalzar K., Charton J.E., Guzzardi M., Décaillet C., Grau M., Dörken B. Malt1-dependent RelB cleavage promotes canonical NF-kappaB activation in lymphocytes and lymphoma cell lines. Proc. Natl. Acad. Sci. USA. 2011;108:14596–14601. doi: 10.1073/pnas.1105020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara H., Ishihara C., Takeuchi A., Imanishi T., Xue L., Morris S.W., Inui M., Takai T., Shibuya A., Saijo S. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat. Immunol. 2007;8:619–629. doi: 10.1038/ni1466. [DOI] [PubMed] [Google Scholar]
- Helft J., Böttcher J., Chakravarty P., Zelenay S., Huotari J., Schraml B.U., Goubau D., Reis e Sousa C. GM-CSF Mouse Bone Marrow Cultures Comprise a Heterogeneous Population of CD11c(+)MHCII(+) Macrophages and Dendritic Cells. Immunity. 2015;42:1197–1211. doi: 10.1016/j.immuni.2015.05.018. [DOI] [PubMed] [Google Scholar]
- Hise A.G., Tomalka J., Ganesan S., Patel K., Hall B.A., Brown G.D., Fitzgerald K.A. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe. 2009;5:487–497. doi: 10.1016/j.chom.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S.N., Park C., Park S.J., Lee C.K., Ye B.D., Kim Y.S., Lee S., Chae J., Kim J.I., Kim Y.H., IBD Study Group of the Korean Association for the Study of Intestinal Diseases (KASID) Deep resequencing of 131 Crohn’s disease associated genes in pooled DNA confirmed three reported variants and identified eight novel variants. Gut. 2016;65:788–796. doi: 10.1136/gutjnl-2014-308617. [DOI] [PubMed] [Google Scholar]
- Ishikawa E., Ishikawa T., Morita Y.S., Toyonaga K., Yamada H., Takeuchi O., Kinoshita T., Akira S., Yoshikai Y., Yamasaki S. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J. Exp. Med. 2009;206:2879–2888. doi: 10.1084/jem.20091750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger M., van der Lee R., Cheng S.C., Johnson M.D., Kumar V., Ng A., Plantinga T.S., Smeekens S.P., Oosting M., Wang X. The RIG-I-like helicase receptor MDA5 (IFIH1) is involved in the host defense against Candida infections. Eur. J. Clin. Microbiol. Infect. Dis. 2015;34:963–974. doi: 10.1007/s10096-014-2309-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski M., Marsland B.J., Gehrig J., Held W., Favre S., Luther S.A., Perroud M., Golshayan D., Gaide O., Thome M. Malt1 protease inactivation efficiently dampens immune responses but causes spontaneous autoimmunity. EMBO J. 2014;33:2765–2781. doi: 10.15252/embj.201488987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhingran A., Mar K.B., Kumasaka D.K., Knoblaugh S.E., Ngo L.Y., Segal B.H., Iwakura Y., Lowell C.A., Hamerman J.A., Lin X., Hohl T.M. Tracing conidial fate and measuring host cell antifungal activity using a reporter of microbial viability in the lung. Cell Rep. 2012;2:1762–1773. doi: 10.1016/j.celrep.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia X.M., Tang B., Zhu L.L., Liu Y.H., Zhao X.Q., Gorjestani S., Hsu Y.M., Yang L., Guan J.H., Xu G.T., Lin X. CARD9 mediates Dectin-1-induced ERK activation by linking Ras-GRF1 to H-Ras for antifungal immunity. J. Exp. Med. 2014;211:2307–2321. doi: 10.1084/jem.20132349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerrigan A.M., Brown G.D. Syk-coupled C-type lectins in immunity. Trends Immunol. 2011;32:151–156. doi: 10.1016/j.it.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryluk K., Li Y., Scolari F., Sanna-Cherchi S., Choi M., Verbitsky M., Fasel D., Lata S., Prakash S., Shapiro S. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat. Genet. 2014;46:1187–1196. doi: 10.1038/ng.3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeibundGut-Landmann S., Gross O., Robinson M.J., Osorio F., Slack E.C., Tsoni S.V., Schweighoffer E., Tybulewicz V., Brown G.D., Ruland J., Reis e Sousa C. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- Leitges M., Mayr M., Braun U., Mayr U., Li C., Pfister G., Ghaffari-Tabrizi N., Baier G., Hu Y., Xu Q. Exacerbated vein graft arteriosclerosis in protein kinase Cdelta-null mice. J. Clin. Invest. 2001;108:1505–1512. doi: 10.1172/JCI12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Utomo A., Cullere X., Choi M.M., Milner D.A., Jr., Venkatesh D., Yun S.H., Mayadas T.N. The β-glucan receptor Dectin-1 activates the integrin Mac-1 in neutrophils via Vav protein signaling to promote Candida albicans clearance. Cell Host Microbe. 2011;10:603–615. doi: 10.1016/j.chom.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobato-Pascual A., Saether P.C., Fossum S., Dissen E., Daws M.R. Mincle, the receptor for mycobacterial cord factor, forms a functional receptor complex with MCL and FcεRI-γ. Eur. J. Immunol. 2013;43:3167–3174. doi: 10.1002/eji.201343752. [DOI] [PubMed] [Google Scholar]
- Miyake Y., Toyonaga K., Mori D., Kakuta S., Hoshino Y., Oyamada A., Yamada H., Ono K., Suyama M., Iwakura Y. C-type lectin MCL is an FcRγ-coupled receptor that mediates the adjuvanticity of mycobacterial cord factor. Immunity. 2013;38:1050–1062. doi: 10.1016/j.immuni.2013.03.010. [DOI] [PubMed] [Google Scholar]
- Mócsai A., Ruland J., Tybulewicz V.L. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat. Rev. Immunol. 2010;10:387–402. doi: 10.1038/nri2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio F., Reis e Sousa C. Myeloid C-type lectin receptors in pathogen recognition and host defense. Immunity. 2011;34:651–664. doi: 10.1016/j.immuni.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Pérez de Diego R., Sánchez-Ramón S., López-Collazo E., Martínez-Barricarte R., Cubillos-Zapata C., Ferreira Cerdán A., Casanova J.L., Puel A. Genetic errors of the human caspase recruitment domain-B-cell lymphoma 10-mucosa-associated lymphoid tissue lymphoma-translocation gene 1 (CBM) complex: Molecular, immunologic, and clinical heterogeneity. J. Allergy Clin. Immunol. 2015;136:1139–1149. doi: 10.1016/j.jaci.2015.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter M., Gross O., Kays S., Ruland J., Nimmerjahn F., Saijo S., Tschopp J., Layland L.E., Prazeres da Costa C. Schistosoma mansoni triggers Dectin-2, which activates the Nlrp3 inflammasome and alters adaptive immune responses. Proc. Natl. Acad. Sci. USA. 2010;107:20459–20464. doi: 10.1073/pnas.1010337107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.J., Osorio F., Rosas M., Freitas R.P., Schweighoffer E., Gross O., Verbeek J.S., Ruland J., Tybulewicz V., Brown G.D. Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J. Exp. Med. 2009;206:2037–2051. doi: 10.1084/jem.20082818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S., Ruland J. Caspase recruitment domain-containing protein 9 signaling in innate immunity and inflammation. Trends Immunol. 2013;34:243–250. doi: 10.1016/j.it.2013.02.006. [DOI] [PubMed] [Google Scholar]
- Ruland J., Duncan G.S., Wakeham A., Mak T.W. Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity. 2003;19:749–758. doi: 10.1016/s1074-7613(03)00293-0. [DOI] [PubMed] [Google Scholar]
- Saijo S., Fujikado N., Furuta T., Chung S.H., Kotaki H., Seki K., Sudo K., Akira S., Adachi Y., Ohno N. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat. Immunol. 2007;8:39–46. doi: 10.1038/ni1425. [DOI] [PubMed] [Google Scholar]
- Saijo S., Ikeda S., Yamabe K., Kakuta S., Ishigame H., Akitsu A., Fujikado N., Kusaka T., Kubo S., Chung S.H. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity. 2010;32:681–691. doi: 10.1016/j.immuni.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Sancho D., Reis e Sousa C. Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu. Rev. Immunol. 2012;30:491–529. doi: 10.1146/annurev-immunol-031210-101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K., Yang X.L., Yudate T., Chung J.S., Wu J., Luby-Phelps K., Kimberly R.P., Underhill D., Cruz P.D., Jr., Ariizumi K. Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor gamma chain to induce innate immune responses. J. Biol. Chem. 2006;281:38854–38866. doi: 10.1074/jbc.M606542200. [DOI] [PubMed] [Google Scholar]
- Sauzeau V., Sevilla M.A., Rivas-Elena J.V., de Alava E., Montero M.J., López-Novoa J.M., Bustelo X.R. Vav3 proto-oncogene deficiency leads to sympathetic hyperactivity and cardiovascular dysfunction. Nat. Med. 2006;12:841–845. doi: 10.1038/nm1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenen H., Bodendorfer B., Hitchens K., Manzanero S., Werninghaus K., Nimmerjahn F., Agger E.M., Stenger S., Andersen P., Ruland J. Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J. Immunol. 2010;184:2756–2760. doi: 10.4049/jimmunol.0904013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah V.B., Ozment-Skelton T.R., Williams D.L., Keshvara L. Vav1 and PI3K are required for phagocytosis of beta-glucan and subsequent superoxide generation by microglia. Mol. Immunol. 2009;46:1845–1853. doi: 10.1016/j.molimm.2009.01.014. [DOI] [PubMed] [Google Scholar]
- Sokol H., Conway K.L., Zhang M., Choi M., Morin B., Cao Z., Villablanca E.J., Li C., Wijmenga C., Yun S.H. Card9 mediates intestinal epithelial cell restitution, T-helper 17 responses, and control of bacterial infection in mice. Gastroenterology. 2013;145:591–601.e3. doi: 10.1053/j.gastro.2013.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurrell D.R., Luckashenak N.A., Minney D.C., Chaplin A., Penninger J.M., Liwski R.S., Clements J.L., West K.A. Vav1 regulates the migration and adhesion of dendritic cells. J. Immunol. 2009;183:310–318. doi: 10.4049/jimmunol.0802096. [DOI] [PubMed] [Google Scholar]
- Strasser D., Neumann K., Bergmann H., Marakalala M.J., Guler R., Rojowska A., Hopfner K.P., Brombacher F., Urlaub H., Baier G. Syk kinase-coupled C-type lectin receptors engage protein kinase C-δ to elicit Card9 adaptor-mediated innate immunity. Immunity. 2012;36:32–42. doi: 10.1016/j.immuni.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strijbis K., Tafesse F.G., Fairn G.D., Witte M.D., Dougan S.K., Watson N., Spooner E., Esteban A., Vyas V.K., Fink G.R. Bruton’s Tyrosine Kinase (BTK) and Vav1 contribute to Dectin1-dependent phagocytosis of Candida albicans in macrophages. PLoS Pathog. 2013;9:e1003446. doi: 10.1371/journal.ppat.1003446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O., Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Taylor P.R., Tsoni S.V., Willment J.A., Dennehy K.M., Rosas M., Findon H., Haynes K., Steele C., Botto M., Gordon S., Brown G.D. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat. Immunol. 2007;8:31–38. doi: 10.1038/ni1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner M., Mee P.J., Walters A.E., Quinn M.E., Mellor A.L., Zamoyska R., Tybulewicz V.L. A requirement for the Rho-family GTP exchange factor Vav in positive and negative selection of thymocytes. Immunity. 1997;7:451–460. doi: 10.1016/s1074-7613(00)80367-2. [DOI] [PubMed] [Google Scholar]
- Tybulewicz V.L. Vav-family proteins in T-cell signalling. Curr. Opin. Immunol. 2005;17:267–274. doi: 10.1016/j.coi.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S., Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- Vonk A.G., Netea M.G., van Krieken J.H., Iwakura Y., van der Meer J.W., Kullberg B.J. Endogenous interleukin (IL)-1 alpha and IL-1 beta are crucial for host defense against disseminated candidiasis. J. Infect. Dis. 2006;193:1419–1426. doi: 10.1086/503363. [DOI] [PubMed] [Google Scholar]
- Wells C.A., Salvage-Jones J.A., Li X., Hitchens K., Butcher S., Murray R.Z., Beckhouse A.G., Lo Y.L., Manzanero S., Cobbold C. The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J. Immunol. 2008;180:7404–7413. doi: 10.4049/jimmunol.180.11.7404. [DOI] [PubMed] [Google Scholar]
- Whitney P.G., Bär E., Osorio F., Rogers N.C., Schraml B.U., Deddouche S., LeibundGut-Landmann S., Reis e Sousa C. Syk signaling in dendritic cells orchestrates innate resistance to systemic fungal infection. PLoS Pathog. 2014;10:e1004276. doi: 10.1371/journal.ppat.1004276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S., Huo J., Lee K.G., Kurosaki T., Lam K.P. Phospholipase Cgamma2 is critical for Dectin-1-mediated Ca2+ flux and cytokine production in dendritic cells. J. Biol. Chem. 2009;284:7038–7046. doi: 10.1074/jbc.M806650200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H., Nakamura Y., Sato K., Takahashi Y., Nomura T., Miyasaka T., Ishii K., Hara H., Yamamoto N., Kanno E. Defect of CARD9 leads to impaired accumulation of gamma interferon-producing memory phenotype T cells in lungs and increased susceptibility to pulmonary infection with Cryptococcus neoformans. Infect. Immun. 2014;82:1606–1615. doi: 10.1128/IAI.01089-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.