

Abstract
Investigations during the last 10 years have revealed a group of disorders mediated by antibodies against ion channels and synaptic receptors, which cause both neurologic and psychiatric symptoms. In this review, I discuss the process of discovery and immunologic triggers of these disorders, and use anti-NMDA receptor encephalitis to emphasize the importance of understanding the underlying physiopathologic mechanisms in those diseases. A better knowledge of these mechanisms reveals points of convergence with other disorders (e.g., schizophrenia), suggests treatment strategies beyond immunotherapy, and is helping us understand how memories are formed and retrieved.
For many years, the only known disorders that associated with autoantibodies against ion channels or receptors were myasthenia gravis and the Lambert-Eaton myasthenic syndrome (LEMS). In these predominantly B-cell autoimmune disorders, the antibodies target the acetylcholine receptor or voltage-gated calcium channels, resulting in reversible physiopathologic alterations that mediate the patients' symptoms.1 In contrast, the only known CNS disorders that associated with neuronal autoantibodies were the paraneoplastic syndromes. In these syndromes the antibodies target cytoplasmic or nuclear proteins, the pathogenic mechanisms are believed to be mediated by cytotoxic T-cells (instead of B cells), and the symptoms are frequently irreversible.2 Keeping in mind these 2 different groups of disorders and the large number of patients with encephalopathies for which the cause was (and still is3) unknown, some of us wondered whether a subgroup of CNS diseases could be mediated by antibodies against cell surface or synaptic proteins in a manner similar to the myasthenic syndromes. In my experience, the answer started to be revealed in December 2003 after we saw a young woman with encephalitis of unknown cause who had been in the intensive care unit for several weeks. She was brought to the hospital for the acute onset of change in behavior and prominent psychiatric symptoms that progressed rapidly to unconsciousness and central hypoventilation. All diagnostic investigations had been negative except for the presence of a small ovarian teratoma that was believed to be unrelated to her disease and, owing to her poor clinical condition, it was not removed. She was given empiric immunotherapy and eventually recovered. The triad of encephalitis with prominent psychiatric symptoms and hypoventilation in a young woman with an ovarian teratoma and the recovery with immunotherapy were crucial in linking her clinical picture to that of another 3 young women with an identical syndrome who also had ovarian teratomas. These 3 young woman had been seen in other centers by colleagues who were also intrigued by the clinical picture and had sent serum and CSF samples to my laboratory (1 of the patients, who was ventilator-dependent, died a few months later). Despite the remarkable syndrome resemblance among the 4 patients, and the clinical and CSF features suggesting an immune-mediated encephalitis, their serum and CSF were negative for all neuronal antibodies known at that time. It took 6 months to optimize the tissue processing4 until it showed a unique pattern of neuropil reactivity with the patients' samples.5 The identity of the antigen (NMDA receptor [NMDAR]) was obtained 2 years later (nowadays, the entire discovery process would take just a few weeks).6 As soon as the syndrome and a diagnostic test became available, the number of referrals grew quickly, leading to the diagnosis of 100 patients in less than 1 year.7 This was a large number for a clinical investigator used to the scarcity of patients with paraneoplastic syndromes. Even more surprising was the reason of the referrals: instead of being asked to search for antibodies with the hope of finding an answer for a mystery syndrome, the physicians, having recognized the similarity of the patient's syndrome with that reported in anti-NMDAR encephalitis, were asking for confirmatory antibody studies.
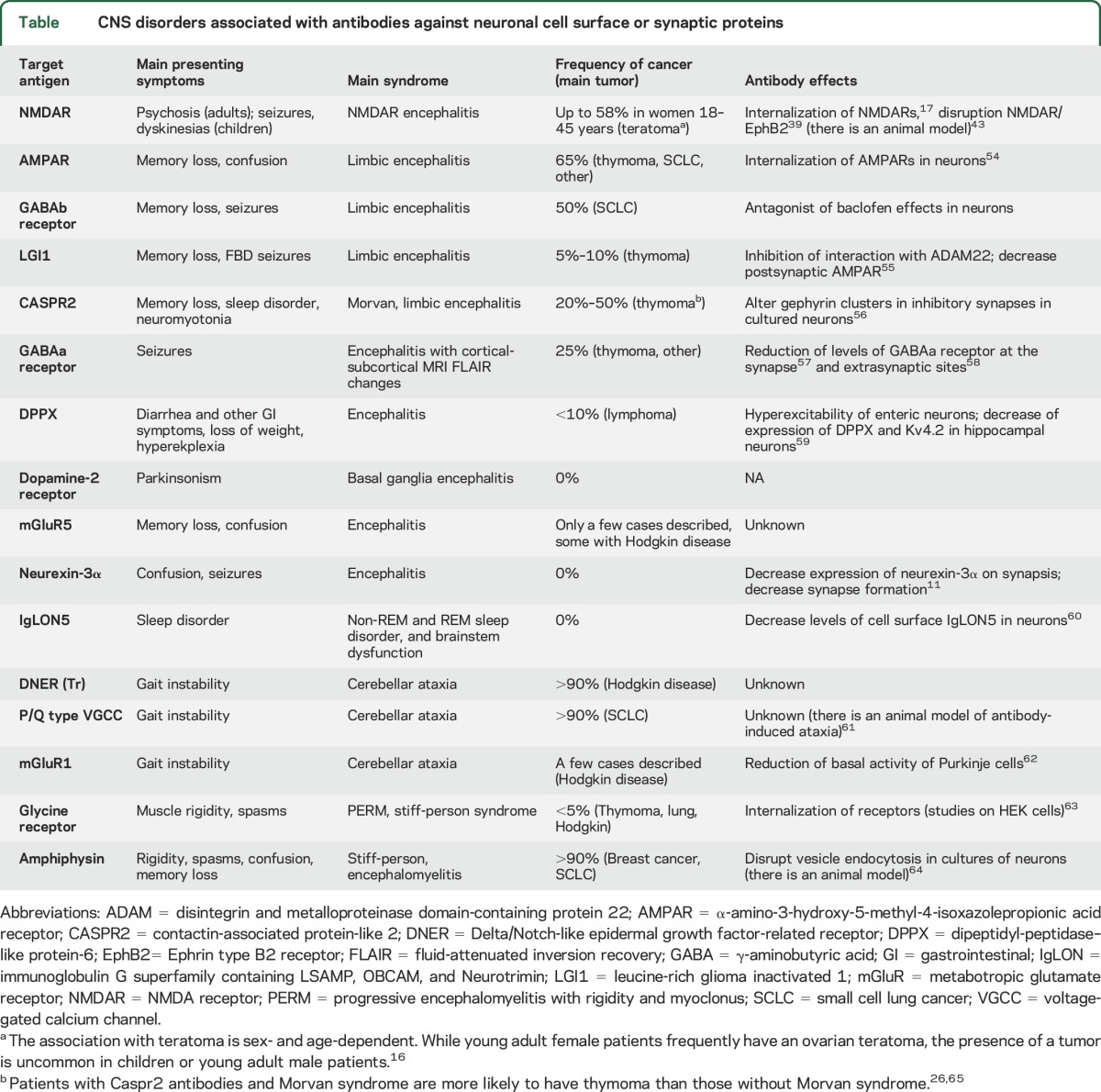
There are currently 16 known disorders with immunoglobulin G (IgG) autoantibodies against cell surface or synaptic proteins, 12 of them manifesting as autoimmune encephalitis (table). The identification of these disorders has changed the diagnostic and treatment approach to many neurologic and psychiatric syndromes that were previously considered idiopathic or not even suspected to be immune-mediated (a position paper on the clinical diagnosis of autoimmune encephalitis was published recently8). These clinical advances sent us back to the laboratory with the goal of investigating the physiopathologic mechanisms that underlie these disorders. Overall, these studies illustrate how lessons learned from the bedside have unique power to enlighten translational research. Using anti-NMDAR encephalitis to exemplify the emerging field of autoimmune neurology, I discuss recent advances in autoimmune encephalitis with potential implications for therapies and, more broadly, for a better understanding of how the brain works.
Table.
CNS disorders associated with antibodies against neuronal cell surface or synaptic proteins
THE PROCESS OF DISCOVERY
The discovery of autoimmune disorders of the synapse usually starts as described above, with a patient who has an unclassifiable syndrome and all routine investigations for a cause are negative.9 In this setting, the presence of one or more features suggesting an immune-mediated mechanism, including rapid onset of symptoms, CSF or MRI findings compatible with an inflammatory process, identification of immunologic triggers (such as tumors or viruses), or clinical improvement after empiric immunotherapy, leads to testing for the presence of autoantibodies in serum or CSF. Figure 1 shows a characteristic pattern of brain neuropil reactivity of the autoantibodies of one such patient suggesting that the target is located on the cell surface or synapse. The cell surface location of the epitopes can be confirmed by examining the reactivity of patients' antibodies with live cultured neurons (figure 1C), and the identity of the antigen can then be obtained by immunoprecipitation and mass spectrometry.9 This approach served to identify most of the autoantigens shown in the table and figure 2.10 Since 2007, when the first target of autoimmune encephalitis was characterized molecularly (NMDAR),6 the rate of discovery has been almost 1 new autoantigen per year, the most recent being neurexin-3α.11
Figure 1. Reactivity of an autoantibody against a neuronal cell-surface antigen compared with the reactivity of an autoantibody against an intracellular antigen.

Section of rat hippocampus immunostained with an autoantibody against a synaptic NMDA receptor (NMDAR) from a patient with anti-NMDAR encephalitis (A, B) compared with the reactivity of an autoantibody against an intracellular neuronal protein (Hu) from a patient with paraneoplastic encephalitis (D, E). The boxed areas are shown in detail in (B) and (E). The demonstration that the target antigen is on the neuronal cell surface is provided by immunocytochemistry with live neurons (obtained from dissociated rat hippocampus): the NMDAR antibodies show intense reactivity with the cognate receptor on the cell surface (C) whereas the Hu antibodies do not show reactivity because they do not reach the intracellular target (F). Scale bars: A, B = 500 μm; C, D = 20 μm; E, F = 10 μm. Adapted from Lancaster et al.,66 with permission.
Figure 2. Brain reactivity of autoantibodies against neuronal cell surface or synaptic proteins.

Sagittal and coronal sections of rat brain immunostained with 9 different autoantibodies from patients with different types of autoimmune encephalitis. For each antibody, the pattern of neuropil immunostaining is highly suggestive that the antigen is on the neuronal cell surface (figure 1). Scale bars: all panels = 2 mm. AMPAR = α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; CASPR2 = contactin-associated protein-like 2; DPPX = dipeptidyl-peptidase–like protein-6; GABA = γ-aminobutyric acid; LGI1 = leucine-rich glioma inactivated 1; mGluR = metabotropic glutamate receptor; NMDAR = NMDA receptor.
A HUMAN MODEL OF IMMUNE DEPLETION OF NMDAR
Patients with anti-NMDAR encephalitis usually develop a predictable syndrome with stages of symptom progression and resolution that resemble those caused by noncompetitive antagonists of the NMDAR such as ketamine or phencyclidine (for a review on this topic, see reference 12). At low doses, NMDAR antagonists cause illusionary perceptions, ideas of reference, and paranoid thoughts, along with impaired performance on tasks requiring executive function.13 This low-dose drug effect would correspond to milder forms of anti-NMDAR encephalitis, best represented at disease onset, or months later during recovery when psychiatric symptoms similar to those at presentation may occur.14 Higher doses of NMDAR antagonists manifest with psychosis, agitation, memory disturbance, stereotyped or repetitive motor behaviors, and decreased responsiveness to pain,15 and at very high doses patients develop dissociative anesthesia, profound unresponsiveness with catatonia, and coma.15 These high-dose drug effects would correspond to the severe stages of anti-NMDAR encephalitis, characterized by dyskinesias, paradoxical responses (e.g., resisting eye opening but unresponsiveness to pain stimuli), catatonia, autonomic dysfunction, hypoventilation, and coma.16 When cultures of dissociated rat hippocampal neurons are exposed to patients' antibodies, the effects on the density of NMDAR correlate strongly and inversely with CSF NMDAR antibody titers,17 decreasing the availability of these receptors in a manner similar to when they are bound by noncompetitive antagonists.
TRIGGERS OF SYNAPTIC AUTOIMMUNITY: TUMORS AND VIRUSES
Two outstanding questions in anti-NMDAR and other autoimmune encephalitis refer to the triggers of the disease and the prolonged duration of symptoms. Figure 3 shows several facts and hypotheses related to the triggers of anti-NMDAR encephalitis. Approximately 50% of young women with this disease have an ovarian teratoma that contains nervous tissue16; in children and men, the frequency of tumors is lower and the histology different (e.g., older men and women have carcinomas instead of teratomas).16,18,19 It is postulated that antigen released by apoptotic tumor cells is taken up by antigen-presenting cells, and then processed and presented to the immunologic system at the regional lymph nodes where memory B cells are generated and the antibody production by plasma cells is initiated. After crossing the blood-brain barrier (BBB) or reaching the brain through the choroid plexus, the memory B cells would undergo restimulation, antigen-driven affinity maturation, clonal expansion, and maturation into antibody-producing plasma cells, the presence of which has been demonstrated in studies of brain tissue from patients.20 Plasma cells are long-lived (for several months to years21), refractory to the most frequently used immunotherapies (plasma exchange, IV immunoglobulin, rituximab), and protected by the BBB from systemically administered drugs. These plasma cells lead to a prolonged synthesis of antibodies within the CNS, demonstrated by a relatively high concentration of antibodies in CSF compared to serum (intrathecal synthesis).7,18 CSF antibodies remain detectable for at least as long as the patients have active disease or substantial neurologic deficits.16,22 In some patients, the CSF and serum antibodies are detectable (albeit at lower titers) several months or years after clinical recovery.23
Figure 3. Immunologic triggers in anti–NMDA receptor (NMDAR) encephalitis.

The figure describes 2 known immunologic triggers of anti-NMDAR encephalitis: a tumor (usually ovarian teratoma) and herpes simplex encephalitis. The underlying multistep process between either of these triggers and the CNS production of antibodies is unknown, but the latter has been confirmed in studies demonstrating intrathecal synthesis of antibodies and CNS plasma cells. It is postulated that NMDAR expressed in nervous tissue contained in the tumor, or released by viral-induced neuronal destruction, is either in soluble form or loaded in antigen-presenting cells transported to the regional lymph nodes (e.g., pelvic-abdominal in case of ovarian teratoma or deep cervical lymph nodes in case of herpes encephalitis) where it is presented to the immunologic system. Naive B cells exposed to NMDAR by antigen-presenting cells, and with cooperation of CD4+ T cells, become antigen-experienced memory B cells, differentiating into antibody-producing plasma cells. Memory B cells reach the brain crossing the BBB or the choroidal plexus. In the brain, these B cells undergo restimulation, antigen-driven affinity maturation, clonal expansion, and differentiation into antibody-producing plasma cells. Of note, in approximately 50% of the patients the immunologic trigger of the disease is unknown. The model of tumor as a trigger of autoimmune encephalitis would be similar for other types of encephalitis (table); in tumors other than teratoma (e.g., without nervous tissue within the tumor itself), the antigen is aberrantly expressed by the neoplastic cells. APC = antigen-presenting cell; TFH = follicular helper T cell.
The contribution to the disease of systemic NMDAR antibodies is unclear. Although antibodies may potentially reach the brain through a disrupted BBB, or at lower amounts through an intact BBB (serum: CSF IgG concentration 500:1), high intrathecal synthesis of antibodies,7,24 presence of CNS plasma cells,20 little clinical evidence of BBB disruption, and low or absent serum antibody levels in some patients with severe deficits23 suggest a minor contribution of serum antibodies compared to the intra-CNS produced antibodies. Moreover, CSF clonally expanded plasma cells that produce disease-relevant NMDAR antibodies were recently identified by different investigators using recombinant antibody technology25 (Goebels et al., 2016, unpublished). In cultured neurons, these antibodies show pathogenic effects,25 confirming previously reported findings using CSF NMDAR antibodies from patients with the disorder.17 In addition, the infusion of one of these antibodies in the cerebroventricular system of mice produced a dramatic reduction of memory and synaptic NMDAR that were similar to those observed in a model (described below) in which patients' CSF antibodies were infused (Dalmau and Goebels, 2016, unpublished).
Autoimmune encephalitis with antibodies against other neuronal cell surface proteins may also associate with tumors. The frequency of this association and tumor histology vary according to the antibody (table). It is likely that the tumor-related triggering mechanisms are similar to those discussed above (figure 3). Tumors of the thymus are not involved in anti-NMDAR encephalitis but they often occur with other disorders (e.g., Caspr2,26,27 AMPA receptor antibody–associated encephalitis28). In these cases, an alteration of the immunoregulatory function of the thymus per se may facilitate the autoimmune response, as occurs in myasthenia gravis.29
An infrequent trigger of autoimmune encephalitis is herpes simplex encephalitis (HSE).30 Approximately 20% of patients with HSE develop antibodies against the NMDAR, which in children associate with change of behavior and choreoathetosis,30,31 previously described as choreoathetosis post-HSE.32,33 In contrast, in adults the predominant symptoms are psychiatric and cognitive, and despite being highly disruptive they are often misdiagnosed as sequelae of the viral infection.34 Patients with anti-NMDAR encephalitis post-HSE are less responsive to treatment than those with other triggers (teratoma or unknown) and their CSF may show additional autoantibodies.30 A fascinating aspect of this complication is that the triggering mechanism is initiated by herpes simplex virus within the CNS providing a milieu of neuronal degeneration and extensive inflammatory infiltrates that lead to antigen presentation (most likely at the deep cervical nodes) and generation of antigen-experienced memory B cells and CNS plasma cells (figure 3). Preliminary data from a study in which patients with HSE are prospectively followed confirm that approximately 20% develop NMDAR antibodies34; the antibodies are usually identified first in the CSF and later in the serum. Antibodies against the GABAa receptor (M. Spatola, MD, and J. Dalmau, 2016, unpublished), dopamine receptor,31 and other unknown antigens (often coexisting with NMDAR antibodies)30 may also occur in some patients. It is unclear whether infections other than HSE are similarly able to initiate autoimmune encephalitis. Research on how viral encephalitis leads to synaptic autoimmunity may help to answer the fundamental question of how and where B cells are activated, reach the CNS, and differentiate into plasma cells.
The use of immune checkpoint inhibitors to enhance antitumor immune responses in patients with melanoma or other cancers,35 and less frequently other medical interventions causing immune dysregulation, such as organ transplantation,36 may associate with autoimmune encephalitis.
ANTIBODY EFFECTS ON SYNAPSES, PLASTICITY, AND BEHAVIOR: IMPLICATIONS FOR THERAPIES
Brain biopsy or autopsy studies of patients with anti-NMDAR encephalitis showed mild inflammatory infiltrates, absent or mild neuronal loss, microglial activation, and deposits of IgG without complement.7 In addition, the pattern of brain IgG immunostaining resembled that observed with rat brain immunostained with patients' antibodies. These findings and the substantial clinical recovery of most patients after immunotherapy suggested that the disorder resulted from neuronal dysfunction caused by the antibodies rather than by irreversible neuronal degeneration as usually occurs with cytotoxic T-cell-mediated disorders.37
Findings using cultured neurons showed that the antibodies caused selective crosslinking and internalization of NMDARs in both excitatory and inhibitory hippocampal neurons, and produced a reduction of NMDAR-mediated synaptic currents.17,38 These effects correlated with the antibody titers, were reversed after the antibodies were removed, and did not affect cell survival.17 At the synapse, the antibodies disrupt the interaction between NMDAR and Ephrin type B2 receptor (EphB2).39 The EphB2 receptor is a member of a family of receptor tyrosine kinases that modulate long-term potentiation (LTP) through their interaction with NMDAR and stabilization and clustering of this receptor in the postsynaptic membrane.40,41 The disruption of this interaction results in displacement of NMDAR to extrasynaptic sites followed by internalization.39 These findings suggested that activation of EphB2 with a soluble form of its ligand (ephrin B2) could antagonize the pathogenicity of patients' antibodies.
Data from these studies showed that anti-NMDAR encephalitis fulfilled most of the Witebsky criteria for antibody-mediated disease42 but the transfer of symptoms to animals was missing. This was recently accomplished in a model of chronic cerebroventricular infusion of patients’ CSF antibodies.43 Mice underwent placement of ventricular catheters connected to osmotic pumps that continuously delivered patients' or control CSF for 14 days. Animals infused with patients' but not control CSF developed progressive memory deficits and anhedonic and depressive-like behaviors (figure 4). The memory deficits gradually worsened during the infusion of CSF antibodies and all symptoms gradually resolved after the infusion stopped. Brain tissue studies showed progressive binding of human NMDAR antibodies to neurons, mainly in the hippocampus, along with a progressive decrease of the density of synaptic NMDAR clusters. These effects occurred in parallel with memory and other behavioral deficits and improved after the infusion of patients' antibodies stopped, leading to restoration of NMDAR levels and reversibility of the symptoms (figure 4).43 Together, these findings showed a definite link between antibody-mediated reduction of NMDAR and memory and behavioral deficits.
Figure 4. Cerebroventricular infusion of patients' NMDA receptor (NMDAR) antibodies causes memory and behavioral deficits in mice.

Mice underwent placement of bilateral ventricular catheters connected to subcutaneous osmotic pumps that during 14 days infused CSF from patients with NMDAR antibodies or control CSF. At several timepoints during and after the infusion, animals underwent behavioral testing or were killed to determine the binding and effects of the antibodies. (A) Animals infused with patients' CSF antibodies developed severe memory impairment demonstrated by the novel object recognition test (gray circles, n = 8 mice) compared with animals infused with control CSF (white circles, n = 10 mice). Data presented as standard error of the mean. Significance assessed by 2-way analysis of variance with α-error of 0.05 and post hoc testing with Sidak-Holm adjustment, ***p < 0.001. This memory deficit along with anhedonia-like behavior (not shown here) were accompanied by a progressive accumulation of brain-bound NMDAR antibodies (predominantly in the hippocampus) that was maximal on day 18 (B, C). Quantitative analysis of the NMDAR clusters in 15 areas of the hippocampus (only 1 per brain is shown in [D], which correspond to the small squares [a] and [b] in [C], day 18) demonstrates a significant reduction of density of total cell-surface NMDAR clusters and synaptic NMDAR clusters (defined by colocalization with PSD95) in animals infused with patients' CSF antibodies (D). (E) Quantitative analysis of synaptic NMDAR cluster density in the 2 groups of animals (dark gray: infused with patients' CSF; light gray: infused with control CSF; 5 animals per group). Data presented as standard error of the mean. Significance assessed by 2-way analysis of variance with α-error of 0.05 (*) and post hoc testing with Sidak-Holm adjustment ($), *$p < 0.05. Note that all antibody effects including memory deficits, accumulation of brain-bound human NMDAR antibodies, and reduction of levels of NMDAR became maximal on day 18, and gradually recovered 10 days after stopping the antibody infusion. Scale bars: B = 2 mm; C = 200 μm. Reproduced from Planagumà et al.43 with permission.
This model has recently been used to further investigate the effects of patient antibodies on synaptic plasticity and determine whether ephrin B2 (the ligand of EphB2) could antagonize the pathogenicity of the antibodies. Mice were infused with patient or control CSF with or without ephrin B2, and subsequently assessed for symptoms, presence of brain-bound antibodies, and alteration of the levels of NMDAR. Synaptic plasticity was determined in acute brain sections; the Schaffer collateral pathway was stimulated and the field excitatory postsynaptic potentials were recorded in the CA1 region of the hippocampus, showing that patient NMDAR antibodies caused a striking impairment of LTP.44 The coinfusion of ephrin B2 with patients' antibodies prevented the pathogenic effects of the latter in all the investigated paradigms used to assess memory, depressive-like behavior, density of synaptic NMDAR and EphB2, and long-term synaptic plasticity.44 Preliminary studies showed that animals infused with patients' antibodies had an increase of dendritic spine density in the CA1 region of the hippocampus that was not observed in mice coinfused with ephrin B2 (Geis et al., 2016, unpublished). This structural remodeling may represent a compensatory mechanism similar to that reported in models of NMDAR hypofunction mediated by ketamine.45 Overall, these studies demonstrate that patients' antibodies cause a prominent impairment of hippocampal networks involved in long-term synaptic plasticity, and suggest a potential role of ephrin B2 analogs in the treatment of patients with anti-NMDAR encephalitis (discussed below).
The approach of combining immunotherapy with drugs antagonizing the effects of autoantibodies would be similar to that used in myasthenia gravis and LEMS, where a better understanding of the alterations produced by the autoantibodies led to the use of drugs (e.g., anticholinesterases, 3,4-diaminopyridine) to antagonize their effects. One envisions for anti-NMDAR and other autoimmune encephalitis a similar strategy combining immunotherapy with small molecules able to cross the BBB and antagonize the antibody effects. This would potentially result in faster control of symptoms and shorter process of recovery.
A similar approach was recently postulated for encephalitis associated with antibodies against the inhibitory GABAb receptor.46 In contrast to NMDAR, the GABAb receptor is not a channel but a metabotropic receptor that has robust inhibitory effects on synaptic transmission. The receptor has 2 subunits: the B1 that binds GABA and the B2 that activates G proteins intracellularly and modulates ion channels and cell signaling pathways. In cultures of dissociated rat hippocampal neurons, these form synapses with each other and spontaneously produce synaptic currents and action potentials that are powerfully attenuated by the application of baclofen. Treatment of cultures of neurons with patients' antibodies (which bind the B1 subunit) abrogates the inhibitory effects of baclofen on excitability. In preliminary studies, it was found that an agonist of the B2 subunit that bypasses the effects of patients' antibodies was able to decrease antibody-mediated hyperexcitability, suggesting a strategy to control the refractory seizures and other symptoms of the disease (Lancaster et al., 2016, unpublished).
Our knowledge of the pathogenic effects of other neuronal cell surface autoantibodies is more limited, due in part to their recent discovery. For at least 11 of them, the pathogenicity has been suggested in studies using cultured neurons or heterologous cells expressing the antigens (table).
ANTI-NMDAR ENCEPHALITIS AND THE NMDAR HYPOFUNCTION MODEL OF SCHIZOPHRENIA: TWO MECHANISMS CONVERGING ON NMDAR NETWORKS
To fully understand the clinical manifestations of antibody-mediated disorders of the synapse, it is necessary to consider the function of the target antigens in brain synaptic networks or circuitry. When this is done, it reveals points of convergence with other disorders, such as schizophrenia. One of the leading theories of schizophrenia is based on data showing hypofunction of NMDAR, which may actually underlie the hyperdopaminergic state typical of this disorder.47 Antagonists of the NMDAR result in positive (hallucinations, delusions, hyperactivity) and negative (decreased motivation, flat affect, deficits of memory and learning) symptoms that resemble not only anti-NMDAR encephalitis, but also schizophrenia.13,15 In addition, there is electrophysiologic, neuroimaging, genetic, and postmortem evidence that patients with schizophrenia have hypofunction of the NMDAR system, as shown in animal models of psychoses (reviewed in reference 12). Figure 5 is a simplified representation of NMDAR-related cognitive networks including 3 nodes: subiculum of the hippocampus (involved in declarative memory), dorsolateral prefrontal cortex (executive function, working memory), and ventral tegmental region (facilitates episodic and working memory, motivation).12 In this network, a decrease of availability of NMDAR (genetic, pharmacologic, or autoimmune) affecting the inhibitory neurons at each of these nodes has important functional consequences, leading to increased pyramidal firing and downstream signaling, lessening the normal inhibitory tone over the ventral tegmental region dopaminergic neurons. As a result, there is an increased production of dopamine, typically observed in psychoses, accompanied by impaired working memory related to abnormal functioning of NMDAR-bearing parvalbumin-positive GABAergic interneurons of the prefrontal cortex.47,48 An increase in pyramidal activity has also been demonstrated with NMDAR ablation restricted to the frontal pyramidal neurons.49 Consequently, a decrease of NMDAR function does not implicate a decrease of extracellular glutamate; on the contrary, as a result of a decrease of GABAergic inhibitory activity or signaling pathways modulating NMDAR, there is an increase of extracellular glutamate, as suggested in schizophrenia47,50 and demonstrated in the frontal cortex and hippocampus of rats after local administration of patients' NMDAR antibodies.51
Figure 5. NMDA receptor (NMDAR)–related cognitive networks and effect of NMDAR hypofunction.

This simplified diagram shows some of the cognitive networks that are affected by NMDAR hypofunction. There are 3 critical nodes: (1) the subiculum, effector region of the hippocampus; (2) the dorsolateral prefrontal cortex (DLPFC), which supports executive function and working memory; and (3) the ventral tegmental area (VTA), which facilitates episodic and working memory, and motivation. (A) At each of these nodes, glutamatergic excitatory inputs (mainly from lateral hypothalamus) to pyramidal or dopaminergic neurons provide collaterals to NMDAR-bearing GABAergic interneurons, which in turn inhibit excessive pyramidal firing. GABAergic interneurons in the subiculum and DLPFC are parvalbumin positive (PV), and in the DLPFC are characterized as the fast-spiking cells, which are involved in the generation of synchronous gamma oscillations. In the VTA, GABAergic interneurons also contain NMDAR and are glutamic acid decarboxylase (GAD)–positive. Dopaminergic VTA neurons (DN) project to the nucleus accumbens (NAc), DLPFC, and hippocampus (not shown). The NAc inhibits the globus pallidus medialis (GPm), which in turn tonically inhibits VTA dopaminergic neurons. (B) NMDAR hypofunction (one of the leading theories of schizophrenia) is associated with increased pyramidal firing, which increases the inhibitory activity of the NAc over the GPm and lessens the inhibitory tone over the VTA dopaminergic neurons. This leads to an increased production of dopamine, as found in psychoses, along with impaired working memory related to the altered function of PV interneurons (adapted from Masdeu et al.,12 with permission).
FUTURE STUDIES: AUTOANTIBODIES TO EXPLORE BRAIN FUNCTION
Different approaches to model NMDAR hypofunction (genetic, pharmacologic, or immunologic) and differences in the region where the NMDAR are predominantly depleted (general, corticolimbic, or hippocampal-parahippocampal) influence the resulting phenotype, which also varies among animal species.15,43 In the mouse model of cerebroventricular infusion of NMDAR antibodies, the region predominantly affected is the hippocampus, leading to impairment of long-term synaptic plasticity and memory deficits.43,44 These findings are not surprising considering the critical role of NMDAR in synaptic plasticity, learning, and memory.52 We can now take advantage of this human-related model (instead of genetic or pharmacologic blockade of the receptor) to determine the role of hippocampal NMDAR networks in the formation and retrieval of long-term memories, and how these networks change during periods of health and disease (e.g., while animals have symptoms and during the process of recovery). Technological advances, such as calcium imaging visualization of the firing of large ensembles of neurons via mini-microscopes implanted in the hippocampus, allow assessment of hippocampal networks in freely behaving mice performing memory tasks (data not shown).53 Ongoing studies applying these techniques to the indicated mouse model will also improve our understanding of the NMDAR-related circuit alterations underlying some of the symptoms observed in patients with anti-NMDAR encephalitis, such as their profound amnesia of the disease.
In just a decade, we have learned of a new category of diseases mediated by antibodies against cell surface and synaptic proteins. Syndrome discovery continues, and as a result the diagnosis and treatment of these patients have improved. The surprise has been in the large number of disorders revealed in a short period of time, suggesting that there are still others to be discovered. Additional excitement comes with the recognition that many of the immune targets are proteins or receptors with critical roles in memory, behavior, cognition, and psychosis. As shown here, we are learning the physiopathology of these disorders, and this will lead to therapies and to a better understanding of how the brain works.
ACKNOWLEDGMENT
The author thanks Eric Lancaster, MD, PhD (Department of Neurology, University of Pennsylvania, Philadelphia), Christian Geis, MD, PhD (Department of Neurology, Jena University Hospital, Jena, Germany), and Norbert Goebels, MD, PhD (Department Neurology, University of Düsseldorf, Germany), for providing useful comments and unpublished data.
GLOSSARY
- BBB
blood–brain barrier
- EphB2
Ephrin type B2 receptor
- FLAIR
fluid-attenuated inversion recovery
- HSE
herpes simplex encephalitis
- IgG
immunoglobulin G
- LEMS
Lambert-Eaton myasthenic syndrome
- LTP
long-term potentiation
- NMDAR
NMDA receptor
AUTHOR CONTRIBUTIONS
Josep O. Dalmau: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data, study supervision, obtaining funding.
STUDY FUNDING
This study was supported in part by NIH RO1NS077851; Fondo de Investigaciones Sanitarias, FEDER (FIS 14/00203) and CIBERER, Instituto de Salud Carlos III, Madrid, Spain; and Fundació CELLEX.
DISCLOSURE
J. Dalmau has a research grant from Euroimmun and receives royalties from patents for the use of Ma2, NMDA receptor, and GABAb receptor as autoantibody tests. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Drachman DB, Adams RN, Josifek LF, Self SG. Functional activities of autoantibodies to acetylcholine receptors and the clinical severity of myasthenia gravis. N Engl J Med 1982;307:769–775. [DOI] [PubMed] [Google Scholar]
- 2.Darnell RB, Posner JB. Paraneoplastic syndromes involving the nervous system. N Engl J Med 2003;349:1543–1554. [DOI] [PubMed] [Google Scholar]
- 3.Vora NM, Holman RC, Mehal JM, Steiner CA, Blanton J, Sejvar J. Burden of encephalitis-associated hospitalizations in the United States, 1998-2010. Neurology 2014;82:443–451. [DOI] [PubMed] [Google Scholar]
- 4.Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain 2005;128:1764–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vitaliani R, Mason W, Ances B, Zwerdling T, Jiang Z, Dalmau J. Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol 2005;58:594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalmau J, Tuzun E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007;61:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008;7:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 2016;15:391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009;65:424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lancaster E, Dalmau J. Neuronal autoantigens-pathogenesis, associated disorders and antibody testing. Nat Rev Neurol 2012;8:380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gresa-Arribas N, Planaguma J, Petit-Pedrol M, et al. Human neurexin-3alpha antibodies associate with encephalitis and alter synapse development. Neurology 2016;86:2235–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Masdeu JC, Dalmau J, Berman KF. NMDA receptor internalization by autoantibodies: a reversible mechanism underlying psychosis? Trends Neurosci Epub 2016 Mar 22. doi: 10.1016/j.tins.2016.02.006. [DOI] [PMC free article] [PubMed]
- 13.Krystal JH, Karper LP, Seibyl JP, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 1994;51:199–214. [DOI] [PubMed] [Google Scholar]
- 14.Kayser MS, Titulaer MJ, Gresa-Arribas N, Dalmau J. Frequency and characteristics of isolated psychiatric episodes in anti-N-methyl-d-aspartate receptor encephalitis. JAMA Neurol 2013;70:1133–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 1991;148:1301–1308. [DOI] [PubMed] [Google Scholar]
- 16.Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes EG, Peng X, Gleichman AJ, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci 2010;30:5866–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010;133:1655–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Titulaer MJ, McCracken L, Gabilondo I, et al. Late-onset anti-NMDA receptor encephalitis. Neurology 2013;81:1058–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez-Hernandez E, Horvath J, Shiloh-Malawsky Y, Sangha N, Martinez-Lage M, Dalmau J. Analysis of complement and plasma cells in the brain of patients with anti-NMDAR encephalitis. Neurology 2011;77:589–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bortnick A, Allman D. What is and what should always have been: long-lived plasma cells induced by T cell-independent antigens. J Immunol 2013;190:5913–5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas A, Rauschkolb P, Gresa-Arribas N, Schned A, Dalmau JO, Fadul CE. Anti-N-methyl-D-aspartate receptor encephalitis: a patient with refractory illness after 25 months of intensive immunotherapy. JAMA Neurol 2013;70:1566–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol 2014;13:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pruss H, Dalmau J, Harms L, et al. Retrospective analysis of NMDA receptor antibodies in encephalitis of unknown origin. Neurology 2010;75:1735–1739. [DOI] [PubMed] [Google Scholar]
- 25.Kreye J, Wenke NK, Chayka M, et al. Human cerebrospinal fluid monoclonal N-methyl-D-aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain 2016;139:2641–2652. [DOI] [PubMed] [Google Scholar]
- 26.Irani SR, Pettingill P, Kleopa KA, et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol 2012;72:241–255. [DOI] [PubMed] [Google Scholar]
- 27.van Sonderen A, Arino H, Petit-Pedrol M, et al. The clinical spectrum of caspr2-antibody associated disease. Neurology 2016;87:521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoftberger R, van Sonderen A, Leypoldt F, et al. Encephalitis and AMPA receptor antibodies: novel findings in a case series of 22 patients. Neurology 2015;84:2403–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marx A, Muller-Hermelink HK, Strobel P. The role of thymomas in the development of myasthenia gravis. Ann NY Acad Sci 2003;998:223–236. [DOI] [PubMed] [Google Scholar]
- 30.Armangue T, Leypoldt F, Malaga I, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol 2014;75:317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohammad SS, Sinclair K, Pillai S, et al. Herpes simplex encephalitis relapse with chorea is associated with autoantibodies to N-methyl-D-aspartate receptor or dopamine-2 receptor. Mov Disord 2014;29:117–122. [DOI] [PubMed] [Google Scholar]
- 32.Wang HS, Kuo MF, Huang SC, Chou ML. Choreoathetosis as an initial sign of relapsing of herpes simplex encephalitis. Pediatr Neurol 1994;11:341–345. [DOI] [PubMed] [Google Scholar]
- 33.De Tiege X, Rozenberg F, Des Portes V, et al. Herpes simplex encephalitis relapses in children: differentiation of two neurologic entities. Neurology 2003;61:241–243. [DOI] [PubMed] [Google Scholar]
- 34.Armangue T, Moris G, Cantarin-Extremera V, et al. Autoimmune post-herpes simplex encephalitis of adults and teenagers. Neurology 2015;85:1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams TJ, Benavides DR, Patrice KA, et al. Association of autoimmune encephalitis with combined immune checkpoint inhibitor treatment for metastatic cancer. JAMA Neurol 2016;73:928–933. [DOI] [PubMed] [Google Scholar]
- 36.Zhao CZ, Erickson J, Dalmau J. Clinical reasoning: agitation and psychosis in a patient after renal transplantation. Neurology 2012;79:e41–e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bien CG, Vincent A, Barnett MH, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain 2012;135:1622–1638. [DOI] [PubMed] [Google Scholar]
- 38.Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol 2014;76:108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mikasova L, De Rossi P, Bouchet D, et al. Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain 2012;135:1606–1621. [DOI] [PubMed] [Google Scholar]
- 40.Dalva MB, Takasu MA, Lin MZ, et al. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell 2000;103:945–956. [DOI] [PubMed] [Google Scholar]
- 41.Henderson JT, Georgiou J, Jia Z, et al. The receptor tyrosine kinase EphB2 regulates NMDA-dependent synaptic function. Neuron 2001;32:1041–1056. [DOI] [PubMed] [Google Scholar]
- 42.Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky's postulates revisited). Immunol Today 1993;14:426–430. [DOI] [PubMed] [Google Scholar]
- 43.Planagumà J, Leypoldt F, Mannara F, et al. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain 2015;138:94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Planagumà J, Haselmann H, Mannara F, et al. Ephrin-B2 prevents NMDA receptor antibody effects on memory and neuroplasticity. Ann Neurol 2016;80:388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zunszain PA, Horowitz MA, Cattaneo A, Lupi MM, Pariante CM. Ketamine: synaptogenesis, immunomodulation and glycogen synthase kinase-3 as underlying mechanisms of its antidepressant properties. Mol Psychiatry 2013;18:1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lancaster E, Lai M, Peng X, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010;9:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 2012;37:4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lisman JE, Coyle JT, Green RW, et al. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci 2008;31:234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tatard-Leitman VM, Jutzeler CR, Suh J, et al. Pyramidal cell selective ablation of N-methyl-D-aspartate receptor 1 causes increase in cellular and network excitability. Biol Psychiatry 2015;77:556–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poels EM, Kegeles LS, Kantrowitz JT, et al. Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Mol Psychiatry 2014;19:20–29. [DOI] [PubMed] [Google Scholar]
- 51.Manto M, Dalmau J, Didelot A, Rogemond V, Honnorat J. In vivo effects of antibodies from patients with anti-NMDA receptor encephalitis: further evidence of synaptic glutamatergic dysfunction. Orphanet J Rare Dis 2010;5:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kentros C, Hargreaves E, Hawkins RD, Kandel ER, Shapiro M, Muller RV. Abolition of long-term stability of new hippocampal place cell maps by NMDA receptor blockade. Science 1998;280:2121–2126. [DOI] [PubMed] [Google Scholar]
- 53.Ziv Y, Burns LD, Cocker ED, et al. Long-term dynamics of CA1 hippocampal place codes. Nat Neurosci 2013;16:264–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peng X, Hughes EG, Moscato EH, Parsons TD, Dalmau J, Balice-Gordon RJ. Cellular plasticity induced by anti-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor encephalitis antibodies. Ann Neurol 2015;77:381–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ohkawa T, Fukata Y, Yamasaki M, et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci 2013;33:18161–18174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pinatel D, Hivert B, Boucraut J, et al. Inhibitory axons are targeted in hippocampal cell culture by anti-Caspr2 autoantibodies associated with limbic encephalitis. Front Cell Neurosci 2015;9:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petit-Pedrol M, Armangue T, Peng X, et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol 2014;13:276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ohkawa T, Satake S, Yokoi N, et al. Identification and characterization of GABA(A) receptor autoantibodies in autoimmune encephalitis. J Neurosci 2014;34:8151–8163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Piepgras J, Holtje M, Michel K, et al. Anti-DPPX encephalitis: pathogenic effects of antibodies on gut and brain neurons. Neurology 2015;85:890–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sabater L, Planagumà J, Dalmau J, Graus F. Cellular investigations with human antibodies associated with anti-IgLON5 syndrome. J Neuroinflammation (in press 2016). [DOI] [PMC free article] [PubMed]
- 61.Martin-Garcia E, Mannara F, Gutierrez-Cuesta J, et al. Intrathecal injection of P/Q type voltage-gated calcium channel antibodies from paraneoplastic cerebellar degeneration cause ataxia in mice. J Neuroimmunol 2013;261:53–59. [DOI] [PubMed] [Google Scholar]
- 62.Sillevis SP, Kinoshita A, De Leeuw B, et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med 2000;342:21–27. [DOI] [PubMed] [Google Scholar]
- 63.Carvajal-Gonzalez A, Leite MI, Waters P, et al. Glycine receptor antibodies in PERM and related syndromes: characteristics, clinical features and outcomes. Brain 2014;137:2178–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Werner C, Pauli M, Doose S, et al. Human autoantibodies to amphiphysin induce defective presynaptic vesicle dynamics and composition. Brain 2016;139:365–379. [DOI] [PubMed] [Google Scholar]
- 65.Lancaster E, Huijbers MG, Bar V, et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol 2011;69:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011;77:179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]