

Abstract
Objective:
To evaluate the long-term benefit and safety of everolimus for the treatment of medically refractory epilepsy in patients with tuberous sclerosis complex (TSC).
Methods:
Everolimus was titrated over 4 weeks and continued an additional 8 weeks in a prospective, open-label, phase I/II clinical trial design. Participants demonstrating initial benefit continued treatment until study completion (48 months). The primary endpoint was percentage of patients with a ≥50% reduction in seizure frequency compared to baseline. Secondary endpoints assessed absolute seizure frequency, adverse events (AEs), behavior, and quality of life.
Results:
Of the 20 participants who completed the initial study phase, 18 continued extended treatment. Fourteen of 18 (78%) participants completed the study, all but 1 of whom reported ≥50% reduction in seizure frequency at 48 months. All participants reported at least 1 AE, the vast majority (94%) of which were graded mild or moderate severity. Improvements in behavior and quality of life were also observed, but failed to achieve statistical significance at 48 months.
Conclusions:
Improved seizure control was maintained for 4 years in the majority of patients with TSC with medically refractory epilepsy treated with everolimus. Long-term treatment with everolimus is safe and well-tolerated in this population. Everolimus may be a therapeutic option for refractory epilepsy in TSC.
Classification of evidence:
This study provides Class IV evidence that for patients with TSC with medically refractory epilepsy everolimus improves seizure control.
More than 80% of individuals with tuberous sclerosis complex (TSC) develop epilepsy, usually within the first year of life.1 Epilepsy in an estimated third of these individuals is refractory to conventional anticonvulsant medications,2 heightening the urgency for development of effective treatments. TSC1 and TSC2, the causative genes for TSC, regulate the protein kinase mammalian target of rapamycin complex 1 (mTORC1). Pharmacologic inhibitors of mTORC1 are now approved to treat multiple clinical aspects of TSC, including subependymal giant cell astrocytomas (SEGA),3,4 renal angiomyolipomas,5 and pulmonary lymphangioleiomyomatosis.6 mTORC1 inhibitors also have been reported to be effective for treatment of topical angiofibromas and cardiac rhabdomyomas in TSC.7,8
Preclinical studies in TSC mouse models demonstrate that mTORC1 inhibitors effectively prevent seizures.9,10 TSC patients in early (open-label) clinical trials to treat SEGA with everolimus reported reduced frequency of daily seizures and increased rates of seizure freedom,4,11–13 but seizure benefit could not be demonstrated in more recent (placebo-controlled, randomized) phase III SEGA clinical trials.3 Therefore, clinical trials to directly assess the safety and benefit of everolimus treatment of epilepsy, independent of SEGA, are needed.
Previously, we reported that everolimus treatment for as little as 12 weeks is well-tolerated, reduces seizure frequency, and improves behavior and quality of life in patients with TSC with medically refractory epilepsy.14 Subsequent studies have yielded similar results.15,16 Epilepsy is a disabling, longer-term manifestation of TSC, yet long-term outcomes have yet to be assessed. Here we report the 4-year, final analysis of the first prospective human clinical trial for patients with TSC with refractory epilepsy treated with everolimus.
METHODS
Study design and procedures.
The design of our prospective, nonrandomized, open-label study has been described previously.14 To be eligible for participation, participants were at least 2 years of age, with a confirmed diagnosis of TSC,17 and averaging 2 or more seizures per week in the month prior to enrollment despite being adequately trialed on at least 2 approved anticonvulsant therapies. Prior treatment with an mTOR inhibitor was not allowed.
The initial phase (main study) consisted of 16 weeks, divided into 4 weeks baseline observation prior to initiating treatment with everolimus, followed by a 12-week treatment period where everolimus was titrated to target ranges for 4 weeks followed by maintenance therapy for an additional 8 weeks. Based on seizure diaries, patients demonstrating benefit and tolerability during the main study were eligible to continue treatment up to an additional 48 months. Participants began treatment with everolimus between May 2010 and August 2011, with the last participant completing treatment in December 2015.
Everolimus was initiated at 5 mg/m2/d, rounded to the nearest 2.5 milligram, and dosed once daily. A blood trough level was measured 2 and 4 weeks after initiation, and adjusted for a target range between 5 and 15 ng/mL. During the extension phase, everolimus blood trough levels were measured every 3 months. Participants experiencing significant side effects or demonstrated benefit at doses lower than the target range could continue lower dosing at clinician discretion. Everolimus treatment during extension continued as long as no significant toxicity occurred and benefit was demonstrated at least 1 of every 3 months prior to scheduled quarterly assessments.
During the initial 4-week observation, 4-week titration, and 8-week maintenance treatment periods, concurrent anticonvulsant treatments were not allowed to change. Rescue medication use was monitored and if used more than 7 days cumulatively during any month, the participant was considered treatment failure and exited from the study. During the extension phase, clinicians could adjust concurrent seizure medications and treatments, with the rationale that patients with improved seizure control resulting from everolimus treatment could potentially reduce/wean therapies no longer needed. Furthermore, we anticipated that dosage/medication adjustments over the 4-year extension treatment phase were likely to be clinically necessary as a result of patient growth, changes in comorbid medical conditions, hormonal changes, or illness.
Seizure frequency was evaluated using patient diaries capturing seizure description and number of occurrences daily. Seizure diaries alone were used to determine eligibility to enter the extension phase and continue everolimus treatment up to an additional 48 months. Loss of efficacy was defined as <25% reduction in seizure frequency for 3 consecutive months of treatment with everolimus. In the final analysis, events recorded in seizure diaries found later be nonepileptic in etiology when 24-hour video EEGs were analyzed were not counted in determining seizure frequency at baseline or follow-up or to classify participants as responders (≥50% reduction in seizure frequency), partial responders (25%–49% reduction), or nonresponders (<25% reduction) in the final analysis.
Endpoints.
The primary efficacy endpoint was the percentage of patients demonstrating clinical response, defined as a 50% or greater reduction in seizure frequency compared to the pretreatment 4-week observation period (responders). Secondary efficacy endpoints included the relative reduction in number of seizures. Quality of life and behavioral assessments were performed every 6 months during the extension phase, using the Quality of Life for Children with Epilepsy Parent Form (QOLCE)18 and the Nisonger Child Behavioral Rating Form (NCBRF),19 respectively. Adverse events (AEs) were collected continuously throughout the study, using Common Terminology Criteria for Adverse Events (CTCAE) version 3.0. Each AE was categorized by type, severity, intervention (if indicated), outcome, and relation to everolimus treatment. By protocol, any infections were considered related to treatment in this open-label study design, consistent with earlier similar studies in TSC.4,11,14,20 EEG conducted at baseline and at the end of the initial treatment phase was not continued during the extension phase.
Statistical analysis and methodology.
Statistical significance between individual time points was determined using paired t test or Wilcoxon signed rank test, depending on distribution. For repeated measures over time, one-way repeated-measures analysis of variance (RM-ANOVA) (Holm-Sidak) or RM-ANOVA on ranks (Dunnett) was used, again depending on distribution. Pearson correlation coefficient was employed to compare efficacy to everolimus dosing and trough levels. No corrections were made for missing observations during the extension phase. All statistical analysis utilized SigmaPlot 13.0 (Systat Software, San Jose, CA). To more readily align comparative analysis by month and better match clinical practice, epochs were calculated differently in the current analysis (calendar months consisting of 28–31 days each) than in the original study report (fixed length of 28 days each).14
Standard protocol approvals, registrations, and patient consents.
Study participants were recruited from Cincinnati Children's Hospital Medical Center (n = 10) and Texas Children's Hospital (n = 10). The institutional review board at each institution approved the protocol prior to initiation and annually thereafter. An independent Data Safety Monitoring Board reviewed study progress at regular intervals. Written informed consent was obtained for each participant. The clinical trial was divided into 2 phases. The study was registered at clinicaltrials.gov (NCT01070316).
Classification of evidence.
This interventional study provides Class IV evidence that prolonged everolimus treatment up to 4 years is effective for treating medically refractory epilepsy in patients with TSC. The primary research question was “Does adjunctive treatment with everolimus (target trough level 5–15 ng/mL) in patients with TSC with medically refractory epilepsy reduce seizure frequency long term?” Fourteen of 18 participants who initiated long-term treatment continued treatment to the final time point of 48 months, 93% of whom reported ≥50% reduction in seizures compared to baseline. Average seizure frequency at 48 months in this group was reduced by 77% (95% confidence interval −48% to −108%; p = 0.002).
RESULTS
Twenty-three participants were enrolled, 20 of whom were eligible to begin initial treatment during the 12-week main study period. The median age at enrollment was 8.0 years (range 2.0–21.3 years), with equal distribution of male and female participants (10:10). Genetic testing had been performed in 7 participants, all of which revealed mutation in TSC2. All patients had failed multiple different antiseizure regimens prior to enrollment and nearly all were on multiple conventional antiepileptic drugs (AEDs) at time of treatment initiation (median 2 concurrent AEDs, range 1–4). Four had undergone focal epilepsy surgery resection previously; 5 had vagus nerve stimulators. During the 4-week baseline observation period, the median total number of seizures was 34.5 (range 8–297). Seizures were characterized by their clinical ictal semiology, with the knowledge that seizures in TSC are almost exclusively of partial onset, often with rapid bilateral synchrony and at times poorly localizable on scalp EEG. Clinical focal-onset seizures were the most common (69% of seizures overall). At the end of the main study phase, seizures overall were reduced by 69% compared to baseline. The largest reduction was observed with focal-onset seizures (83% reduction, 13 of 15 participants). By comparison, clinical generalized seizures were reduced by 41% (6 of 9 participants).
Twelve participants were responders (≥50% reduction) and 4 participants were near-responders (25%–49% reduction) at the end of the initial treatment phase. Two additional participants qualified as near-responders for extended treatment based on seizure diaries at the time of eligibility determination, but were reclassified as nonresponders after later EEG analysis revealed that some events reported by caregivers as seizures were nonepileptic in etiology. In total, 18 participants entered the extension phase and continued treatment with everolimus (figure 1).
Figure 1. Patient flow and response to extended treatment with everolimus over 48 months.

Left column shows the number of participants who initiated treatment, when individual participants discontinued treatment, and reason for treatment discontinuation during the extension phase of the study. Middle column shows how many participants entered and continued treatment each year based on meeting treatment eligibility criteria based on patient-reported seizure frequency (seizure diaries). Right column shows the number of patients on treatment at each time point classified as responders (≥50% reduction compared to baseline) or near-responders (25%–49% reduction compared to baseline) in which EEG-confirmed nonepileptic events reported as seizures were excluded.
Entering extension, the median number of seizures (any type) per month was 7 (range 0–46). Again, focal-onset seizures were more common, present in 11 participants, whereas generalized-onset seizures were present in 7 participants, nonepileptic events in 4 participants, and 1 participant each reporting epileptic spasms and seizures that could not be classified by description as to whether onset was focal or generalized. Focal-onset seizures were most likely to be complex partial in nature, with or without secondary generalization, whereas seizures with generalized onset were most likely to be tonic in nature.
Fourteen of the 18 participants (78%) entering extension completed the full 4 years of continued treatment with everolimus (figure 1). Combined continuous treatment duration (main and extension phases combined) was 50.9 months (range 2.7–53.9). Three participants lost efficacy and by protocol were required to discontinue treatment during extension, at months 10, 13, and 36. Another participant withdrew consent after 40 months of treatment. Everolimus continued to reduce seizure frequency, compared to baseline, throughout the extension phase. Responders (≥50% reduction in seizures) accounted for 76%, 75%, 80%, and 93% of those continuing treatment after 12, 24, 36, and 48 months, respectively. Partial responders accounted for 6%, 13%, 13%, and 0% at the same timepoints. Overall, the average number of seizures per month was reduced by 72%–81% throughout the extension phase (figure 2). Whereas no participants were able to go a whole month without seizures at baseline, several participants reported no seizures at 12 months (n = 5), 24 months (n = 4), 36 months (n = 7), and 48 months (n = 5). However, significant individual patient variability was observed over time, including occasional exacerbations in seizure frequency despite continued treatment with everolimus (figure 3).
Figure 2. Overall response to extended treatment with everolimus.

Monthly average number of seizures for all participants over time (±SD), starting at baseline and continuing to 48 months (n = 18).
Figure 3. Individual response to extended treatment with everolimus.

Change in seizure frequency compared to baseline for each participant, starting at baseline and continuing to 48 months. Shaded areas indicate the timing of treatment discontinuation for participants not completing the full 48-month treatment period.
As in the main study phase, target everolimus dosing aimed to achieve serum trough levels between 5 and 15 ng/mL during extension. Upon start of the extension treatment phase, the actual median daily dose was 0.34 mg/kg/d (range 0.06–1.02), resulting in a median measured trough level of 6.1 ng/mL (range 1.5–16.1). Throughout extension, the median daily dose and corresponding serum trough levels narrowed but did not change significantly (0.47–0.56 mg/kg/d and 7.4–10.8 ng/mL, respectively). We observed a weak but statistically significant relationship between seizure frequency and everolimus dose (r = −0.3394, p < 0.001) and everolimus trough level (r = −0.1606, p = 0.045).
One participant was weaned off all daily seizure medication during the extension phase and maintained seizure control exclusively with everolimus. The remainder of participants completing the extension phase continued on at least 1 conventional AED. Excluding AEDs used primarily as rescue medications (diazepam, clonazepam, midazolam, and lorazepam), the overall number of concurrent AEDs remained unchanged (median = 2, range 0–4) in the cohort treated for the full 48 months. Individually, most participants continued on the same AEDs (n = 7). Two participants were on the same number of AEDs but in different combination, 2 participants reduced the number of AEDs, and 3 participants increased the number of AEDs. Everolimus was most often combined with vigabatrin (n = 5), valproic acid (n = 5), lacosamide (n = 4), lamotrigine (n = 4), or clobazam (n = 3) in participants classified as responders at the end of the 48-month treatment period.
Overall, quality of life measured by the QOLCE composite score improved an average of 14% (43.7 ± 13.4 at baseline compared to 52.0 ± 17.8 after 48 months). Positive changes in stigma, self-rated quality of life, attention/concentration, anxiety, language, and general health matched those identified as significant in the initial treatment period,14 but failed to reach statistical significance due to individual variation and smaller cohort size at the end of the extension phase. Trends in behavior improvement in both positive and negative domains were also observed after 48 months of treatment, as measured by the NCBRF, but similarly did not reach statistical significance.
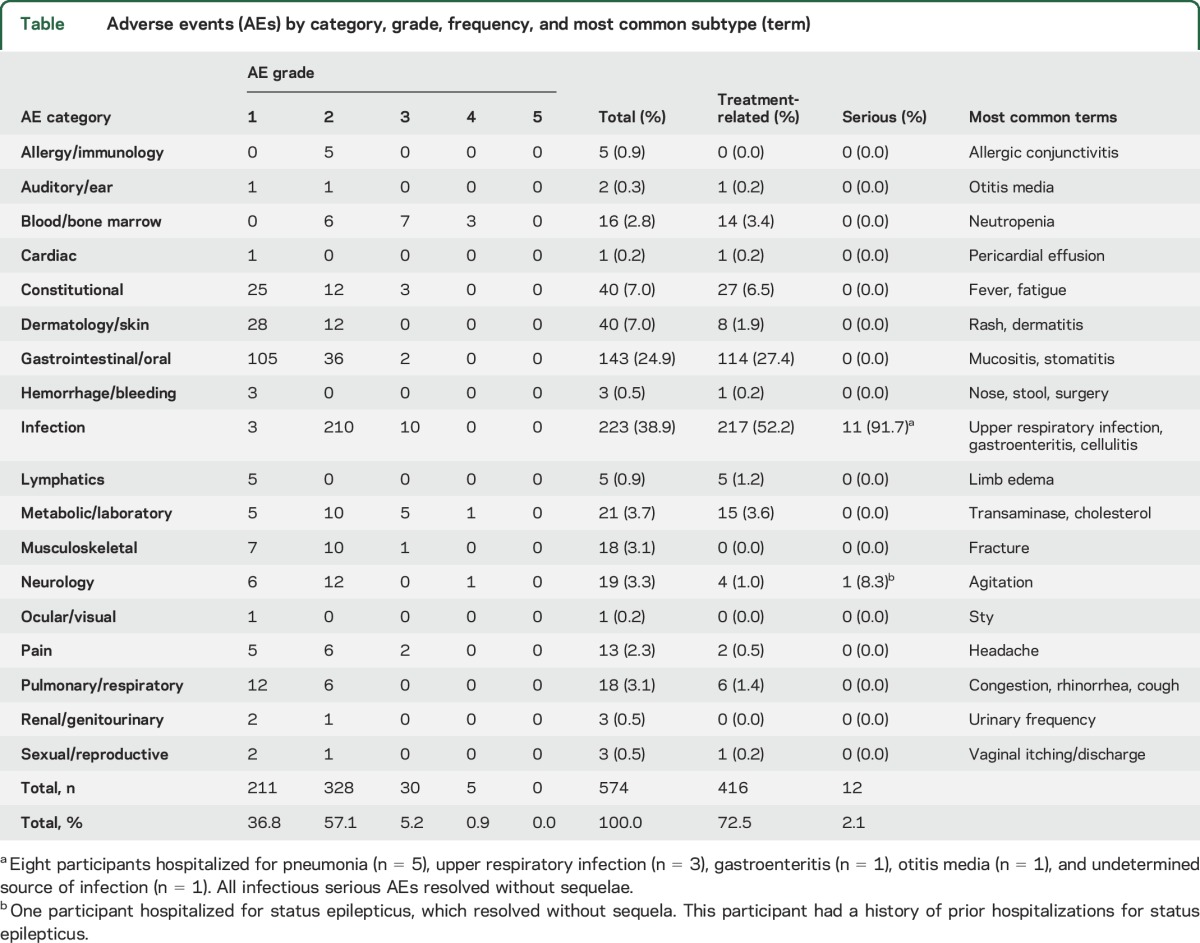
Consistent with earlier published reports,11,13 everolimus treatment for extended periods was well-tolerated. A total of 574 AEs were reported during the combined main and extension treatment period (table). A total of 72.5% were considered treatment-related. Infections were the most common, the majority (95%) of which were commonly encountered, community-acquired infections that required no treatment or were effectively treated with a single course of antibiotic. Gastrointestinal complaints, including aphthous ulcers or stomatitis, were also frequent. Most AEs were mild or moderate (CTCAE grade 1 or 2) in severity (94% of all reported AE). Serious AEs (SAEs) were rare, comprising 12 events over 4+ years of treatment exposure, or 2% of all AEs. All SAEs were the result of hospitalization for observation or treatment, resolved without any sequelae, and participants resumed treatment with everolimus following resolution of the SAE.
Table.
Adverse events (AEs) by category, grade, frequency, and most common subtype (term)
DISCUSSION
This open-label trial of everolimus for intractable epilepsy in TSC demonstrates persistent efficacy in seizure control with up to 4 years of continued treatment. Seizure frequency was reduced and AEs were the same as those observed during initial treatment and tended to decrease in frequency over time, consistent with studies of everolimus for treatment of SEGA in tuberous sclerosis.11,13,21 Infections were most common, and by protocol all were considered treatment-related as immune-related risks were largely unknown at the time of study initiation. Placebo-controlled studies in TSC for SEGA, angiomyolipoma, and lymphangioleiomyomatosis have since been published that reveal nearly identical infection rates in patients treated with mTORC1 inhibitors and placebo controls.3,5,6 Thus most infections in the present study, including several of the precautionary hospitalizations reported as SAEs, in reality more likely reflect common, community-acquired infections such as upper respiratory infection that have been estimated to occur 4–6 times/y in children and 2–4 times/y in young adults22 rather than everolimus exposure specifically.
We found that partial seizures with or without secondary generalization were primarily responsible for everolimus treatment-related improvement in seizure frequency. In TSC, all primary seizure types have been reported to occur but partial seizures are predominant.1 Infantile spasms, frequently coexisting with partial seizures, are also common during infancy.1,23 Due to the unclear safety profile of everolimus treatment in infants with TSC, individuals under the age of 2 years were excluded from our study. Hence, whether or not everolimus treatment at younger ages would have similar benefit, when infantile spasms are most prevalent in TSC, still needs to be determined. Recent studies have shown that earlier treatment initiation with the anticonvulsant vigabatrin for infantile spasms, even before onset of clinical seizures, may favor improved clinical outcome in TSC.24,25 Similarly, earlier initiation of everolimus in TSC could be of greater benefit than waiting until clinical seizures emerge and epileptogenic processes have progressed to the point of irreversibility and intractability. Potential age-dependent effects also might explain why when everolimus was evaluated in 117 patients aged 0–65 years for the treatment of SEGA, no seizure benefit could be detected,3 but in a subanalysis of participants under 3 years of age, seizure prevention (3/8) or reduction in seizure frequency >50% (3/8) was reported in the majority of participants.12
Traditional AEDs work by binding to transmembrane proteins in neurons or glia to facilitate or inhibit the flow of ions through transmembrane channels or to act upon receptors that have secondary metabolic effects on the synapse and thereby reduce neuronal excitability.26,27 In contrast, mTORC1 inhibitors such as everolimus have no such direct effects. mTORC1 inhibitors inactivate mTORC1 by displacing its cofactor Raptor from its binding site,28 resulting in inhibition of protein translation mediated by 4EBP1 and S6 kinase. Everolimus treatment would hope to improve CNS anatomic and functional abnormalities in TSC, but the exact mechanism or mechanisms through which these lead to seizure reduction in TSC requires continued investigation. Alteration in neuronal excitability, structural reorganization of synaptic junctions, changes in the extracellular microenvironment, large network connectivity changes, myelination, inflammation, and metabolism—all of which are important to normal CNS maturation and function and dependent on normal regulation of the mTORC1 pathway—could be involved.29
mTORC1 activation is not exclusive to epilepsy associated with TSC. Genes responsible for regulation of mTORC1 have been identified in epileptic patients with isolated focal cortical dysplasia IIb,30,31 hemimegencephaly,32 and syndromic epilepsies caused by DEPDEC5,32,33 PTEN,34 and STRADA.35 Loss of mTORC1 regulation has been identified in animal models of acquired epilepsies, including temporal lobe epilepsy and posttraumatic epilepsy, which are responsive to sirolimus treatment.36,37 Human patients with early-onset intractable epilepsy caused by STRADA mutation demonstrate marked reduction in seizure frequency when treated with sirolimus.35 Thus targeting the mTOR pathway with mTORC1 inhibitors such as everolimus may have application well beyond epilepsy associated solely with TSC.
The primary limitations of our study are the small study size and open-label design. As the first study of its kind when initiated in 2010, safety concerns and funding limitations prevented a larger, more robust clinical trial design. We were able to demonstrate long-term safety and overall seizure benefit prospectively over 4 years of treatment. The small sample size prevents us from assessing potential independent contributors to treatment response, including age, prior epilepsy treatments and clinical features, and individual concurrent anticonvulsant medication combinations. We similarly are unable to determine if improvements in neurobehavior or quality of life are the direct result of improved seizure control or an independent effect of mTORC1 inhibition unrelated to epilepsy.
GLOSSARY
- AE
adverse event
- AED
antiepileptic drug
- CTCAE
Common Terminology Criteria for Adverse Events
- mTORC1
mammalian target of rapamycin complex 1
- NCBRF
Nisonger Child Behavioral Rating Form
- QOLCE
Quality of Life for Children with Epilepsy Parent Form
- RM-ANOVA
repeated-measures analysis of variance
- SAE
serious adverse event
- SEGA
subependymal giant cell astrocytoma
- TSC
tuberous sclerosis complex
AUTHOR CONTRIBUTIONS
Darcy A. Krueger: study concept and design, data acquisition, analysis and interpretation of data, manuscript preparation and editing. Angus A. Wilfong: study concept and design, data acquisition, analysis and interpretation of data, manuscript preparation and editing. Christina M. Talley: data acquisition, manuscript preparation and editing. Maxwell Mays: data acquisition, manuscript preparation and editing. Karen Agricola: data acquisition, manuscript preparation and editing. Cindy Tudor: data acquisition, manuscript preparation and editing. Jamie Capal: data acquisition, manuscript preparation and editing. Katherine Holland-Bouley: study concept and design, analysis and interpretation of data, manuscript preparation and editing. David Neal Franz: study concept and design, data acquisition, analysis and interpretation of data, manuscript preparation and editing.
STUDY FUNDING
This study was funded by Novartis Pharmaceuticals, the Clack Foundation, and Cincinnati Children's Research Foundation. This study utilized clinical research facilities and resources supported by the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health Grant (8UL1-TR000077).
DISCLOSURE
D. Krueger has received consulting and speaking fees and travel expenses from Novartis and additional research support from the National Institute of Neurologic Disorders and Stroke of the NIH (U01-NS082320, U54-NS092090, P20-NS080199), the Tuberous Sclerosis Alliance, the Van Andel Research Institute, Novartis, and Upsher-Smith Pharmaceuticals. A. Wilfong has received consulting fees from Lundbdeck, Supernus, and Cyberonics; speaking fees from Cyberonics; royalties from Up to Date; and additional research support from Novartis, NINDS, Moody Foundation, UCB, Pfizer, Upsher-Smith, Impax, and Glaxo-Smith-Kline. M. Mays, C. Talley, K. Agricola, C. Tudor, J. Capal, and K. Holland-Bouley report no disclosures relevant to the manuscript. D. Franz has received consultant fees and travel expenses from Novartis and additional research support from Novartis. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia 2010;51:1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krueger DA, Franz DN. Current management of tuberous sclerosis complex. Pediatr Drugs 2008;10:299–313. [DOI] [PubMed] [Google Scholar]
- 3.Franz D, Belousova E, Sparagana S, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2013;381:125–132. [DOI] [PubMed] [Google Scholar]
- 4.Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. New Engl J Med 2010;363:1801–1811. [DOI] [PubMed] [Google Scholar]
- 5.Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2013;381:817–824. [DOI] [PubMed] [Google Scholar]
- 6.McCormack FX, Inoue Y, Moss J, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. New Engl J Med 2011;364:1595–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demir HA, Ekici F, Yazal Erdem A, Emir S, Tunc B. Everolimus: a challenging drug in the treatment of multifocal inoperable cardiac rhabdomyoma. Pediatrics 2012;130:e243–e247. [DOI] [PubMed] [Google Scholar]
- 8.Koenig MK, Hebert AA, Roberson J, et al. Topical rapamycin therapy to alleviate the cutaneous manifestations of tuberous sclerosis complex: a double-blind, randomized, controlled trial to evaluate the safety and efficacy of topically applied rapamycin. Drugs R D 2012;12:121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meikle L, Pollizzi K, Egnor A, et al. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci 2008;28:5422–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol 2008;63:444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franz DN, Agricola K, Mays M, et al. Everolimus for subependymal giant cell astrocytoma: 5-year final analysis. Ann Neurol 2015;78:929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kotulska K, Chmielewski D, Borkowska J, et al. Long-term effect of everolimus on epilepsy and growth in children under 3 years of age treated for subependymal giant cell astrocytoma associated with tuberous sclerosis complex. Eur J Paediatr Neurol 2013;17:479–485. [DOI] [PubMed] [Google Scholar]
- 13.Krueger DA, Care MM, Agricola K, Tudor C, Mays M, Franz DN. Everolimus long-term safety and efficacy in subependymal giant cell astrocytoma. Neurology 2013;80:574–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krueger DA, Wilfong AA, Holland-Bouley K, et al. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol 2013;74:679–687. [DOI] [PubMed] [Google Scholar]
- 15.Cardamone M, Flanagan D, Mowat D, Kennedy SE, Chopra M, Lawson JA. Mammalian target of rapamycin inhibitors for intractable epilepsy and subependymal giant cell astrocytomas in tuberous sclerosis complex. J Pediatr 2014;164:1195–1200. [DOI] [PubMed] [Google Scholar]
- 16.Wiegand G, May T, Ostertag P, Boor R, Stephani U, Franz D. Everolimus in tuberous sclerosis patients with intractable epilepsy: a treatment option? Eur J Paediatr Neurol 2013;17:631–638. [DOI] [PubMed] [Google Scholar]
- 17.Northrup H, Krueger DA. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 international tuberous sclerosis complex consensus conference. Pediatr Neurol 2013;49:243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sabaz M, Lawson JA, Cairns DR, et al. Validation of the quality of life in childhood epilepsy questionnaire in American epilepsy patients. Epilepsy Behav 2003;4:680–691. [DOI] [PubMed] [Google Scholar]
- 19.Aman MG, Tasse MJ, Rojahn J, Hammer D. The Nisonger CBRF: a child behavior rating form for children with developmental disabilities. Res Dev Disabil 1996;17:41–57. [DOI] [PubMed] [Google Scholar]
- 20.Bissler JJ, McCormack FX, Young LR, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. New Engl J Med 2008;358:140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franz DN, Belousova E, Sparagana S, et al. Everolimus for subependymal giant cell astrocytoma in patients with tuberous sclerosis complex: 2-year open-label extension of the randomised EXIST-1 study. Lancet Oncol 2014;15:1513–1520. [DOI] [PubMed] [Google Scholar]
- 22.Monto AS, Ullman BM. Acute respiratory illness in an American community: The Tecumseh study. JAMA 1974;227:164–169. [PubMed] [Google Scholar]
- 23.Wu JY, Peters JM, Goyal M, et al. Clinical electroencephalographic biomarker for impending epilepsy in asymptomatic tuberous sclerosis complex infants. Pediatr Neurol 2016;54:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jóźwiak S, Kotulska K, Domańska-Pakieła D, et al. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur J Paediatric Neurol 2011;15:424–431. [DOI] [PubMed] [Google Scholar]
- 25.Bombardieri R, Pinci M, Moavero R, Cerminara C, Curatolo P. Early control of seizures improves long-term outcome in children with tuberous sclerosis complex. Eur J Paediatric Neurol 2010;14:146–149. [DOI] [PubMed] [Google Scholar]
- 26.Lason W, Chlebicka M, Rejdak K. Research advances in basic mechanisms of seizures and antiepileptic drug action. Pharmacol Rep 2013;65:787–801. [DOI] [PubMed] [Google Scholar]
- 27.Vajda FJ, Eadie MJ. The clinical pharmacology of traditional antiepileptic drugs. Epileptic Disord 2014;16:395–408. [DOI] [PubMed] [Google Scholar]
- 28.MacKeigan JP, Krueger DA. Differentiating the mTOR inhibitors everolimus and sirolimus in the treatment of tuberous sclerosis complex. Neuro Oncol 2015;17:1550–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryther RC, Wong M. Mammalian target of rapamycin (mTOR) inhibition: potential for antiseizure, antiepileptogenic, and epileptostatic therapy. Curr Neurol Neurosci Rep 2012;12:410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim JS, Kim WI, Kang HC, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med 2015;21:395–400. [DOI] [PubMed] [Google Scholar]
- 31.Nakashima M, Saitsu H, Takei N, et al. Somatic Mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann Neurol 2015;78:375–386. [DOI] [PubMed] [Google Scholar]
- 32.D'Gama AM, Geng Y, Couto JA, et al. Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann Neurol 2015;77:720–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scheffer IE, Heron SE, Regan BM, et al. Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol 2014;75:782–787. [DOI] [PubMed] [Google Scholar]
- 34.Elia M, Amato C, Bottitta M, et al. An atypical patient with Cowden syndrome and PTEN gene mutation presenting with cortical malformation and focal epilepsy. Brain Dev 2012;34:873–876. [DOI] [PubMed] [Google Scholar]
- 35.Parker WE, Orlova KA, Parker WH, et al. Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Sci translational Med 2013;5:182. ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo D, Zeng L, Brody DL, Wong M. Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PLoS One 2013;8:e64078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci 2009;29:6964–6972. [DOI] [PMC free article] [PubMed] [Google Scholar]