

Abstract
Acidic microenvironment is a common feature of solid tumors. We have previously shown that neuron specific acid-sensing ion channel 1 (ASIC1) is expressed in breast cancer, and it is responsible for acidosis-induced cellular signaling through AKT, leading to nuclear factor-κB (NF-κB) activation, and cell invasion and metastasis. However, AKT is frequently activated in cancer. Thus, a key question is whether ASIC1-mediated cell signaling still takes place in the cancer cells carrying constitutively active AKT. In the present study, we show that among four prostate cancer cell lines tested, 22Rv1 cells express the highest level of phosphorylated AKT that is not impacted by acidosis. However, acidosis can still induce NF-κB activation during which extracellular signal-regulated kinase (ERK) serves as an alternative pathway for ASIC-mediated cell signaling. Inhibition of ERK by chemical inhibitors or small interfering RNAs suppresses the acidosis-induced NF-κB activity through regulation of the inhibitory subunit IκBα phosphorylation. Furthermore, suppression of ASIC1-mediated generation of reactive oxygen species (ROS) by ROS scavengers, such as glutathione or N-acetyl-cysteine causes a decrease in ERK phosphorylation and degradation of IκBα. Finally, ASIC1 is upregulated in a subset of prostate cancer cases and ASIC1 knockout by CRISPR/Cas9 significantly suppresses cell invasion, and castration resistance both in vitro and in vivo. Together, these results support the significance of ASIC1-ROS-ERK-IκBα-NF-κB axis in prostate tumorigenesis, especially in the constitutively active AKT background.
Introduction
Hypoxic conditions, disorganized tumor vasculature, heterogeneous blood flow and increased glycolysis (Warburg effect) are a common feature for solid tumors, which often lead to an acidotic condition (acidosis).1 As low intracellular pH (pHi) is harmful, tumor cells have to employ various mechanisms to remove intracellular acids in order to maintain physiological pHi. These include V-type ATPases, Na+/H+ exchangers and those facilitated by carbonic anhydrases such as CA2, CA9 and CA12.2 As a result, the extracellular pH (pHe) becomes acidic. For example, pHe of solid tumors could reach 6.2–6.8.3 In some extreme cases, interstitial pH values could even be as low as 5.8.4 Although such acidic extracellular conditions are often harmful to normal cells, tumor cells seem to have adapted well and furthermore, tumor cells can even use acidosis as a signal to promote their invasion and metastasis.1 We recently reported that acid-sensing ion channels (ASICs) are expressed in a subset of breast cancer cases and breast cancer cell lines.5 More importantly, ASIC1 plays a critical role in breast cancer cell invasion and metastasis, which is in part through activation of AKT and nuclear factor-κB (NF-κB).
AKT is a key downstream target of phosphoinositide 3-kinase (PI3K)-mediated signaling pathway and it plays an important role in regulation of diverse cellular processes such as cell survival, cell cycle progression, metabolism and angiogenesis.6 Inappropriate activation of AKT has been reported in many types of human diseases, including cancer. Several known factors control AKT activity positively or negatively; a notable negative regulator is the tumor suppressor PTEN that is frequently mutated or deleted in cancer. In addition, reactive oxygen species (ROS) can deactivate several phosphatases, including PTEN, leading to activation of AKT.7 Activated AKT is capable of phosphorylating IκBα kinase (IKK), which is a kinase for IκBα, a direct inhibitor of NF-κB. Thus, AKT can serve as an upstream molecule for NF-κB.
NF-κB is a ubiquitously expressed pleiotropic transcription factor that can be activated in response to a number of stimuli, including acidosis.8, 9, 10 Under normal conditions, NF-κB stays in the cytoplasm as a heterotrimeric complex consisting of the subunits p50, p65 and the inhibitory subunit IκBα. In response to inducing stimuli, IκBα undergoes phosphorylation, ubiquitination and proteolytic degradation. The p65 subunit then undergoes phosphorylation and moves into the nucleus, where it binds to specific DNA sequence and can activate the transcription of hundreds of genes.11 In addition to AKT, IKK can also catalyze phosphorylation of IκBα. Aberrant regulation of NF-κB and the signaling pathways that control its activity often leads to inflammation, drug/radiation resistance and tumorigenic potential of cancer cells.12 As an AKT effector, NF-κB is critical to the acidosis-induced tumor initiation, progression and invasion.5 However, a question remains as to whether acidosis can still induce the cell signaling and impact different aspects of tumorigenesis in the highly active AKT background.
In the present study, we show that ERK, one of mitogen-activated protein kinases (MAPKs), serves as an alternative pathway through which ASIC1 and NF-κB are connected, especially in cancer cells carrying constitutively active AKT, leading to tumor cell growth and invasion.
Results
Acidosis induces NF-κB activity independent of AKT status
We recently showed that acidosis induces activation of AKT and NF-κB.13 However, AKT is often highly activated in cancer due to various mechanisms, including PTEN mutation or deletion. Hence, we asked whether acidosis-induced cell signaling still takes place. To this end, we first examined the pAKT level in prostate cancer cell lines 22Rv1, LNCaP, DU-145 and PC-3. It is evident that the pAKT level is highest in 22Rv1 cells among them (Figure 1a). Next, we determined the effect of acidosis on AKT activity. As acidosis induces AKT activation as early as 5 min after exposure to acidosis (pH 6.6) and the pAKT level reached a peak at 2 h,13 we treated cells at pH 6.6 for up to 2 h. Although we detected an increase in pAKT in LNCaP, DU-145 and PC-3, no AKT activation was seen in 22Rv1 cells (Figure 1b). For instance, in LNCaP cells AKT was induced at as early as 30 min; in PC-3 cells this AKT induction was seen at 2 h after exposure to acidosis. These results suggest that induction of AKT by acidosis is dynamic in different cell lines and different cell signaling pathways may be involved in this process. Next, we determined whether NF-κB is activated in response to acidosis because NF-κB is a key factor responsible for cell growth, invasion and metastasis. As shown in Figure 1c, not all cell lines responded to acidosis in the same way. For instance, in DU-145 cells NF-κB activation was seen at 1 h, whereas in PC-3 we detected NF-κB activation at 30 min. NF-κB activation was not obvious in LNCaP cells. Of interest was 22Rv1 with activation of NF-κB at as early as 30 min despite of a high level of base line AKT activity, suggesting that acidosis can still impact tumorigenesis in the cancer cells with constitutively active AKT.
Figure 1.

Acidosis induces nuclear translocation of NF-κB. (a), Expression of pAKT in 22Rv1, DU-145, LNCaP and PC-3 cells, as detected by western blot. (b) Detection of acidosis-induced AKT in selected prostate cancer cells. Note that no AKT activation is seen in 22Rv1 cells. (c) Nuclear localization of NF-κB in prostate cancer cells in response to acidosis. ‘Time 0 h' indicates that the cells were cultured at pH 7.4.
ASIC1 is required for acidosis-induced cell signaling
As ASIC1 has been shown to be required for acidosis-induced cell signaling in breast cancer,5 we determined expressions of ASIC1 in these four prostate cancer cell lines. As shown in Figure 2a, two of these cell lines (22Rv1 and LNCaP) expressed a significant level of ASIC1; moreover, the ASIC1 level was much higher in 22Rv1 cells than in LNCaP cells. A similar trend was also seen at the messenger RNA level (Supplementary Figure S1). To determine the role of ASIC1 in acidosis-induced cell signaling, we treated the cells with specific ASIC1 inhibitor psalmotoxin 1 that was able to block acidosis-induced NF-κB activation in 22Rv1 cells (Figure 2b). Hence, we chose 22Rv1 cells to characterize the ASIC1-mediated cell signaling. First, we knocked out ASIC1 by CRISPR/Cas9 technology and dual guide RNA (gRNA) approach targeting exon 2 and 3 of ASIC15 and obtained several knockout (KO) clones (Figure 2c). We chose two clones for further characterization. Although ASIC1 KO had no effect on the pAKT level in response to acidosis (Figure 2d), ASIC1 was critical to the acidosis-induced NF-κB activation (Figure 2e). There was little induction of NF-κB in either KO clone as compared with gRNA control. To further determine the role of ASIC1 in acidosis-induced NF-κB activation, we performed a rescue experiment, that is, re-expression of Myc-tagged ASIC1 in KO cells (Figure 2f). As expected, NF-κB activation was restored by ASIC1 (Figure 2g), demonstrating a critical role for ASIC1 in acidosis-mediated cell signaling. These results suggest that there may exist an alternative pathway bypassing AKT for acidosis-induced NF-κB activation.
Figure 2.

ASIC1 is required for acidosis-induced nuclear translocation of NF-κB. (a) Detection of ASIC1 expression in prostate cancer cell lines by western blot. (b) Suppression of NF-κB activity by ASIC1 inhibitor psalmotoxin 1 in response to acidosis. (c) Knockout (KO) of ASIC1. (d) ASIC1 KO has no effect on pAKT. Cells were treated at pH 6.6 for 2 h. (e) ASIC1 KO suppresses nuclear translocation of NF-κB. Cells were treated at pH 6.6 for 2 h. (f) Detection of ASIC1 (ASIC1-Myc-tag) in KO cells after re-expression. (g) Re-expression of ASIC1 restores the ability to activate NF-κB in response to acidosis. Cells were treated at pH 6.6 for 2 h.
ERK is involved in ASIC1-mediated cell signaling
IκBα serves as a NF-κB inhibitor and degradation of IκBα leads to nuclear translocation of NF-κB. Consistent with NF-κB activation, we detected the degradation of IκBα at as early as 30 min after acidosis (Figure 3a). To search for further upstream signaling molecules, we tested ERK that, like AKT, is also involved in a variety of important cellular processes such as proliferation, differentiation, motility, stress response, apoptosis and survival in response to stimuli such as mitogens, cytokines, growth factors and environmental stress. Once activation, ERK can regulate targets in the cytosol or translocate to the nucleus to regulate gene expression.14 As expected, we detected induction of ERK in response to acidosis in a time-dependent manner (Figure 3b). Of interest, ASIC1 is required for this acidosis-induced ERK activation. For instance, ASIC1 KO suppressed this induction (Figure 3c). To further determine the role of ERK in acidosis-induced NF-κB activation, we suppressed ERK by ERK inhibitors U0126 and PD98059. Both inhibitors blocked acidosis-induced NF-κB activation (Figure 3d; Supplementary Figure S2). The inhibition of NF-κB activity was seen at 5 μm for both inhibitors. Similarly, ERK small interfering RNAs (siRNAs) also suppressed NF-κB activation (Figure 3e). These results suggest a critical role for ERK in ASIC1-mediated cell signaling in response to acidosis.
Figure 3.

ERK is required for the acidosis-induced nuclear localization of NF-κB in 22Rv1 cells. For acidosis treatment, cells were cultured at pH 6.6 for 2 h before the cells were harvested. (a) Acidosis-induced nuclear localization of NF-κB is associated with a decrease in the IκBα level. (b) Acidosis induces pERK. (c) ASIC1 KO suppresses acidosis-induced ERK activation. (d) Suppression of ERK activity by ERK inhibitors U0126 and PD98059. Cells were treated with inhibitors at indicated concentrations for 1 h before acidosis. (e) Suppression of ERK activity by ERK siRNAs reduces nuclear localization of NF-κB.
ERK is required for IκBα phosphorylation through ROS
Moreover, this acidosis-induced IκBα degradation was also blocked by ERK inhibitors (Figure 4a). Like other types of stress, acidosis induces phosphorylation of IκBα15 that leads to IκBα degradation.16 In consistent with this finding, ERK inhibitors suppressed phosphorylation of IκBα (Figure 4a). Similarly, ERK siRNAs also blocked the acidosis-induced phosphorylation of IκBα such that the level of IκBα was restored (Figure 4b). As acidosis can cause generation of ROS,17, 18 we tested the role of ROS in acidosis-induced activation of ERK. We treated the cells with two ROS scavengers glutathione (GSH) and N-acetyl-cysteine (NAC). As shown in Figure 4c, both GSH and NAC reduced the level of pERK induced by acidosis. Moreover, both GSH and NAC reduced the nuclear p65 level (Figure 4d).
Figure 4.

Acidosis induces phosphorylation of IκBα through ERK in 22Rv1 cells. For acidosis treatment, cells were cultured at pH 6.6 for 2 h before the cells were collected. (a) Suppression of ERK by ERK inhibitors reduces acidosis-induced IκBα degradation and IκBα phosphorylation. Cells were treated with U0126 and PD98059 at indicated concentrations for 1 h before acidosis. (b) Suppression of ERK activity by ERK siRNAs reduces acidosis-induced IκBα degradation and IκBα phosphorylation. (c) Both GSH and NAC suppress ERK activity. (d) Both GSH and NAC suppress nuclear localization of NF-κB.
Acidosis increases invasion ability of 22Rv1 cells through ASIC1
Given the importance to ERK and NF-κB in cancer, we tested the effect of ASIC1 KO on cell invasion by transwell invasion assays. There was an increase in cell invasion by acidosis, and ASIC1 KO significantly reduced acidosis-induced invasive cell number as compared with gRNA control (Figure 5a, left). For example, for gRNA control, average invasive cell number was <50 for pH 7.4; this number was increased to over 100 for pH 6.6. In the ASIC1 KO group, the invasive cell number was under 20 for both pH 6.6 and pH 7.4 (Figure 5a, right). Rescue experiments revealed that ASIC1 was able to restore the ability to increase invasiveness (Figure 5b). In consistent with these findings, we found that as expected, acidosis increased expression of Twist and Snail (Figures 5c and d), both of which have been implicated in cell invasion and metastasis.19 In contrast, there was no increase of Twist and Snail in ASIC1 KO cells in response to acidosis.
Figure 5.

ASIC1 promotes acidosis-induced cell invasion and expression of Twist and Snail. (a) ASIC1 KO suppresses acidosis-induced cell invasion. Invasion assay was detailed in Materials and methods. Invasive cell number is the average of the number from five fields. (b) Re-expression of ASIC1 in KO cells restores the invasion ability. (c, d) ASIC1 KO suppresses acidosis-induced Twist and Snail, respectively. Cells were cultured at pH 6.6 for 2 h before the cells were collected for RNA isolation and quantitative PCR with reverse transcription. Values are mean±s.e. (n=3), *P<0.05.
ASIC1 promotes tumor cell growth and castration resistance
As ASIC1 is at top of this ASIC1-ROS-ERK-IκBα-NF-κB axis, we determined the consequence of ASIC1 KO. Cell growth assays indicated that ASIC1 slightly suppressed cell growth in regular medium (Supplementary Figure S3). However, in androgen-free medium ASIC1 KO significantly suppress cell growth, implicating ASIC1 in castration resistance (Figure 6a; Supplementary Figure S3). Therefore, we focused on the effect of ASIC1 on castration resistance. Rescue experiments further confirmed that re-expression of ASIC1 restored cell growth in androgen-free medium (Figure 6a). To further determine the role of ASIC1 in castration resistance, we performed xenograft animal tumor models using castrated severe combined immunodeficiency male mice. As shown in Figures 6b and c, ASIC1 KO suppressed tumor growth and tumor weight. Finally, by interrogating The Cancer Genome Atlas prostate adenocarcinoma data set at cBioPortal (http://www.cbioportal.org/),20, 21 we found that ASIC1 is upregulated in a subset of patient populations (6% among 498 cases; Figure 6e). Furthermore, when all four ASIC genes (ASIC1–4) were combined, they were upregulated in 18% of 498 cases (Supplementary Figure S4). Together, these results suggest that ASIC1 plays a role in prostate tumorigenesis and castration resistance.
Figure 6.

ASIC1 is required for cell growth in androgen-free medium and tumor growth in castrated male mice. (a), ASIC1 KO (#23) suppresses cell growth; re-expression of ASIC1 increases cell growth in androgen-free medium. Values are mean±s.e. (n=3), *P<0.05. (b) ASIC1 KO (#23) causes tumor growth inhibition. Two tumors for pH 6.6 gRNA control group were collected at day 19 and another two tumors for pH 7.4 gRNA control group were collected at day 23 because they reached 2 cm in diameter; (c) ASIC1 KO (#23) reduces tumor load. Two tumors for pH 6.6 gRNA control group and two for pH 7.4 gRNA control group harvested early were not pictured here (left), but they were included in the calculation of average tumor weight (right). The rest tumors were harvested at day 33 after tumor cell injection. Values are mean±s.e. (n=7), *P<0.05. (d) Analysis of prostate adenocarcinoma data set (The Cancer Genome Atlas, Provisional) at cBioPortal (http://www.cbioportal.org/) indicates 6% of cases with upregulation of ASIC1. Query Language (OQL) setting was ‘ASIC1: EXP>1.5'.
Discussion
AKT and ERK are two well-known signaling molecules downstream of receptor tyrosine kinase, and play a critical role in cell growth, proliferation and survival in response to growth factors or stress. Although AKT and ERK pathways can cross talk each other, they are relatively independent. For instance, AKT signaling involves PI3K, whereas ERK signaling is through RAS/RAF. However, they converge on survival signals. Thus, it is important to determine how they respond to environmental stimuli in different cell cellular content.
Through a long period of co-evolution with the host, tumor cells have adapted to acidic microenvironment and as such they become more aggressive.22, 23, 24 Early studies have implicated ASICs in the growth and migration of glioma cells.25, 26 For instance, acidosis causes activation of AKT and NF-κB, and generation of ROS;8, 10, 13 expression of ASIC1 and ASIC2 could induce glioma cation current, whereas suppression of this conductance decreases glioma growth and cell migration.25 Our recent study indicates that ASIC1 is critical to breast cancer invasion and metastasis through the ROS-AKT-NF-κB pathway.5 However, little is known whether there is any alternative acidosis-induced pathway, leading to NF-κB activation and cell invasion. The present study provides evidence that ASIC1 plays a critical role in response to acidosis in prostate cancer. In particular, our study suggests that ERK serves as a key player in ASIC1-mediated cell signaling in the highly active AKT background.
It is well known that tumor cells often carry highly active AKT activity that can be attributed to many factors, including deletion or mutation of PTEN27 or mutations in the PI3K genes.28 In this scenario, acidosis-induced cell signaling can take alternative pathways such as MAPK pathway. Like AKT, MAPKs are a highly conserved family of serine/threonine protein kinases involved in a variety of important cellular processes, such as proliferation, differentiation, motility, stress response, apoptosis, and survival. Extracellular stimuli, such as mitogens, cytokines, growth factors and environmental stress, cause a cascade of MAPK signaling through phosphorylation of MAP3K, MAP2K (for example, mitogen-activated protein kinase kinase (MEK)) and MAPK (for example, ERK). ERK serves as a substrate for MEK. Once activation by MEK, ERK can regulate a variety of downstream targets. Several lines of evidence from this study suggest that ERK plays a critical role in acidosis-induced NF-κB activation in cancer cells carrying a highly active AKT. (1) Acidosis activates ERK in a time-dependent manner; (2) ERK inhibitors or ERK siRNAs suppress the acidosis-induced NF-κB activation; and (3) ERK inhibitors or ERK siRNAs suppress the acidosis-induced IκBα phosphorylation. These findings are in line with the report that ASIC1 is also required for ERK activation in glioma cells.29
Our previous study suggests the involvement of acidosis-induced ROS production in breast cancer cells, leading to activation of NF-κB.13 As the present study reveals that both GSH and NAC suppress the acidosis-induced ERK activation, and subsequently inhibit nuclear localization of NF-κB, it may suggest that ROS acts at the upstream of ERK. Although the precise underlying mechanism still remains to be determined yet, it is possible that ROS may activate MAPK pathways through oxidative modifications of MAPK signaling proteins and inactivation and/or degradation of MAP kinase phosphatases.30 In support of this notion, ROS can alter protein structure and function by modifying critical amino-acid residues of proteins.31 Similarly, PTEN is a phosphatase and it can undergo inactivation (oxidation) by ROS.32 We have shown that GSH and NAC can block ROS-induced PTEN inactivation in response to acidosis.13 In addition, oxidant H2O2 may activate MAPK pathways via activation of growth factor receptors in several cell types.33
Although AKT and ERK are distinct pathways, they also cross talk each other. In particular, they not only negatively regulate each other's activity, but they cross activate each other.34 More importantly, they can share same substrates. For instance, both AKT and ERK can activate ER by phosphorylation in breast cancer.35, 36 In support of this notion is our finding that ASIC1-mediated NF-κB activation can bypass AKT through ERK, especially in the case that AKT is constitutively activated. They converge on NF-κB activation regardless of AKT status.
The clinical significance of ASIC1 is supported by the findings that ASIC1 is upregulated in a subset of prostate cancer cases. Furthermore, ASIC1 also contributes to castration resistance. For example, although ASIC1 KO suppresses cell growth in normal medium, this suppression is more significant under androgen deprivation. However, the underlying mechanism still remains to be determined. Accumulating evidence indicates that multiple factors can contribute to castration resistance, including androgen receptor, c-Myc overexpression, upregulation of PI3K/AKT and RAS/MAPK.37 ERK is frequently activated in prostate cancer samples, especially metastatic samples.38 Overexpression of nuclear receptor coactivator 2 along with loss of PTEN can result in activation of ERK, promoting tumor malignance in murine prostate tumor model.39 Thus, it is conceivable that ERK activation may contribute to castration resistance especially when tumor microenvironment becomes acidic.
In summary, our results suggest that ERK serves as an alternative pathway in ASIC1-mediated cell signaling in response to acidosis under constitutively active AKT background (Figure 7). ASIC1 sits at the top of this cascade. Acidic tumor microenvironment can induce ROS. Although IKK is a well-known factor that can phosphorylate IκBα, other factors such as AKT can also regulate IκBα activity. The present study suggests that in the presence of highly active AKT, tumor cells can bypass AKT and acidosis-induced ROS may activate ERK (phosphorylation) possibly through deactivation of certain phosphatases. Subsequently, the activated ERK may facilitate IκBα phosphorylation directly or indirectly, leading to nuclear localization of NF-κB. As a cancer promoter, NF-κB can activate a larger number of genes40 important to cell growth, migration, invasion and metastasis. Therefore, this ASIC1-ROS-ERK-IκBα-NF-κB axis may provide an opportunity for intervention in cancer therapy.
Figure 7.
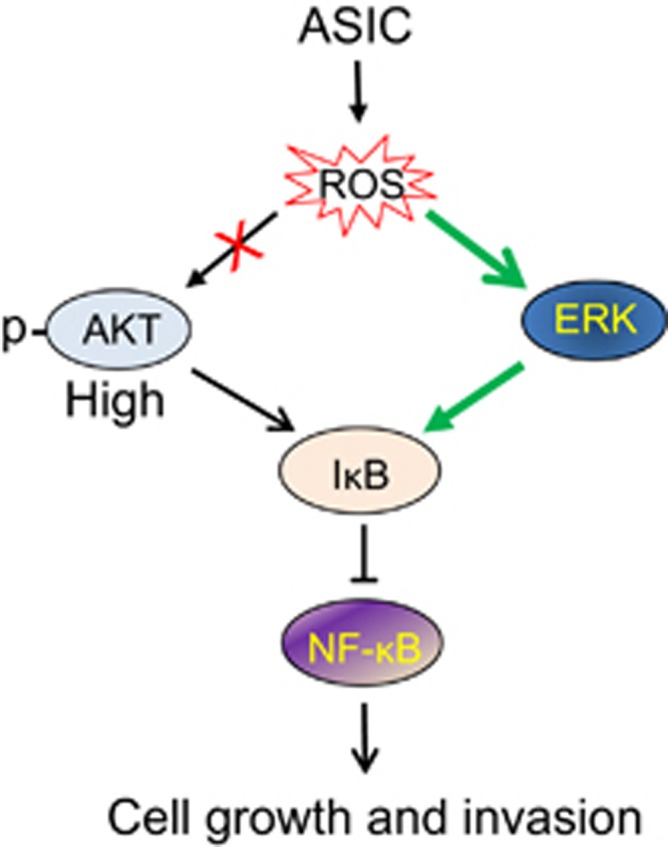
A working model for acidosis-induced NF-κB activation through ERK in the highly active AKT background. See text for explanation.
Materials and methods
Regents
Sources of primary antibodies were: p65 (D14E12), p-IĸBα (5A5), IĸBα (L35A5), pERK (D13.14.4E), ERK (137F5), pAKT (D9E), AKT (C67E7), p-IKKα (16A6) and PARP (46D11) from Cell Signaling (Danvers, MA, USA); ASIC1 (clone N271/44) from UC Davis (Davis, CA, USA), sold by Antibodies Inc (Davis, CA, USA); Myc-tag from Applied Biological Materials (ABM; Richmond, BC, Canada); α-tubulin, GAPDH and ERK (66192-1-Ig) from Proteintech (Rosemont, IL, USA); and hnRNP I (SH54) from Santa Cruz Biotechnology (Dallas, TX, USA). Secondary antibodies conjugated with IRDye 800CW or IRDye 680 were purchased from LI-COR Biosciences (Lincoln, NE, USA). PCR primers were purchased from IDT (Coralville, IA, USA). ERK siRNAs, U0126 and PD98059 were purchased from Cell Signaling. GSH and NAC were obtained from Sigma-Aldrich (St. Louis, MO, USA). Psalmotoxin 1 was purchased from Peptides International (Louisville, KY).
Cell culture
Prostate cancer cell lines 22RV1, DU-145, LNCaP and PC-3, and HEK293T were obtained from American Type Culture Collection (Manassas, VA, USA) and were authenticated by DDC Medical (http://www.ddcmedical.com) using the short tandem repeat profiling method. 22RV1, DU-145, LNCaP and PC-3 cells were cultured in RPMI-1640 medium with 10% fetal bovine serum (FBS) and 2 mM glutamine. HEK293T cells were cultured in DMEM with 10% FBS. For androgen-deprivation cells were grown in phenol free RPMI-1640 supplemented with charcoal stripped 5% FBS (Sigma-Aldrich). All media contained 2 mM glutamine, 100 units of penicillin per ml and 100 mg of streptomycin per ml. The pH of the acidic medium was adjusted to 6.6 with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and 1,4-piperazinediethanesulfonic acid (PIPES).
Plasmid construction
KO constructs using the dual gRNA approach41 targeting ASIC1 exon 2 and 3, and donor for ASIC1 were described previously.5 ASIC1 expression vector used for rescue experiments was constructed by cloning ASIC1-coding region into pCDH-Myc by Cold Fusion kit (System Biosciences, Mountain View, CA, USA). All PCR products were verified by DNA sequencing.
Transfection
Transfection of siRNAs was carried out using RNAfectin from ABM (Richmond, BC, Canada) according to the manufacturer's instructions. In brief, cells were seeded at 20–30% density in 12-well plates the day before transfection. Next day, control siRNAs or target gene siRNAs were mixed with RNAfectin and then added to cell culture at 100 nM. Sixteen hours later, the transfection medium was replaced with fresh medium.
Invasion assay
Invasion assay was used for measuring the invasion ability of 22Rv1 cells in BioCoat invasion system from BD Biosciences (San Jose, CA). The cells were cultured in either acidic or normal medium for 2 h. Then, cells (2 × 104) in 500 μl serum-free medium were added to the up chambers and the low chambers contained medium with 10% FBS. After 24 h, cells in the upper membrane were wiped away, and the cells in the lower membrane were immersed in 95% ethanol followed by stain with crystal violet. Cells were counted in five fields.
Cell growth assay
22Rv1 cells (2 × 104) were seeded in 12-well plates under either normal medium or androgen-free medium. The cells were collected, trypan blue stained and counted at day 4 as described.42 The relative cell growth was calculated by fold changes of cell numbers.
Real time quantitative PCR with reverse transcription
Total RNA was isolated using Direct-zol RNA MiniPrep (Zymo Research, Irvine, CA, USA) per the manufacturer's protocol and 0.5 μg RNA was used to synthesize complementary DNA by RevertAid Reverse Transcriptase (Fisher Scientific, Pittsburgh, PA, USA) with random primer mix (New England BioLabs, Ipswich, MA, USA) in 20 μl reaction. The resultant complementary DNA was used for quantitative PCR reactions. To specifically detect expression of Twist and Snail, we used the SYBR Green method with primers described in Supplementary Table 1. β-actin was used as an internal control. Delta-delta Ct values were used to determine their relative expression as fold changes, as previously described.41
Western blot analysis
Cytoplasmic, nuclear or whole cellular extract were prepared to detect levels of proteins, and western blot analysis was carried out as described earlier.13
Animal work
The animal studies were conducted in accordance with National Institutes of Health animal use guidelines and the experimental protocol approved by the UMMC's Animal Care and Use Committee. Male severe combined immunodeficiency mice at 5–6 week old purchased from Charles River (Wilmington, MA, USA) were first castrated, and 1 week later tumor cells were injected subcutaneously into these mice with 1 million cells containing 50% matrigel per spot. Four groups of mice consisted of ASIC1 KO or vector control cells grown at pH 7.4 and pH 6.6, respectively, for 2 h before they were collected. Tumor growth was monitored every other 2 days and collected at day 33 after injection. Tumor volumes were calculated as: (π/6) × (length × width2).
Statistical analysis
Comparisons between groups were analyzed using the Student's t-test (two groups) or a one-way analysis of variance followed by post hoc Tukey test (multiple groups). The Welch's correction of t-test was performed when the variances in two samples were unequal. Two-sided P<0.05 were considered as significant.
Acknowledgments
This work was supported by grants no. 81402100 from National Natural Science Foundation of China (BC); no. Q201408 from the Foundation of Health and Family Planning Commission of Jiangsu Province (BC); no. SH2014026 and SH2016031 from the Social Development Foundation of Zhenjiang (BC), and grant LY15C050002 from the Natural Science Foundation of Zhejiang Province (XD) and National Institutes of Health grant R01 CA154989 (YM).
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on the Oncogenesis website (http://www.nature.com/oncsis).
Supplementary Material
References
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer 2004; 4: 891–899. [DOI] [PubMed] [Google Scholar]
- Damaghi M, Wojtkowiak JW, Gillies RJ. pH sensing and regulation in cancer. Front Physiol 2013; 4: 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashim AI, Zhang X, Wojtkowiak JW, Martinez GV, Gillies RJ. Imaging pH and metastasis. NMR Biomed 2011; 24: 582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res 1989; 49: 4373–4384. [PubMed] [Google Scholar]
- Gupta SC, Singh R, Asters M, Liu J, Zhang X, Pabbidi MR et al. Regulation of breast tumorigenesis through acid sensors. Oncogene 2015; 35: 4102–4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev 2004; 30: 193–204. [DOI] [PubMed] [Google Scholar]
- Gough DR, Cotter TG. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis 2011; 2: e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppicelli S, Bianchini F, Contena C, Tombaccini D, Calorini L. Acidic pH via NF-kappaB favours VEGF-C expression in human melanoma cells. Clin Exp Metastasis 2013; 30: 957–967. [DOI] [PubMed] [Google Scholar]
- Shi Q, Le X, Wang B, Xiong Q, Abbruzzese JL, Xie K. Regulation of interleukin-8 expression by cellular pH in human pancreatic adenocarcinoma cells. J Interferon Cytokine Res 2000; 20: 1023–1028. [DOI] [PubMed] [Google Scholar]
- Xu L, Fidler IJ. Acidic pH-induced elevation in interleukin 8 expression by human ovarian carcinoma cells. Cancer Res 2000; 60: 4610–4616. [PubMed] [Google Scholar]
- Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010; 464: 1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell 2004; 6: 203–208. [DOI] [PubMed] [Google Scholar]
- Gupta SC, Singh R, Pochampally R, Watabe K, Mo YY. Acidosis promotes invasiveness of breast cancer cells through ROS-AKT-NF-kappaB pathway. Oncotarget 2014; 5: 12070–12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov A, Zehorai E, Procaccia S, Seger R. The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim Biophys Acta 2011; 1813: 1619–1633. [DOI] [PubMed] [Google Scholar]
- Dong L, Li Z, Leffler NR, Asch AS, Chi JT, Yang LV. Acidosis activation of the proton-sensing GPR4 receptor stimulates vascular endothelial cell inflammatory responses revealed by transcriptome analysis. PLoS ONE 2013; 8: e61991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci 2005; 30: 43–52. [DOI] [PubMed] [Google Scholar]
- Lamonte G, Tang X, Chen JL, Wu J, Ding CK, Keenan MM et al. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab 2013; 1: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemann A, Schneider B, Ihling A, Nowak M, Sauvant C, Thews O et al. Acidic environment leads to ROS-induced MAPK signaling in cancer cells. PLoS ONE 2011; 6: e22445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConkey DJ, Choi W, Marquis L, Martin F, Williams MB, Shah J et al. Role of epithelial-to-mesenchymal transition (EMT) in drug sensitivity and metastasis in bladder cancer. Cancer Metastasis Rev 2009; 28: 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbey CK, Borowsky AD, McGoldrick ET, Gregg JP, Maglione JE, Cardiff RD et al. In vivo positron-emission tomography imaging of progression and transformation in a mouse model of mammary neoplasia. Proc Natl Acad Sci USA 2004; 101: 11438–11443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res 2013; 73: 1524–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res 2009; 69: 2260–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdiev BK, Xia J, McLean LA, Markert JM, Gillespie GY, Mapstone TB et al. Acid-sensing ion channels in malignant gliomas. J Biol Chem 2003; 278: 15023–15034. [DOI] [PubMed] [Google Scholar]
- Bubien JK, Keeton DA, Fuller CM, Gillespie GY, Reddy AT, Mapstone TB et al. Malignant human gliomas express an amiloride-sensitive Na+ conductance. Am J Physiol 1999; 276: C1405–C1410. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Escudero I, Oliver MD, Andres-Pons A, Molina M, Cid VJ, Pulido R. A comprehensive functional analysis of PTEN mutations: implications in tumor- and autism-related syndromes. Hum Mol Genet 2011; 20: 4132–4142. [DOI] [PubMed] [Google Scholar]
- Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008; 27: 5497–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooj AK, McNicholas CM, Bartoszewski R, Bebok Z, Benos DJ, Fuller CM. Glioma-specific cation conductance regulates migration and cell cycle progression. J Biol Chem 2012; 287: 4053–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J Signal Transduction 2011; 2011: 792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol 2000; 279: L1005–L1028. [DOI] [PubMed] [Google Scholar]
- Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem 2002; 277: 20336–20342. [DOI] [PubMed] [Google Scholar]
- Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem 1996; 271: 4138–4142. [DOI] [PubMed] [Google Scholar]
- Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci 2011; 36: 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem 2001; 276: 9817–9824. [DOI] [PubMed] [Google Scholar]
- Chen D, Washbrook E, Sarwar N, Bates GJ, Pace PE, Thirunuvakkarasu V et al. Phosphorylation of human estrogen receptor alpha at serine 118 by two distinct signal transduction pathways revealed by phosphorylation-specific antisera. Oncogene 2002; 21: 4921–4931. [DOI] [PubMed] [Google Scholar]
- Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013; 32: 5501–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland DJ, Kobayashi N, Ruscetti M, Zhi A, Tran LM, Huang J et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res 2012; 72: 1878–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Lee HJ, Wu SP, Lin SC, Lanz RB, Creighton CJ et al. Androgen deprivation-induced NCoA2 promotes metastatic and castration-resistant prostate cancer. J Clin Invest 2014; 124: 5013–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer 2013; 12: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TT, Zhou N, Huang J, Koirala P, Xu M, Fung R et al. Targeting non-coding RNAs with the CRISPR/Cas9 system in human cell lines. Nucleic Acids Res 2015; 43: e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdeva M, Wu H, Ru P, Hwang L, Trieu V, Mo YY. MicroRNA-101-mediated Akt activation and estrogen-independent growth. Oncogene 2011; 30: 822–831. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.