

Abstract
Protein acetylation catalyzed by specific histone acetyltransferases (HATs) is an essential post-translational modification (PTM) and involved in the regulation a broad spectrum of biological processes in eukaryotes. Although several ten thousands of acetylation sites have been experimentally identified, the upstream HATs for most of the sites are unclear. Thus, the identification of HAT-specific acetylation sites is fundamental for understanding the regulatory mechanisms of protein acetylation. In this work, we first collected 702 known HAT-specific acetylation sites of 205 proteins from the literature and public data resources, and a motif-based analysis demonstrated that different types of HATs exhibit similar but considerably distinct sequence preferences for substrate recognition. Using 544 human HAT-specific sites for training, we constructed a highly useful tool of GPS-PAIL for the prediction of HAT-specific sites for up to seven HATs, including CREBBP, EP300, HAT1, KAT2A, KAT2B, KAT5 and KAT8. The prediction accuracy of GPS-PAIL was critically evaluated, with a satisfying performance. Using GPS-PAIL, we also performed a large-scale prediction of potential HATs for known acetylation sites identified from high-throughput experiments in nine eukaryotes. Both online service and local packages were implemented, and GPS-PAIL is freely available at: http://pail.biocuckoo.org.
As one of the most important and ubiquitous post-translational modifications (PTMs) in proteins, the lysine acetylation catalyzed by histone acetyltransferases (HATs) or lysine acetyltransferases (KATs) reversibly regulates a large number of biological processes, such as transcriptional regulation, metabolism and autophagy1,2,3,4,5,6,7. The dysregulation of site-specific HAT-substrate relations is frequently associated with human diseases such as cancers2,3,8,9. In eukaryotes, numerous HATs have been classified into three major families including p300/CBP, GCN5-related N-acetyltransferases (GNATs) and MYST proteins1,2,3,10,11. Different HATs can recognize overlapping but distinct substrates1,11,12. Most HATs exist in multisubunit complexes in vivo by physically interacting with non-catalytic proteins, which are also involved in recognizing substrates and synergistically determine the specificity together with HATs2,3. In this regard, the identification of HAT-specific acetylation sites in proteins is fundamental for understanding the molecular mechanisms and regulatory roles of lysine acetylation.
Previously, systematic identification of protein acetylation sites or “acetylome” was a great challenge, due to the technical limitation4,13. For example, in 2006, Kim et al. used an anti-acetyllysine antibody to purify acetyl-peptides and only detected 388 acetylation sites of 195 proteins from human HeLa cells and mouse liver mitochondria4. Recently, advances in the development of high-throughput mass spectrometry (HTP-MS) and highly potent anti-acetyllysine antibodies have greatly improved the acetylomic profiling. For example, in 2009, Choudhary et al. identified ~3,600 lysine acetylation sites in 1,750 proteins from a human acute myeloid leukemia cell line7. Later, Zhao et al. detected >1,300 acetyl-peptides of 1,047 proteins human liver tissues, and further demonstrated a number of metabolic enzymes to be regulated by acetylation5. More recently, Svinkina et al. totally identified and quantified more than 10,000 acetyl-peptides in over 3,000 proteins from Jurkat cells treated with or without suberoylanilide hydroxamic acid (SAHA)14. In our database of compendium of protein lysine modifications (CPLM), we manually curated known acetylation information and totally collected 20,088 acetylated substrates with 58,563 sites15. Although more and more acetylation sites were experimentally characterized, the regulatory HATs for most of sites remain to be dissected.
In contrast with labor-intensive and time-consuming experiments, computational prediction of lysine acetylation sites from protein sequences is also helpful to generate highly useful information for further experimental consideration. In 2006, we used 246 non-redundant lysine acetylation sites of 89 proteins as the training data set, and developed the first tool of PAIL for accurately predicting acetylation sites in proteins16. Later, Basu et al. prepared two training data sets containing 51 and 73 known acetylation sites respectively, and designed an alternative software package of PredMod17. In 2010, Gnad et al. compiled a much larger training data set with 3,600 human lysine acetylation sites from a large-scale study7, and adopted the support vector machines (SVMs) algorithm to predict acetylation sites18. To date, there have been at least a dozen of additional computational programs constructed for the accurate prediction of general lysine acetylation sites, such as LysAcet19, N-Ace20, EnsemblePail21, BPBPHKA22, PLMLA23,24, PSKAcePred25, KAcePred26, LAceP27, SSPKA28, AceK29, iPTM-mLys30 and KA-predictor31. However, none of them can predict HAT-specific sites. In 2012, Li et al. collected 267 and 82 sites modified by CBP/p300 and GCN5/PCAF HATs, respectively11,32. Using this training data set, they developed the first tool of ASEB to accurately predict HAT- or KAT-specific acetylation sites in the family level11,32. They further predicted and experimentally validated that MBD1 and MTA1 are exclusively acetylated by p300 but not PCAF, whereas DNA polymerase β (Pol-β) and DDB1 are specifically modified by PCAF but not p30011.
In this study, we aimed to develop a highly useful tool to predict HAT-specific lysine acetylation sites in the individual HAT level. First, we manually collected 702 experimentally identified HAT-specific sites of 205 proteins for seven well-characterized HATs, including CREBBP, EP300, HAT1, KAT2A, KAT2B, KAT5 and KAT8. In our data set, there were 544 and 158 HAT-specific acetylation sites in 160 human and 45 non-human proteins, respectively. A previously established algorithm of Group-Based Prediction System (GPS)33 was adopted and further improved for training a computational model for each HAT, by using human HAT-specific sites as the training data set. Then GPS-PAIL was constructed, whereas its prediction accuracy was critically evaluated with the leave-one-out (LOO) validation and n-fold cross-validations. We also compared GPS-PAIL with the existing tool of ASEB11,32, using non-human HAT-specific sites as a testing data set. We also used GPS-PAIL to perform a large-scale prediction of potential HATs for acetylation sites identified from high-throughput experiments in eukaryotes. Both online service and local packages of GPS-PAIL were implemented and could be accessed at http://pail.biocuckoo.org/.
Results
Sequence preferences around different types of HAT-specific acetylation sites
From the scientific literature and public data resources11,15,32, we totally collected 702 non-redundant HAT-specific acetylation sites of 205 protein protein substrates for seven HATs (Table 1, Supplementary Tables S1 and S2). The numbers of collected substrates and acetylation sites were summarized for each HAT, whereas the keywords used for searching HAT-specific acetylation sites were also present (Table 1). For convenience, the standard gene names in UniProt database34 were adopted. CREBBP and EP300, usually called as CBP and p300, belong to the p300/CBP family1,10. HAT1, KAT2A and KAT2B, also named as KAT1, GCN5 and PCAF, are key members of the GNAT family1,3,12. Also, KAT5 and KAT8, also called as Tip60 and MOF/MYST1, are essential HATs of the MYST family3,35.
Table 1. A summary of the numbers of acetylated substrates and sites for seven types of HATs curated from the literature.
| HAT | Number | Keywords | |
|---|---|---|---|
| Substrate | Site | ||
| CREBBP | 71 | 248 | CREB-binding protein acetylation; CREBBP acetylation; CBP acetylation |
| EP300 | 138 | 496 | EP300 acetylation; P300 acetylation |
| HAT1 | 12 | 15 | KAT1 acetylation; HAT1 acetylation |
| KAT2A | 24 | 69 | KAT2A acetylation; GCN5 acetylation; GCN5L2 acetylation |
| KAT2B | 39 | 109 | KAT2B acetylation; PCAF acetylation |
| KAT5 | 15 | 32 | KAT5 acetylation; HTATIP acetylation; TIP60 acetylation |
| KAT8 | 8 | 10 | KAT8 acetylation; MOF acetylation; MYST1 acetylation |
The keywords used to search PubMed for the collection of HAT-specific acetylation sites were shown.
Previously, it was demonstrated that different types of HATs can acetylate overlapping but distinct substrates1,11,12. For example, both CREBBP and KAT2B acetylate Ku70 at K542 in vivo to inhibit the Bax-mediated apoptosis12, whereas several proteins such as MBD1 and MTA1 are preferentially acetylated by p300 but not PCAF11. Thus, different HATs exhibit mutual but still distinct specificity for the substrate recognition, and we speculated whether there are potentially different sequence preferences around different types of HAT-specific sites. To address this problem, here we used pLogo36, a convenient tool for the visualization of sequence logos, to analyze the amino acid occurrence around different types of HAT-specific sites (Fig. 1). The sequence logo of HAT1 was not drawn due to the data limitation.
Figure 1. The amino acid frequencies of different types of HAT-specific lysine acetylation sites were analyzed and visualized by pLogo36.

From the results, although the K residue is significantly over-represented in +3, +4 and +5 positions for both CREBBP- and EP300-specific acetylation sites, the G and S residues are enriched in −1 and +1 positions for CREBBP, whereas A and K residues prefer to occur at −1 and +1 positions for EP300, respectively (Fig. 1). For the GNAT family, a G residue preferentially occur at −2 position for both KAT2A and KAT2B, while the K and G residues are over-represented at −4 positions of KAT2A and KAT2B, respectively (Fig. 1). In addition, the residues of G, K and K prefer to occur at −5, −4 and −3 positions of KAT5-specific sites, while the residues of G, G and A preferentially occur at −3, −2 and −1 positions of KAT8-specific sites. However, the R and K residues are enriched at +3 and +4 positions for both KAT5 and KAT8, respectively (Fig. 1). Taken together, our results demonstrated that different types of HAT-specific sites have considerably similar but distinct sequence preferences.
Development of GPS-PAIL for the prediction of HAT-specific lysine acetylation sites
Since different HATs have distinct sequence specificities for the substrate modifications, here we aimed to develop a highly useful tool to predict HAT-specific acetylation sites from protein sequences, and improved a previously established algorithm of GPS 2.233 to train a computational model for each HAT, respectively. We used 544 human HAT-specific acetylation sites of 160 protein substrates as the training data set. For a convenient usage, both online service and stand-alone packages of GPS-PAIL were provided, with a user-friendly interface. GPS-PAIL can predict HAT-specific acetylation sites for seven HATs including CREBBP, EP300, HAT1, KAT2A, KAT2B, KAT5 and KAT8.
The online service of GPS-PAIL was implemented in PHP and JavaScript. Also, two web services, IUPred37 and NetSurfP38 were integrated for the prediction of protein structural features, such as disorder regions, secondary structures and surface accessibilities. The website of GPS-PAIL was extensively tested on various web browsers such as Internet Explorer, Mozilla Firefox and Google Chrome to provide a robust service. For the usage of GPS-PAIL, here we chose the protein sequence of human p53 as an example (Fig. 2). The input of the online service contained three parts, including the HAT types, the protein sequences, and the thresholds (Fig. 2a). One or multiple HATs can be selected by clicking the checkboxes, while four threshold options including “High”, “Medium” and “Low” and “All” were provided in the lower panel. In GPS-PAIL, the “High”, “Medium” and “Low” thresholds were selected with Sp values of ~95%, ~90 and 85%, respectively. The “All” option will generate a predicted score for each lysine residues with no stringency. One or multiple protein sequences can be directly input or uploaded through a sequence file in FASTA format. Furthermore, users can transfer to the “comprehensive” mode by clicking the “here >>” link, to perform the predictions of secondary structures and surface accessibilities of given proteins (Fig. 2a).
Figure 2. The user interface of GPS-PAIL online service and local packages.

(a) As an example, the protein sequence of human p53 can be directly inputted for the prediction of HAT-specific acetylation sites. (b) The detailed predictions will be shown in a tabular format, while additional information such as surface accessibilities, disorder regions and secondary structures will be predicted and presented. A brief summarization of predicted sites will be also shown. (c) For predicting multiple protein sequences, the local packages can be downloaded and used with a higher speed.
After starting the prediction, the website will be redirected into a waiting page and then transferred to the result page (Fig. 2b). The results of p53 contained four sequential parts, including the list of 27 predicted HAT-specific acetylation sites with the HAT information, predicted surface accessibilities and disorder regions, predicted secondary structures, and a summarization of the results. All the results can be downloadable through clicking the “Download” button (Fig. 2b). To ensure the stability of the online service, the input of protein sequences was limited with <2MB, while the large-scale computation can be performed through installing the stand-alone software packages, which were implemented in JAVA and supported for three major operation systems including Windows, Linux and Mac OS (Fig. 2c).
Performance evaluation and a comparison with ASEB
To evaluate the prediction performance and robustness of GPS-PAIL, the training data set was used to perform the LOO validation and n-fold cross-validations. The receiver operating characteristic (ROC) curves were drawn, and the values of area under the curve (AUC) were calculated. Due to the data limitation, only 4- and 6-fold cross-validations were performed for HAT1 and KAT8, whereas 4-, 6-, 8- and 10-fold cross-validations were carried out for remaining HATs (Fig. 3). From the LOO results, AUC values are 0.661, 0.704, 0.998, 0.776, 0.767, 0.544 and 0.981 for CREBBP, EP300, HAT1, KAT2A, KAT2B, KAT5 and KAT8, respectively (Fig. 3). Thus, the prediction accuracies are generally satisfying except KAT5. In addition, the results of n-fold cross-validations are quite similar with the LOO results, suggesting the computational models were trained in a robust manner (Fig. 3).
Figure 3. The LOO validation and n-fold cross-validations were performed for seven HATs including CREBBP, EP300, HAT1, KAT2A, KAT2B, KAT5 and KAT8.

The known human HAT-specific sites were used for training. Due to the data limitation, only 4- and 6-fold cross-validations were carried out for HAT1 and KAT8. AROC values of the LOO validation and n-fold cross-validations were calculated.
Moreover, we used 158 non-human HAT-specific sites as an additional testing data set, and compared GPS-PAIL to ASEB, the first established tool for predicting HAT-specific sites in the family level11,32. For a justified comparison, we directly input the protein sequences of the testing data set to GPS-PAIL and ASEB to calculate the performances (Table 2). We fixed the specificity (Sp) values to be approximately identical and compared the sensitivity (Sn) scores. For convenience, the LOO results of GPS-PAIL on our training data set were also shown (Table 2). Although the accuracies of CREBBP and EP300 in GPS-PAIL were similar with the results of CREBBP/EP300 in ASEB, GPS-PAIL generated a much better performance against ASEB for KAT2A and KAT2B (Table 2). In addition, since more HATs were available for the prediction, GPS-PAIL is more applicable for further dissecting the signaling regulations of site-specific acetylation in proteins.
Table 2. Comparison of GPS-PAIL with ASEB11,32, by using 158 non-human HAT-specific sites of 45 proteins as the testing data set.
| HAT | Positivea | Negative | Pr | Sn | Sp | |
|---|---|---|---|---|---|---|
| GPS-PAIL (LOO) | CREBBP | 167 | 1719 | 16.12 | 38.32 | 80.63 |
| EP300 | 411 | 3525 | 22.81 | 50.61 | 80.03 | |
| HAT1 | 10 | 110 | 62.50 | 100.00 | 94.55 | |
| KAT2A | 32 | 265 | 30.56 | 68.75 | 81.13 | |
| KAT2B | 69 | 954 | 17.17 | 49.28 | 82.81 | |
| KAT5 | 28 | 531 | 39.29 | 39.29 | 96.80 | |
| KAT8 | 6 | 177 | 33.33 | 83.33 | 94.35 | |
| GPS-PAIL (Testing) | CREBBP | 81 | 405 | 28.32 | 39.51 | 80.00 |
| EP300 | 85 | 605 | 25.85 | 44.71 | 81.98 | |
| HAT1 | 5 | 33 | 100.00 | 100.00 | 100.00 | |
| KAT2A | 37 | 430 | 19.23 | 54.05 | 80.47 | |
| KAT2B | 40 | 133 | 47.92 | 57.50 | 81.20 | |
| KAT5 | 4 | 7 | 100.00 | 100.00 | 100.00 | |
| KAT8 | 4 | 139 | 36.36 | 100.00 | 94.96 | |
| ASEB (Testing) | CREBBP/EP300 | 107 | 771 | 22.61 | 42.06 | 80.03 |
| KAT2A/KAT2B | 73 | 522 | 23.08 | 41.10 | 80.84 |
For convenience, the LOO results of GPS-PAIL on our training data set were also provided. a. Positive, the number of positive sites.
Large-scale prediction of potential HATs for acetylomes in eukaryotes
Ab initio prediction of HAT-specific acetylation sites directly from protein sequences will generate too many false positive hits. Thus, in this study we performed a systematic prediction of potential HATs for experimentally identified acetylation sites without the HAT information. Previously, we developed a comprehensive database of CPLM, containing 58,563 known lysine acetylation sites of 20,088 proteins from both eukaryotes and prokaryotes15. Because eukaryotic HATs were generally not conserved in prokaryotes, here we only predict potential HATs for eukaryotic acetylation sites. From CPLM, we totally obtained 44,850 sites in 15,898 proteins for nine eukaryotic species, including Homo sapiens, Mus musculus, Rattus norvegicus, Drosophila melanogaster, Cavia porcellus, Plasmodium falciparum, Toxoplasma gondii, Saccharomyces cerevisiae, and Arabidopsis thaliana (Supplementary Table S3). Before the prediction, we first determined the existence of potential orthologs of seven HATs across the nine organisms. We downloaded the proteome sequences of these species and pairwisely detected orthologs, using the strategy of reciprocal best hits (RBH)39. The orthologs of seven HATs were exactly identified and picked out if available (Fig. 4). From the results, we observed that the seven HATs were not equally conserved in eukaryotes. For example, all seven HATs are encoded in Homo sapiens, Mus musculus and Rattus norvegicus, whereas only HAT1, KAT2A/GCN5, and KAT5/ESA1 are conserved in Saccharomyces cerevisiae (Fig. 4). For each species, only detected HATs were selected for the large-scale predictions.
Figure 4. The potential orthologs of seven HATs among nine eukaryotic organisms were computationally identified with the approach of reciprocal best hits39.

The existent HATs were marked with a black ball.
To greatly reduce false positive predictions, the high threshold in GPS-PAIL was chosen. In the results, we predicted totally 4,344 acetylation sites of 2,764 protein substrates with at least one potential HAT, with an annotated coverage of 9.69% and 17.39% of all acetylation sites and proteins (Fig. 5 and Supplementary Table S3). For different species, the annotated coverage values ranged from 1.56% to 24.72% at the site level. For example, GPS-PAIL only predicted 67 sites of 46 substrates with at least one HAT from 4,284 un-annotated sites of 1,368 proteins in Saccharomyces cerevisiae (Fig. 5 and Supplementary Table S3). However, 14.20% and 24.72% of total acetylation sites were predicted with the HAT information in Homo sapiens and Arabidopsis thaliana, respectively (Fig. 5 and Supplementary Table S3). Thus, our results proposed that GPS-PAIL might be more efficient to predict HAT-specific acetylation sites in mammalians and plants.
Figure 5. A summary of numbers of proteins and acetylation sites predicted with at least one HAT across nine eukaryotes.

The heatmap was drawn with HemI44, and detailed statistics was shown in Supplementary Table S3.
In addition, the distribution of numbers of protein substrates and sites modified by different types of HATs were analyzed (Fig. 6). Among 2,764 potential HAT-specific substrates, 1,939 proteins (70.15%) were predicted to be acetylated by only one HAT, whereas 514 substrates (18.60%) were predicted to be mutually modified by two HATs (Fig. 6a). Only 311 proteins (11.25%) might be regulated by over two HATs (Fig. 6a). In the site level, the results are similar that 3,219 (74.10%) and 752 (17.31) acetylation sites were modified by one and two HATs, respectively (Fig. 6b). The overlaps of predicted substrates and sites for CREBBP, EP300, HAT1, KAT2A and KAT2B were analyzed (Fig. 6c,d), while KAT5 and KAT8 were not included due to the data limitation from predictions. In the protein level, most of substrates were acetylated by only one HAT, while only nine proteins can be regulated by the five HATs (Fig. 6c). In the acetylation site level, the results were similar and no site can be modified by all the five HATs (Fig. 6d). Taken together, our large-scale analyses of predicted acetylated proteins and sites also demonstrated that different HATs recognize mutual but still distinct substrates. The detailed results of 2,764 proteins together with predicted sites and GPS-PAIL scores were shown in Supplementary Table S4.
Figure 6. The distribution of predicted HAT-specific substrates and sites in nine eukaryotes.

The number of potential HATs for (a) predicted proteins and (b) acetylation sites. The overlaps of (c) predicted substrates and (d) sites were shown for CREBBP, EP300, HAT1, KAT2A and KAT2B.
Discussion
HAT- or KAT-mediated acetylation at specific lysine residues of proteins is an essential PTM, conserved in both prokaryotes and eukaryotes, and plays a critical role in the regulation of numerous biological processes and cellular pathways1,2,3,4,5,6,7. Recent advances in the development of the state-of-the-art techniques in acetylomics have enabled to identify and quantify thousands of acetylation sites in a single run5,7,14. Although over 58,000 acetylation sites have been characterized in prokaryotic and eukaryotic species, the regulatory HATs of most of these sites still remain to be elucidated. Previously, we and others developed about 15 computational programs to predict general acetylation sites from protein sequences, with a satisfying accuracy7,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31. However, the prediction of HAT-specific acetylation sites was still unavailable until the release of ASEB11,32, which clearly demonstrated that different types of HATs could modify distinct protein substrates11,32. Since ASEB only predicted HAT-specific sites in the family level, with only two predictors such as CBP/p300 and GCN5/PCAF, the prediction of specific acetylation sites for individual HATs is still a great challenge.
In this work, we first collected 702 known HAT-specific acetylation sites in 205 proteins for seven HATs including CREBBP, EP300, HAT1, KAT2A, KAT2B, KAT5 and KAT8, from the scientific literature and public data resources such as CPLM15 and ASEB11,32. The sequence preferences of different types of HAT-specific sites were analyzed, while the results demonstrated that different HATs recognize similar but considerably distinct sequence motifs for the substrate recognition. Using known human HAT-specific sites as the training data set, we further developed GPS-PAIL for the prediction of HAT-specific sites in the single HAT level, while both online service and local packages were implemented. We critically evaluated the prediction performance of GPS-PAIL by using the LOO validation and n-fold cross-validations. By a comparison with ASEB using non-human HAT-specific sites as an additional testing data set, GPS-PAIL exhibited at least a comparative accuracy. For HAT1 and KAT8, the values of Pr, Sn and Sp were all equal to 100% on the testing data set. However, the LOO results of HAT1 and KAT8 on the training data set didn’t reach an accuracy of 100% (Table 2). Because there were only 5 and 4 known HAT1- and KAT8-specific acetylation sites in the testing data set, we couldn’t conclude a perfect performance for the two HATs, and further evaluations still remain to be performed when more specific sites were experimentally identified.
Using GPS-PAIL, we performed a large-scale analysis to annotate potential HATs for known acetylation sites in nine eukaryotic organisms. Again, the large-scale prediction proposed that most of protein substrates and sites were acetylated by only one HAT, and the results further supported that different HATs recognize overlapping but still distinct substrates. We also carefully checked the literature and UniProt database34, and all known site-specific HAT-substrate relations (ssHSRs) in the prediction results were pinpointed (Supplementary Table S4). Previously, it was demonstrated that various functional features of proteins, such as gene ontology (GO) annotations and protein-protein interactions (PPIs), were beneficial for the prediction of kinase-specific phosphorylation sites40,41. In this work, the GO information was not used, because the functional diversity of HAT-specific acetylated substrates was high and no particularly significant GO terms were detected from the statistical enrichment analysis. However, the PPIs between HATs and substrates are potentially useful to reduce false positive predictions. From the STRING database42, the pre-integrated PPI data sets for nine species were retrieved, and the site-specific HAT-substrate relations with or without PPIs were shown (Supplementary Table S4). In the results, we observed there were 315 (5.23%) known ssHSRs with experimental evidences and 2,493 (41.41%) predicted ssHSRs with PPIs, respectively (Supplementary Table S4). Interestingly, we found 101 known ssHSRs without the PPI information (Supplementary Table S4). This is because interactions between HATs and substrates are usually transient and dynamic with a weak binding affinity, which might be difficult to be detected by standard PPI screenings or computational predictions.
For the future plan, we will continuously collect experimentally identified HAT-specific acetylation sites if available in the literature. Undoubtedly, a larger training data set will generate a more accurate performance for the prediction. Also, we will further refine and improve the prediction algorithm. For example, we recently developed GPS-SUMO for the prediction of sumoylation sites and SUMO-interaction motifs from protein sequences, with an enhanced version of GPS algorithm43. Currently, the GPS algorithm is still under improvement, and we will test the accuracy of the latest version of GPS algorithm on the prediction of HAT-specific acetylation sites. Taken together, in this study we developed an efficient tool GPS-PAIL to predict HAT-specific acetylation sites for seven HATs, with a satisfying accuracy. The prediction results of potential HATs for known acetylation sites in eukaryotes from the large-scale analysis can also serve as a useful data resource for further experimental consideration.
Methods
Data collection and preparation
First, we collected experimentally identified HAT-specific lysine acetylation sites from the scientific literature if available. For each known HAT2,3, we used its standard gene name, protein name or aliases together with the keyword of “acetylation” to search the PubMed database (Table 1). For example, multiple keyword combinations such as “’CREB-binding protein’ acetylation”, “CREBBP acetylation” and “CBP acetylation” were used to search CREBBP-specific acetylation sites, whereas “KAT5 acetylation”, “Tip60 acetylation” and “HTATIP acetylation” were used to find KAT5-specific sites (Table 1). The known HAT-specific sites in CPLM database15 and ASEB training data set11,32 were also integrated. Only HATs with at least five known specific sites were reserved for the further analysis, and their standard gene names from the UniProt database34 were adopted. Then we mapped all HAT-specific substrates to the primary protein sequences downloaded from the UniProt database34, and pinpointed the exact acetylation position. The redundancy was cleared, and the final data set contained 702 unique HAT-specific acetylation sites in 205 proteins, including 544 human acetylation sites in 160 proteins (Supplementary Table S1) and 158 non-human sites of 45 proteins (Supplementary Table S2).
In this study, the human HAT-specific sites were adopted for training, while non-human sites were used as an additional data set to test the prediction performance. For the preparation of the training data set, we defined an acetylation site peptide ASP(m, n) as an acetyllysine amino acid flanked by m residues upstream and n residues downstream. For each HAT, its experimentally identified acetylation sites were taken as positive data (+), whereas all the other non-acetylated lysine residues in the same proteins were regarded as negative data (−). The training and testing procedures were independently performed for each HAT. For the large-scale prediction of HAT-specific acetylation sites in eukaryotes, we totally obtained 44,850 known but un-annotated acetylation sites in 15,898 proteins of nine species from CPLM15 (Supplementary Table S3).
Performance evaluation
As previously described33, three measurements of Sn, Sp and precision (Pr) were adopted to evaluate the prediction performance. The three measurements were defined as equation (1), (2), and (3):
 |
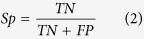 |
 |
The LOO validation and 4-, 6-, 8- and 10-fold cross-validations were performed. The ROC curves were drawn and AROC values were calculated.
Algorithm
Previously, we developed the GPS 2.2 algorithm for the prediction of APC/C recognition motifs such as D-boxes and KEN-boxes proteins33. The algorithm comprises two major parts, including the scoring strategy and performance improvement.
In the former part, based on the basic hypothesis of similar short peptides exhibiting similar biochemical properties with similar functions, we used an amino acid substitution matrix, e.g., BLOSUM62, to evaluate the similarity between two ASP(m, n) peptides of A and B as equation (4):
 |
Score(A[i], B[i]) represents the substitution score of the two residues of A[i] and B[i] in the amino acid substitution matrix at the position i. If S(A, B) <0, we redefined it as S(A, B) = 0. For each HAT, a given ASP(m, n) was then pairwisely compared with each of its known specific acetylation sites to calculate the similarity score. The average value of the substitution scores was taken as the final score for the HAT.
The second part comprises three distinct steps, including motif length selection, weight training and matrix mutation. To monitor the performance improvement, here we fixed the Sp at 90% and compared Sn values of the LOO validation.
Motif length selection
In this step, the combinations of ASP(m, n) (m = 1, …, 30; n = 1, …, 30) were exhaustively tested, while the optimized combination of ASP(m, n) was determined based on the highest LOO result for each HAT, separately.
Weight training
Since different positions can provide different contributions to modification specificity, we refined the substitution score between the two ASP(m, n) peptides A and B was as equation (5):
 |
The wi value denotes the weight of position i. Again, if S′(A, B) < 0, we redefined it as S′(A, B) = 0. Initially, the weight of each position in ASP(m, n) was taken as 1. Then we randomly picked out a weight of any position for +1 or −1, and adopted the manipulation if the LOO performance was increased. The process was continued until the Sn value was not increased any longer.
Matrix mutation
The aim of this step is to generate an optimal or near-optimal scoring matrix. BLOSUM62 was chosen as the initial matrix, and the LOO performance was calculated. Then we improved the Sn though randomly picking out an element of the BLOSUM62 matrix for +1 or −1. The process was repeated until convergence was reached.
During the training, the order of the three steps in performance improvement can be shuffled. To improve the training efficiency, we adopted the simulated annealing (SA) algorithm to optimize the parameters for the steps of Weight Training and Matrix Mutation.
The PPI data sets
The PPIs together with corresponding protein sequences of nine species were downloaded from the STRING database (Version 10, http://string-db.org), which is an integrative data resource for both physical and functional associations among proteins in over 2,000 organisms42. Totally, we obtained 28,386,035 pairs of PPIs in nine species, including 4,274,001, 5,109,107, 5,319,621, 2,176,849, 2,340,229, 2,575,257, 332,297, 939,998 and 5,318,676 PPIs from Homo sapiens, Mus musculus, Rattus norvegicus, Drosophila melanogaster, Cavia porcellus, Plasmodium falciparum, Toxoplasma gondii, Saccharomyces cerevisiae and Arabidopsis thaliana, respectively.
Additional Information
How to cite this article: Deng, W. et al. GPS-PAIL: prediction of lysine acetyltransferase-specific modification sites from protein sequences. Sci. Rep. 6, 39787; doi: 10.1038/srep39787 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Basic Research Program (973 project) (2013CB933900, 2012CB910101 and 2011CB910600), Natural Science Foundation of China (31671360, 81272578, J1103514 and 31501069), and International Science & Technology Cooperation Program of China (2014DFB30020).
Footnotes
Author Contributions Y. Xue and Z.L. designed and supervised experiments. W.D., C.W., Z.L. and Y. Xue performed experiments and data analysis. W.D., C.W. and Z.L. developed the tool. Y.Z., Y. Xu and S.Z. contributed to data analysis. Y. Xue wrote the manuscript with contributions of all authors. All authors reviewed the manuscript.
References
- Schiltz R. L. et al. Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. J Biol Chem 274, 1189–1192 (1999). [DOI] [PubMed] [Google Scholar]
- Yang X. J. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res 32, 959–976 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. J. & Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26, 5310–5318 (2007). [DOI] [PubMed] [Google Scholar]
- Kim S. C. et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 23, 607–618 (2006). [DOI] [PubMed] [Google Scholar]
- Zhao S. et al. Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C. et al. Function and molecular mechanism of acetylation in autophagy regulation. Science 336, 474–477 (2012). [DOI] [PubMed] [Google Scholar]
- Choudhary C. et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 (2009). [DOI] [PubMed] [Google Scholar]
- Li D. et al. Regulation of Kruppel-like factor 6 tumor suppressor activity by acetylation. Cancer Res 65, 9216–9225 (2005). [DOI] [PubMed] [Google Scholar]
- Wang L. et al. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science 333, 765–769 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L. W. & Giordano A. Acetyltransferase machinery conserved in p300/CBP-family proteins. Oncogene 21, 2253–2260 (2002). [DOI] [PubMed] [Google Scholar]
- Li T. et al. Characterization and prediction of lysine (K)-acetyl-transferase specific acetylation sites. Mol Cell Proteomics 11, M111 011080 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen H. Y. et al. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell 13, 627–638 (2004). [DOI] [PubMed] [Google Scholar]
- Smith K. T. & Workman J. L. Introducing the acetylome. Nat Biotechnol 27, 917–919 (2009). [DOI] [PubMed] [Google Scholar]
- Svinkina T. et al. Deep, Quantitative Coverage of the Lysine Acetylome Using Novel Anti-acetyl-lysine Antibodies and an Optimized Proteomic Workflow. Mol Cell Proteomics 14, 2429–2440 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. et al. CPLM: a database of protein lysine modifications. Nucleic Acids Res 42, D531–536 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A., Xue Y., Jin C., Wang M. & Yao X. Prediction of Nepsilon-acetylation on internal lysines implemented in Bayesian Discriminant Method. Biochem Biophys Res Commun 350, 818–824 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A. et al. Proteome-wide prediction of acetylation substrates. Proc Natl Acad Sci USA 106, 13785–13790 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnad F., Ren S., Choudhary C., Cox J. & Mann M. Predicting post-translational lysine acetylation using support vector machines. Bioinformatics 26, 1666–1668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. et al. Improved prediction of lysine acetylation by support vector machines. Protein Pept Lett 16, 977–983 (2009). [DOI] [PubMed] [Google Scholar]
- Lee T. Y. et al. N-Ace: using solvent accessibility and physicochemical properties to identify protein N-acetylation sites. J Comput Chem 31, 2759–2771 (2010). [DOI] [PubMed] [Google Scholar]
- Xu Y., Wang X. B., Ding J., Wu L. Y. & Deng N. Y. Lysine acetylation sites prediction using an ensemble of support vector machine classifiers. J Theor Biol 264, 130–135 (2010). [DOI] [PubMed] [Google Scholar]
- Shao J. et al. Systematic analysis of human lysine acetylation proteins and accurate prediction of human lysine acetylation through bi-relative adapted binomial score Bayes feature representation. Molecular bioSystems 8, 2964–2973 (2012). [DOI] [PubMed] [Google Scholar]
- Shi S. P. et al. PLMLA: prediction of lysine methylation and lysine acetylation by combining multiple features. Molecular bioSystems 8, 1520–1527 (2012). [DOI] [PubMed] [Google Scholar]
- Shi S. P. et al. A method to distinguish between lysine acetylation and lysine methylation from protein sequences. J Theor Biol 310, 223–230 (2012). [DOI] [PubMed] [Google Scholar]
- Suo S. B. et al. Position-specific analysis and prediction for protein lysine acetylation based on multiple features. PloS one 7, e49108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suo S. B. et al. Proteome-wide analysis of amino acid variations that influence protein lysine acetylation. Journal of proteome research 12, 949–958 (2013). [DOI] [PubMed] [Google Scholar]
- Hou T. et al. LAceP: lysine acetylation site prediction using logistic regression classifiers. PloS one 9, e89575 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. Accurate in silico identification of species-specific acetylation sites by integrating protein sequence-derived and functional features. Scientific reports 4, 5765 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C. T., Lee T. Y. & Chen Y. J. An intelligent system for identifying acetylated lysine on histones and nonhistone proteins. BioMed research international 2014, 528650 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W. R., Sun B. Q., Xiao X., Xu Z. C. & Chou K. C. iPTM-mLys: identifying multiple lysine PTM sites and their different types. Bioinformatics (2016). [DOI] [PubMed] [Google Scholar]
- Wuyun Q., Zheng W., Zhang Y., Ruan J. & Hu G. Improved Species-Specific Lysine Acetylation Site Prediction Based on a Large Variety of Features Set. PloS one 11, e0155370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Du Y., Lu M. & Li T. ASEB: a web server for KAT-specific acetylation site prediction. Nucleic Acids Res 40, W376–379 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. et al. GPS-ARM: computational analysis of the APC/C recognition motif by predicting D-boxes and KEN-boxes. PloS one 7, e34370 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res 43, D204–212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z. et al. Functional characterization of TIP60 sumoylation in UV-irradiated DNA damage response. Oncogene 27, 931–941 (2008). [DOI] [PubMed] [Google Scholar]
- O’Shea J. P. et al. pLogo: a probabilistic approach to visualizing sequence motifs. Nature methods 10, 1211–1212 (2013). [DOI] [PubMed] [Google Scholar]
- Dosztanyi Z., Csizmok V., Tompa P. & Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21, 3433–3434 (2005). [DOI] [PubMed] [Google Scholar]
- Petersen B., Petersen T. N., Andersen P., Nielsen M. & Lundegaard C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC structural biology 9, 51 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov R. L., Koonin E. V. & Lipman D. J. A genomic perspective on protein families. Science 278, 631–637 (1997). [DOI] [PubMed] [Google Scholar]
- Fan W. et al. Prediction of protein kinase-specific phosphorylation sites in hierarchical structure using functional information and random forest. Amino acids 46, 1069–1078 (2014). [DOI] [PubMed] [Google Scholar]
- Xu X. et al. Improving the performance of protein kinase identification via high dimensional protein-protein interactions and substrate structure data. Molecular bioSystems 10, 694–702 (2014). [DOI] [PubMed] [Google Scholar]
- Szklarczyk D. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q. et al. GPS-SUMO: a tool for the prediction of sumoylation sites and SUMO-interaction motifs. Nucleic Acids Res 42, W325–330 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W., Wang Y., Liu Z., Cheng H. & Xue Y. HemI: a toolkit for illustrating heatmaps. PloS one 9, e111988 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.