

Abstract
With the genomic revolution and the era of targeted therapy, prognostic and predictive gene signatures are becoming increasingly important in clinical research. They are expected to assist prognosis assessment and therapeutic decision making. Notwithstanding, an evidence-based approach is needed to bring gene signatures from the laboratory to clinical practice. In early breast cancer, multiple prognostic gene signatures are commercially available without having formally reached the highest levels of evidence-based criteria. We discuss specific concepts for developing and validating a prognostic signature and illustrate them with contemporary examples in breast cancer. When a prognostic signature has not been developed for predicting the magnitude of relative treatment benefit through an interaction effect, it may be wishful thinking to test its predictive value. We propose that new gene signatures be built specifically for predicting treatment effects for future patients and outline an approach for this using a cross-validation scheme in a standard phase III trial. Replication in an independent trial remains essential.
Keywords: gene signature, prognostic, predictive, evidence based, clinical utility
introduction
Molecular signatures are becoming increasingly important for anticipating the prognosis of individual patients (‘prognostic’ biomarkers) or for predicting how individual patients will respond to specific treatments (‘predictive’ biomarkers, more generally called ‘treatment-effect modifiers’). A voluminous literature of >150 000 papers documenting thousands of claimed biomarkers has been produced in medicine of which fewer than 100 have been validated for routine clinical practice [1]. Indeed, <20 prognostic or predictive biomarkers are recognized with variable levels of evidence in the 2014 European Society of Medical Oncology (ESMO) clinical practice guidelines for lung, breast, colon and prostate cancer [2].
In early breast cancer, while several clinical prediction models exist based on clinical and pathological (CP) characteristics, such as age, tumor size, nodal status, tumor grade, estrogen receptor, at least six different gene signatures are commercially available (Oncotype DX, MammaPrint, Genomic Grade Index, PAM50, Breast Cancer Index and EndoPredict). The concordance of predicted risk categories of the different gene signatures for individual patients is moderate [3, 4], as illustrated by recent OPTIMA study which evaluated—among others—the two well-known tests Mammaprint (low/high) and Oncotype Dx (≤25 versus >25) on 302 patients in a head-to-head comparison and found a low level of agreement, i.e. a kappa value of 0.40 (95% CI 0.30–0.49) [5]. Of course, even when repeating the same assay twice on a single tumor sample, some inherent degree of inaccuracy would be expected but unlikely to this extent. This has led to a pretty awkward situation where the treatment decision for adjuvant chemotherapy does not depend anymore on the clinician but on the genomic test ordered. Furthermore, according to a European consensus panel, none of these tests reached the highest level of evidence [6] and according to an Evaluation of Genomic Applications in Practice and Prevention (EGAPP) panel, there was only indirect evidence that Oncotype Dx could predict benefit from chemotherapy [7], while an ASCO panel in the United States gave a strong recommendation with high level of evidence that Oncotype Dx may be used to guide decisions on adjuvant systemic chemotherapy for node-negative (N0) ER-positive (ER+), HER2-negative (HER–) breast cancer [8]. This divergence may result from the degree of subjectivity in evidence evaluation or from a different vision of what type of evidence is needed for a gene signature to be clinically useful. In this commentary, we focus on prognostic and predictive gene expression signatures in breast cancer to highlight the difficult path from the laboratory to the clinic, but the concepts are applicable to other omics data.
prognostic versus predictive signature: what's in a name?
Gene signatures can assist clinicians in prognosis assessment and therapeutic decision making. A signature is prognostic if it discriminates well between patients with a good or bad prognosis in the absence of treatment or in the context of a standard therapy. In the top left panel of Figure 1, we show an example of prognostic signature: for untreated patients (dashed lines), the survival profile is very different according to the signature categories, i.e. the likely natural course of the disease can be forecasted thanks to the signature values [9]. On the other hand, whatever the risk group, the relative effect of treatment (solid versus dashed lines) is similar. A signature is called predictive (of the treatment effect) if the relative treatment benefit varies according to signature values. In the bottom panels of Figure 1, one can see that the treatment is beneficial only for low-score patients, while for high score ones it is either less beneficial (left, quantitative interaction) or harmful (right, qualitative interaction). In the case of a quantitative interaction, the magnitude of the relative treatment effect is different, but the effect is in the same direction. In this case, it is not clear that treatment could be withheld in any of the subgroups. In the case of a qualitative interaction, the direction of the treatment effect is different according to signature values. The most appropriate way to identify a predictive gene signature is through an interaction test between the signature and the treatment using data from a trial [10] in which the treatment has been randomly allocated to patients.
Figure 1.

Example of survival curves in experimental (Exp) versus control (Ctrl) arms for patients with a high gene signature score (High score) versus patients with a low gene signature score (Low score) in the case of a prognostic gene signature (top left) or a predictive gene signature, with either quantitative (bottom left) or qualitative (bottom right) interaction.
Results from randomized controlled trials are often difficult to translate into predictions for individual patients, but estimated absolute risk reductions from large randomized trials do still provide the best guidance [10]. Even when there is not a single predictive signature or biomarker for a particular treatment, prognostic signatures or biomarkers can be useful for prognosis and treatment counseling [11]. In early breast cancer, for instance, the proportional risk reduction obtained by chemotherapy does not significantly vary according to CP factors, even in large well-powered meta-analyses [12, 13]. If we assume a 33% relative risk reduction using chemotherapy regimen in an ER+ early breast cancer population [12], we can estimate absolute increases in 10-year predicted survival when adding chemotherapy to endocrine therapy [14]. For example, for an N0 ER+ patient with a 10-year breast cancer-specific survival predictions of 95% with endocrine therapy, the absolute benefit when adding chemotherapy is estimated by 2% which needs to be outweighed against potential side effects of the treatment.
prognostic gene signatures: the evidence-based path from proof of concept to clinical utility
development and validation of a signature
One of the very first steps in the development of a gene signature is finding out how to compute a score based on the biomarkers measured, while the number of biomarkers keeps on increasing with technology advances. Identifying a meaningful prognostic model through high-dimensional regression raises particular challenges from a statistical point of view, including nonidentifiability of the models, instability of selected biomarkers [15], sparse model selection and multiple testing. Several penalized methods exist to perform variable selection in this high-dimensional space [16], while controlling the risk of false positives [17].
Table 1 shows different criteria to evaluate when developing a signature from bench to bedside. The EGAPP initiative has proposed general definitions of analytical and clinical validity, and of clinical utility [18], which are transposed here to the gene signature context. Issues related to the assessment of their analytical validity are beyond the scope of this review. The assessment of the generalizability of a gene signature needs independent validation of its prognostic value in multiple series; this is now well established [19–21] and will not be detailed here.
Table 1.
Evidence-based criteria for a prognostic gene signature in the path from the laboratory to clinical practice
| No. | Concept | Elaboration |
|---|---|---|
| 1 | Proof of concept | Do signature levels differ substantially between patients with and without outcome? |
| 2 | Analytical validity | Signature's ability to accurately and reliably measure the genotype of interest between and within laboratories |
| 3 | Clinical validity | Does the signature predict risk of outcome in multiple external cohorts or nested case–control/case–cohort studies? |
| 4 | Incremental value | Does the signature add enough information to established clinico-pathological prognostic markers or provide a more reproducible measurement of one of them? |
| 5 | Clinical impact | Does the signature change predicted risk sufficiently to change recommended therapy? |
| 6 | Clinical utility | Does use of the signature improve clinical outcome, especially when prospectively used for treatment decisions in a randomized controlled trial? |
| 7 | Cost-effectiveness | Does use of the signature improve clinical outcome sufficiently to justify the additional costs of testing and treatment? |
compare the signature with established clinico-pathological factors
A key issue in assessing the added value of a prognostic signature is to study whether it adds independent prognostic information to the risk determined by a CP model (incremental value). A gene signature could also be of interest if it provides a more reproducible, cheaper or more accurate measurement of an already existing biomarker that has proven clinical utility so that the CP rule could be updated [20]. The Oncotype Dx assay is very successful in the USA with an estimated target market penetration of 50% [22]. One could hypothesize that one of the main reasons for its success is that a proliferation-based signature measured by a single reference laboratory took the place of the histological tumor grading, which has been plagued by a perceived suboptimal between-laboratory reproducibility [23].
To illustrate how to evaluate incremental prognostic value, we used publicly available microarray data of 845 patients (189 pathological complete responses or pCRs) from eight clinical studies that included patients treated by anthracycline-based chemotherapy [24]. We computed two gene signatures: an approximate version of the MammaPrint signature that we denote as proliferation signature and an immune-based gene signature [24]. Because the gene signatures are often derived on different microarray platforms from different laboratories and heterogeneous retrospective patient cohorts, we computed the scores as a weighted average of the genes and scaled each signature within study so that the 2.5% and 97.5% quantiles equaled −1 and +1, respectively [24, 25]. The added value of the two gene signatures was evaluated in logistic regression models after adjustment for CP factors as detailed in Table 2. When using the likelihood ratio test relative to the model with established prognostic factors only [26, 27], both signatures do add significant prognostic information to the CP model and they both add information to each other. We also evaluated the discrimination, i.e. the ability to distinguish patients who had a pCR from those who did not, through the area under the receiver operating characteristic curve (AUC), for the CP model with and without the gene signatures. The AUC of the CP model was already high (0.78; 95% CI 0.75–0.82; Figure 2 and Table 2), illustrating the strong discrimination of the CP factors. Adding both the signatures provided only a slight increase (0.80; 95% CI 0.77–0.83). Therefore, the added discrimination of the gene signatures for pCR is moderate in this neoadjuvant example in breast cancer. This is often the case in applications in medicine as only very strong independent prognostic factors can lead to large increases in predictive accuracy. For survival outcomes, there exist different generalization of the AUC [28, 29] and an alternative measure is an R2-type statistic to compare the extra variation in clinical outcome explained by the gene signature [30]. Of note, also the batch and laboratory effects typically observed play a role in the lack of applicability of many gene signatures in the clinic, for which a fully specified algorithm is needed for a single patient from a random batch or laboratory.
Table 2.
Evaluation of incremental prognostic value of a proliferation and immune gene signature to a standard clinico-pathological (CP) model for pathological complete response (pCR) in 845 early breast cancer patients treated with neoadjuvant anthracycline-based chemotherapy
| Comparison | Likelihood ratio statistic | P-value | AUCa (95% CI) |
|---|---|---|---|
| CP versus null model | 151.4 | <10−16 | 0.78 (0.75–0.82) |
| CPb + proliferation versus CP | 9.4 | 2.2 × 10−3 | 0.79 (0.75–0.82) |
| CP + immune versus CP | 13.8 | 2.0 × 10−4 | 0.79 (0.76–0.83) |
| CP + immune + proliferation versus CP | 26.1 | 2.2 × 10−6 | 0.80 (0.77–0.83) |
| CP + immune + proliferation versus CP + immune | 12.2 | 4.7 × 10−4 | 0.80 (0.77–0.83) |
| CP + immune + proliferation versus CP + proliferation | 16.6 | 4.5 × 10−5 | 0.80 (0.77–0.83) |
aAUC (area under the ROC curve) of the left-sided model in the comparison.
bClinico-pathological logistic model for pathological complete response including treatment (anthracyclines versus anthracyclines plus taxanes), age (≤50 versus >50 years), clinical tumor size (cT0, 1, 2 versus cT3, 4), clinical nodal status (negative versus positive), histologic grade (1, 2 versus 3), ER status (negative versus positive) and HER2 status (negative versus positive) and study effect using publicly available gene expression data of neoadjuvant studies (845 patients, 189 pathological complete responses) as described in [24]; proliferation: approximate version of the MammaPrint gene signature, immune: immune1 signature from [24].
Figure 2.
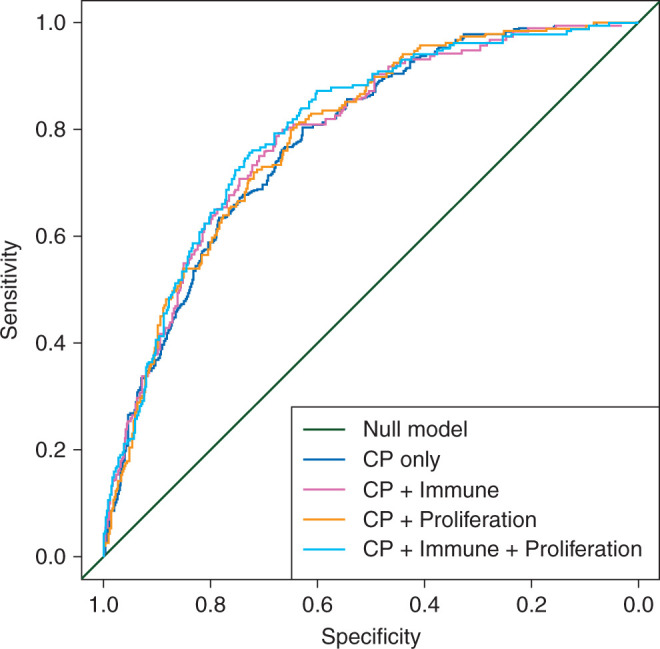
Receiver-operating characteristics curves when adding a proliferation and immune gene signature to a clinico-pathological (CP) model for pathological complete response in 845 early breast cancer patients treated with neoadjuvant anthracycline-based chemotherapy.
In another example of 883 women treated with either tamoxifen or letrozole monotherapy in the Breast International Group 1–98 trial, one of the cited proliferation signatures in breast cancer, the Genomic Grade Index, was prognostic of the distant recurrence-free interval, in addition to the CP model as measured by the likelihood ratio test [31]. Nevertheless, similar results were obtained with centrally reviewed continuous Ki67 by an expert pathologist, which highlights the importance of including all known prognostic factors in the CP model.
In addition to the discrimination, it is also of importance to evaluate the calibration of prediction models that include gene signatures, i.e. the agreement between predicted risk and clinical outcome frequencies [32], In our opinion, very little is known about the calibration of the commercially available gene signatures in early breast cancer, e.g. for patients with a predicted 10-year risk of distant metastasis below 5% with the CP model and with the CP model plus the signature, what is the observed frequency of distant events at 10-years? Adding the gene signature to an established model will also be only of interest if the predicted risk of such patients changes sufficiently compared with the standard CP model to have consequences in terms of treatments. Useful summary measures and graphical displays to evaluate the subtle changes in prediction scores of patients can be found elsewhere [32–34].
clinical trial designs for prognostic signatures
To assess the readiness of omics-based tests for guiding patient care in clinical trials, a useful tool is the checklist developed by the USA National Cancer Institute [35], covering issues related to specimens, assays, mathematical modeling, clinical trial design, and ethical, legal and regulatory aspects. Trial designs evaluating the clinical impact of patients being offered a prognostic gene signature are rather similar to available trial designs for diagnostic tests [36–39]. The operating characteristics of some of the trial designs integrating gene signatures in breast cancer have been discussed previously [40]. In the MINDACT study [41], a randomized trial was setup in the discordant risk population—based on a CP model and the gene signature—to evaluate the capacity of the MammaPrint signature to identify patients in whom chemotherapy can be avoided when the CP model says otherwise. Its primary test statistic is however not based on the randomization, so a prospective cohort study would have been sufficient to answer this objective. If the two prediction models (CP model and gene signature) disagree in 32% of the patients, and if the treatment reduces 10-year mortality in the overall population from 24% to 20%, then the absolute difference in mortality between the two strategies is only 0.5%, and it has been calculated that 50 000 patients would be necessary to identify this mortality difference in a statistically satisfactory manner [42].
In the TAILORx trial [43], women with intermediate Oncotype DX signature risk score were randomized between adjuvant chemotherapy and not, and the primary objective is to evaluate the noninferiority of the control arm compared with the chemotherapy arm. It may seem peculiar to set up a noninferiority trial of standard chemotherapy of which the relative efficacy is already well known. Recently, the data and safety monitoring committee of the TAILORx trial recommended that the results of the unrandomized low-risk group defined by Oncotype Dx be released [44]. After a median follow-up of 6.7 years, the estimated 5-year invasive disease-free survival was 93.8% (95% CI 92.4%–94.9%) in this low-risk group. The question remains whether such a subgroup of patients could not have been identified with a solid CP model.
The big hope behind these trials is that secondary analyses would reveal variation in relative efficacy according to fine-tuned modeling of CP factors and the gene signatures that were missed in prior analyses of historical trials by categorized CP risk groups. On the other hand, one could argue that, if the biological signal was really strong, even a less reliable measurement method (e.g. of ER and Ki67) with a different categorization would already have shown some variation. This may be matter of subjective debate. In both these trials, since the annual rate of distant relapse or deaths is quite low in early breast cancers, a very long follow-up is required to answer the clinical questions [40]. These examples illustrate that, in the context of a relatively good prognosis population and a small absolute treatment benefit (of chemotherapy), developing a randomized controlled trial to demonstrate the clinical utility of a prognostic gene signature is quite challenging and that a cohort study may in several occasions be a more appropriate tool to develop and validate a fine-tuned CP plus gene signature model.
cost-effectiveness
The very last aspect in studying a gene signature is its cost-effectiveness: despite being more prognostic than the CP model alone, a signature could be of limited usefulness if its cost is too high. For illustration, in the population of N0 breast cancer patients, the MammaPrint signature was deemed unlikely to be cost-effective from the French National Insurance perspective [45].
For EGAPP, the cost-effectiveness evaluation is only seen as a contextual factor [18], while for the National Institute for Health and Care Excellence in the UK, the value of diagnostic technologies is based on three main criteria: test accuracy, clinical effectiveness and cost-effectiveness. Specific evidence requirements need to be defined for policymakers and reimbursement agencies to introduce gene signatures or molecular tests into clinical practice from a health economical perspective [2].
‘predictive’ gene signatures as treatment-effect modifiers
The approach of treating broad populations of patients and having large inclusion criteria in clinical trials is based on the assumption that treatment-by-subset interactions are unlikely to occur on mortality end points [46]. On the other hand, increasing knowledge of biology suggests that such interactions are actually more likely to occur than previously thought [47], with ER, HER2, KRAS and EGFR mutations as famous examples. Furthermore, ignoring strong treatment-by-biomarker interactions in a patient population can substantially reduce the statistical power of trials aimed at showing the overall benefit of new treatments [48]. Recently, there have been some attempts to identify gene signatures that are associated with higher benefit of treatments, such as an 8-gene and a 14-gene signature for the degree of trastuzumab benefit in early breast cancer [49, 50]. There is some risk of overoptimism in these two examples since the former was flawed by a well-known error in cross-validation that consists of not retesting all the genes in each of the folds of the cross-validation [51] and the latter used data from another expression platform on a subset of the patient series to perform a first gene selection. Before outlying the approach to develop gene signatures that interact with relative treatment benefit, we have a look again at the prognostic signatures in early breast cancer.
are some of well-known prognostic signatures in early breast cancer also predictive: wishful thinking?
None of the published gene signatures in early breast cancer we studied so far was developed for predicting the relative magnitude of a treatment effect, i.e. they were fitted in the development series using only main effects for prognosis. Nevertheless, a study has claimed that the Oncotype DX signature predicts the magnitude of chemotherapy benefit [52], when including in a subtle manner the patients from the development series [19]. The only truly independent evaluation of Oncotype DX was performed in a subset of 367 patients included in the S8814 trial for node-positive, ER+ postmenopausal breast cancer women, in which the gene signature was tested for interaction with additional chemotherapy prior to tamoxifen [53]. This study showed a significant treatment-by-signature interaction in the first 5 years after inclusion in a Cox regression model (interaction P = 0.03). Nevertheless, once this model also included ER expression, the interaction was no longer statistically significant (P = 0.15), which suggests that there may be some confounding between the gene signature and ER expression; furthermore, the subdivision of the time scale in two periods (before and after 5 years) may or may not have been preplanned. The small number of events in the different subgroups defined by the signature makes it hard to obtain reliable treatment effect estimates. A prospective randomized controlled trial, RxPonder, has been started to replicate this chemotherapy by signature interaction by making the bold assumption that there would exist a qualitative interaction between the chemotherapy and the gene signature on invasive disease-free survival [54].
development of predictive gene signatures in randomized controlled trials
In randomized controlled phase III trials of an experimental treatment versus standard treatment or placebo, it has become common to test multiple candidate predictive biomarkers on baseline tumor or plasma samples for a possible interaction with treatment effect. If the predictive signature is known beforehand, alternative procedures exist to test the effect of the treatment both in the overall trial population as well as in the signature-positive (or -negative) subgroup of patients [40, 55].
Previously, we proposed a global interaction approach for controlling the family-wise type I error of a predictive signature in a randomized controlled trial [25, 56]. The interaction statistic measures the degree of differential treatment effect according to gene signature values. To evaluate the global interaction signal, a permutation procedure was proposed (Figure 3). The underlying idea is that if the biomarkers are not predictive, they are exchangeable between patients in same arm; thus, by repeatedly rearranging the biomarkers of the patients and by computing the interaction statistic for each permutation, the distribution of the global test statistic under the null hypothesis is obtained and can be used to compute the P-value of the statistic observed in the original data. In case of a significant interaction signal, the magnitude of treatment effects within subgroups defined by the signature values will determine whether it has some clinical importance for which a large number of events is needed.
Figure 3.
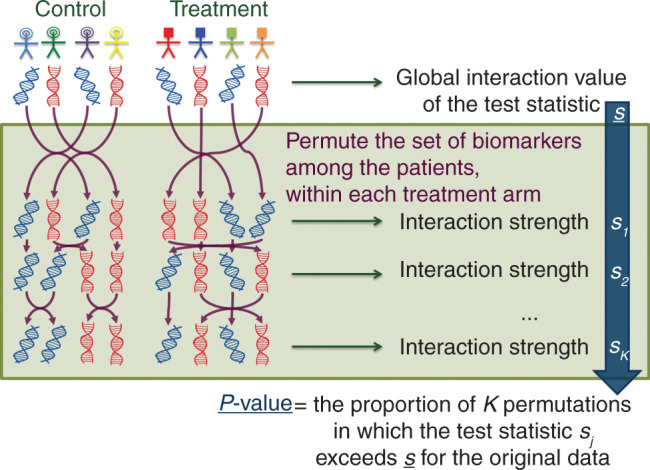
Permutation scheme for computing the P-value of a global interaction test to evaluate the ability of a gene signature to be associated with the magnitude of treatment benefit.
Several strategies have been proposed to identify and validate a signature in a randomized trial using cross-validation techniques to overcome a potential overfitting issue [40, 57–59]. Figure 4 shows the general scheme of cross-validation in this context. The data at hand are divided into K groups, often 10. For each group, the data from the remaining K − 1 groups are used to select the most predictive biomarkers which will make up the signature and coefficients are estimated. Then, the data in the excluded group are used to calculate the signature for left-out patients. The entire model building procedure is iterated over the K folds to obtain a gene signature score for each patient and to evaluate the capacity of the signature to predict the magnitude of treatment effect. The application of the entire gene signature building process to the full randomized controlled trial data leads to an ‘indication’ classifier to use for future patients [60]. An application of this analysis strategy on trials of adjuvant anthracycline-based chemotherapy can be found in [25]. Developing a gene signature requires selecting the biomarkers which are the most predictive and combining them efficiently in the regression model. Those tasks get increasingly complex as the number of biomarkers at hand increases. New statistical developments in this active field of research aim to extend existing selection methods to higher dimensional setting [61]. One of the major matters in this context is achieving the right balance between type I error and power. Once a predictive signature has been successfully identified in a phase III trial, its performances will need to be evaluated in a truly independent trial.
Figure 4.

K-fold cross-validation process to develop a signature and to limit overfitting in the evaluation of the magnitude of treatment benefit according to gene signature values, when only one single randomized controlled clinical trial is available.
conclusions
In this commentary, we have illustrated the challenges in taking a gene signature from bench to bedside for which more clear evidence-based requirements are needed [2].
Clinical trial designs originally proposed for diagnostic tests can be adopted for trials with prognostic gene signatures. We propose to shift from prognostic gene signatures to gene signatures specifically developed on randomized controlled trial data as treatment-effect modifiers. Our approach consists of applying first a global permutation test. If the global test gives a green light, a treatment-modifying gene signature can be developed on the trial data using a particular variable selection method and a cross-validation scheme to estimate treatment effects within gene signature defined subgroups. More research is ongoing on approaches to develop and validate gene signatures in randomized controlled trials. In the era of data sharing of clinical trials, a larger role for meta-analyses of individual patient data can be expected in this context. Last but not least, in clinical trials of gene signatures some of the strongest logistical challenges are to control confounding that can arise through the handling of the specimens, batch effects within and between laboratories, measurement error and tumor heterogeneity.
acknowledgements
We are grateful to the associate editor and the reviewers for their fruitful comments.
funding
NT received a PhD grant from the Foundation Philanthropia Lombard-Odier (no grant number).
disclosures
Dr Michiels served on an advisory board for PAM50 (Prosigna) and received honorarium from Nanostring. All remaining authors have declared no conflicts of interest.
references
- 1. Poste G. Bring on the biomarkers. Nature 2011; 469: 156–157. [DOI] [PubMed] [Google Scholar]
- 2. Schneider D, Bianchini G, Horgan D et al. . Establishing the evidence bar for molecular diagnostics in personalised cancer care. Public Health Genomics 2015; 18: 349–358. [DOI] [PubMed] [Google Scholar]
- 3. Koscielny S. Why most gene expression signatures of tumors have not been useful in the clinic. Sci Transl Med 2010; 2: 14ps12. [DOI] [PubMed] [Google Scholar]
- 4. Dowsett M, Sestak I, Lopez-Knowles E et al. . Comparison of PAM50 risk of recurrence score with oncotype DX and IHC4 for predicting risk of distant recurrence after endocrine therapy. J Clin Oncol 2013; 31: 2783–2790. [DOI] [PubMed] [Google Scholar]
- 5. Bartlett JM, Bayani J, Marshall A et al. . Comparing breast cancer multiparameter tests in the OPTIMA prelim trial: no test is more equal than the others. J Natl Cancer Inst 2016; 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Azim HA Jr, Michiels S, Zagouri F et al. . Utility of prognostic genomic tests in breast cancer practice: The IMPAKT 2012 Working Group Consensus Statement. Ann Oncol 2013; 24: 647–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Evaluation of Genomic Applications in P, Prevention Working G. Recommendations from the Extended group name: Evaluation of Genomic Applications in Practice and Prevention Working Group: does the use of Oncotype DX tumor gene expression profiling to guide treatment decisions improve outcomes in patients with breast cancer? Genet Med 2015; 18: 770–779. [DOI] [PubMed] [Google Scholar]
- 8. Harris LN, Ismaila N, McShane LM, Hayes DF. Use of biomarkers to guide decisions on adjuvant systemic therapy for women with early-stage invasive breast cancer: American Society of Clinical Oncology clinical practice guideline summary. J Oncol Pract 2016; 12: 384–389. [DOI] [PubMed] [Google Scholar]
- 9. Buyse M, Michiels S, Sargent DJ et al. . Integrating biomarkers in clinical trials. Exp Rev Mol Diagn 2011; 11: 171–182. [DOI] [PubMed] [Google Scholar]
- 10. Rothwell PM. Treating individuals 2. Subgroup analysis in randomised controlled trials: importance, indications, and interpretation. Lancet 2005; 365: 176–186. [DOI] [PubMed] [Google Scholar]
- 11. Windeler J. Prognosis - what does the clinician associate with this notion? Stat Med 2000; 19: 425–430. [DOI] [PubMed] [Google Scholar]
- 12. Early Breast Cancer Trialists’ Collaborative Group Peto R, Davies C et al. . Comparisons between different polychemotherapy regimens for early breast cancer: meta-analyses of long-term outcome among 100,000 women in 123 randomised trials. Lancet 2012; 379: 432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Early Breast Cancer Trialists’ Collaborative Group Davies C, Godwin J et al. . Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 2011; 378: 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stewart LA, Parmar MK. The results of a quantitative overview of chemotherapy in advanced ovarian cancer: what can we learn? Bull Cancer 1993; 80: 146–151. [PubMed] [Google Scholar]
- 15. Michiels S, Koscielny S, Hill C. Prediction of cancer outcome with microarrays: a multiple random validation strategy. Lancet 2005; 365: 488–492. [DOI] [PubMed] [Google Scholar]
- 16. Ternes N, Arnedos M, Koscielny S et al. . Statistical methods applied to omics data: predicting response to neoadjuvant therapy in breast cancer. Curr Opin Oncol 2014; 26: 576–583. [DOI] [PubMed] [Google Scholar]
- 17. Ternes N, Rotolo F, Michiels S. Empirical extensions of the lasso penalty to reduce the false discovery rate in high-dimensional Cox regression models. Stat Med 2016; 35: 2561–2573. [DOI] [PubMed] [Google Scholar]
- 18. Teutsch SM, Bradley LA, Palomaki GE et al. . The Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Initiative: methods of the EGAPP Working Group. Genet Med 2009; 11: 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Michiels S, Koscielny S, Hill C. Interpretation of microarray data in cancer. Br J Cancer 2007; 96: 1155–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Michiels S, Kramar A, Koscielny S. Multidimensionality of microarrays: statistical challenges and (im) possible solutions. Mol Oncol 2011; 5: 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simon RM, Paik S, Hayes DF. Use of archived specimens in evaluation of prognostic and predictive biomarkers. J Natl Cancer Inst 2009; 101: 1446–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miller I, Ashton-Chess J, Spolders H et al. . Market access challenges in the EU for high medical value diagnostic tests. Pers Med 2011; 8: 137–148. [DOI] [PubMed] [Google Scholar]
- 23. Rakha EA, Reis-Filho JS, Baehner F et al. . Breast cancer prognostic classification in the molecular era: the role of histological grade. Breast Cancer Res 2010; 12: 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ignatiadis M, Singhal SK, Desmedt C et al. . Gene modules and response to neoadjuvant chemotherapy in breast cancer subtypes: a pooled analysis. J Clin Oncol 2012; 30: 1996–2004. [DOI] [PubMed] [Google Scholar]
- 25. Michiels S, Rotolo F. Evaluation of clinical utility and validation of gene signatures in clinical trials. In Matsui S, Buyse M, Simon R (eds). Design and Analysis of Clinical Trials for Predictive Medicine. Boca Raton, Florida: CRC Press; 2015; 187–203. [Google Scholar]
- 26. Vickers AJ, Cronin AM, Begg CB. One statistical test is sufficient for assessing new predictive markers. BMC Med Res Methodol 2011; 11: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pepe MS, Kerr KF, Longton G, Wang Z. Testing for improvement in prediction model performance. Stat Med 2013; 32: 1467–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pencina MJ, D'Agostino RB Sr, Song L. Quantifying discrimination of Framingham risk functions with different survival C statistics. Stat Med 2012; 31: 1543–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heagerty PJ, Lumley T, Pepe MS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics 2000; 56: 337–344. [DOI] [PubMed] [Google Scholar]
- 30. Dunkler D, Michiels S, Schemper M. Gene expression profiling: does it add predictive accuracy to clinical characteristics in cancer prognosis? Eur J Cancer 2007; 43: 745–751. [DOI] [PubMed] [Google Scholar]
- 31. Ignatiadis M, Azim HA Jr, Desmedt C et al. . The genomic grade assay compared with Ki67 to determine risk of distant breast cancer recurrence. JAMA Oncol 2015; 2: 217–224. [DOI] [PubMed] [Google Scholar]
- 32. McGeechan K, Macaskill P, Irwig L et al. . Assessing new biomarkers and predictive models for use in clinical practice: a clinician's guide. Arch Intern Med 2008; 168: 2304–2310. [DOI] [PubMed] [Google Scholar]
- 33. Steyerberg EW, Pencina MJ, Lingsma HF et al. . Assessing the incremental value of diagnostic and prognostic markers: a review and illustration. Eur J Clin Invest 2012; 42: 216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Steyerberg EW, Vedder MM, Leening MJ et al. . Graphical assessment of incremental value of novel markers in prediction models: from statistical to decision analytical perspectives. Biom J 2015; 57: 556–570. [DOI] [PubMed] [Google Scholar]
- 35. McShane LM, Cavenagh MM, Lively TG et al. . Criteria for the use of omics-based predictors in clinical trials: explanation and elaboration. BMC Med 2013; 11: 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bossuyt PM, Lijmer JG, Mol BW. Randomised comparisons of medical tests: sometimes invalid, not always efficient. Lancet 2000; 356: 1844–1847. [DOI] [PubMed] [Google Scholar]
- 37. de Graaff JC, Ubbink DT, Tijssen JG, Legemate DA. The diagnostic randomized clinical trial is the best solution for management issues in critical limb ischemia. J Clin Epidemiol 2004; 57: 1111–1118. [DOI] [PubMed] [Google Scholar]
- 38. Lu B, Gatsonis C. Efficiency of study designs in diagnostic randomized clinical trials. Stat Med 2013; 32: 1451–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rodger M, Ramsay T, Fergusson D. Diagnostic randomized controlled trials: the final frontier. Trials 2012; 13: 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Buyse M, Michiels S. Omics-based clinical trial designs. Curr Opin Oncol 2013; 25: 289–295. [DOI] [PubMed] [Google Scholar]
- 41. Bogaerts J, Cardoso F, Buyse M et al. . Gene signature evaluation as a prognostic tool: challenges in the design of the MINDACT trial. Nat Clin Pract Oncol 2006; 3: 540–551. [DOI] [PubMed] [Google Scholar]
- 42. Hooper R, Diaz-Ordaz K, Takeda A, Khan K. Comparing diagnostic tests: trials in people with discordant test results. Stat Med 2013; 32: 2443–2456. [DOI] [PubMed] [Google Scholar]
- 43. Sparano JA. TAILORx: trial assigning individualized options for treatment (Rx). Clin Breast Cancer 2006; 7: 347–350. [DOI] [PubMed] [Google Scholar]
- 44. Sparano JA, Gray RJ, Makower DF et al. . Prospective validation of a 21-gene expression assay in breast cancer. N Engl J Med 2015; 373: 2005–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bonastre J, Marguet S, Lueza B et al. . Cost effectiveness of molecular profiling for adjuvant decision making in patients with node-negative breast cancer. J Clin Oncol 2014; 32: 3513–3519. [DOI] [PubMed] [Google Scholar]
- 46. Yusuf S, Collins R, Peto R. Why do we need some large, simple randomized trials? Stat Med 1984; 3: 409–422. [DOI] [PubMed] [Google Scholar]
- 47. Simon R. New challenges for 21st century clinical trials. Clin Trials 2007; 4: 167–169; discussion 173-167. [DOI] [PubMed] [Google Scholar]
- 48. Betensky RA, Louis DN, Cairncross JG. Influence of unrecognized molecular heterogeneity on randomized clinical trials. J Clin Oncol 2002; 20: 2495–2499. [DOI] [PubMed] [Google Scholar]
- 49. Pogue-Geile KL, Kim C, Jeong JH et al. . Predicting degree of benefit from adjuvant trastuzumab in NSABP trial B-31. J Natl Cancer Inst 2013; 105: 1782–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Perez EA, Thompson EA, Ballman KV et al. . Genomic analysis reveals that immune function genes are strongly linked to clinical outcome in the North Central Cancer Treatment Group n9831 Adjuvant Trastuzumab Trial. J Clin Oncol 2015; 33: 701–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dupuy A, Simon RM. Critical review of published microarray studies for cancer outcome and guidelines on statistical analysis and reporting. J Natl Cancer Inst 2007; 99: 147–157. [DOI] [PubMed] [Google Scholar]
- 52. Paik S, Tang G, Shak S et al. . Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol 2006; 24: 3726–3734. [DOI] [PubMed] [Google Scholar]
- 53. Albain KS, Barlow WE, Shak S et al. . Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol 2010; 11: 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Barlow W. Design of a clinical trial for testing the ability of a continuous marker to predict therapy benefit. In Crowley J, Hoering A (eds). Handbook of Statistics in Clinical Oncology, 3rd edition CRC Press; 2012; 293–304. [Google Scholar]
- 55. Freidlin B, Korn EL. Biomarker enrichment strategies: matching trial design to biomarker credentials. Nat Rev Clin Oncol 2014; 11: 81–90. [DOI] [PubMed] [Google Scholar]
- 56. Michiels S, Potthoff RF, George SL. Multiple testing of treatment-effect-modifying biomarkers in a randomized clinical trial with a survival endpoint. Stat Med 2011; 30: 1502–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Matsui S, Simon R, Qu P et al. . Developing and validating continuous genomic signatures in randomized clinical trials for predictive medicine. Clin Cancer Res 2012; 18: 6065–6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Polley MY, Polley EC, Huang EP et al. . Two-stage adaptive cutoff design for building and validating a prognostic biomarker signature. Stat Med 2014; 33: 5097–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Freidlin B, Jiang W, Simon R. The cross-validated adaptive signature design. Clin Cancer Res 2010; 16: 691–698. [DOI] [PubMed] [Google Scholar]
- 60. Simon R. Clinical trials for predictive medicine. Stat Med 2012; 31: 3031–3040. [DOI] [PubMed] [Google Scholar]
- 61. Ternès N, Rotolo F, Heinze G, Michiels S. Identification of biomarker-by-treatment interactions in randomized clinical trials with survival outcomes and high-dimensional spaces. Biom J 2016; 10.1002/bimj.201500234 [DOI] [PMC free article] [PubMed] [Google Scholar]