

Abstract
VEGF is a secreted mitogen associated with angiogenesis and is also a potent vascular permeability factor. The biological role of VEGF in the ischemic brain remains unknown. This study was undertaken to investigate whether VEGF enhances cerebral microvascular perfusion and increases blood-brain barrier (BBB) leakage in the ischemic brain. Using magnetic resonance imaging (MRI), three-dimensional laser-scanning confocal microscope, and functional neurological tests, we measured the effects of administrating recombinant human VEGF165 (rhVEGF165) on angiogenesis, functional neurological outcome, and BBB leakage in a rat model of focal cerebral embolic ischemia. Late (48 hours) administration of rhVEGF165 to the ischemic rats enhanced angiogenesis in the ischemic penumbra and significantly improved neurological recovery. However, early postischemic (1 hour) administration of rhVEGF165 to ischemic rats significantly increased BBB leakage, hemorrhagic transformation, and ischemic lesions. Administration of rhVEGF165 to ischemic rats did not change BBB leakage and cerebral plasma perfusion in the contralateral hemisphere. Our results indicate that VEGF can markedly enhance angiogenesis in the ischemic brain and reduce neurological deficits during stroke recovery and that inhibition of VEGF at the acute stage of stroke may reduce the BBB permeability and the risk of hemorrhagic transformation after focal cerebral ischemia.
Introduction
Functional neurological recovery from stroke commonly occurs, but is often incomplete in patients who survive a stroke (1, 2). Stroke patients with a higher density of blood vessels appear to have reduced morbidity and survive longer (3). Functional imaging of stroke patients shows increased cerebral blood flow and metabolism in tissue surrounding focal brain infarcts (4, 5). These data suggest that restoration of cerebral microvascular circulation is important for functional recovery after a stroke. Late restoration of cerebral microcirculatory flow may involve increases in the collateral circulation and angiogenesis in the penumbra (6, 7). The substances responsible for inducing formation of new blood vessels in the ischemic brain have not been identified conclusively.
VEGF is an angiogenic growth factor that binds to two high-affinity receptors, fms-like tyrosine kinase (flt-1) and the kinase domain region (KDR) (8, 9). Administration of recombinant human VEGF165 (rhVEGF165) improves myocardial perfusion in patients with coronary ischemia and enhances angiogenesis in animal models of myocardial and limb ischemia (10–14). Transient and permanent middle cerebral artery (MCA) occlusion in rats evokes expression of VEGF in the ischemic brain (15–17), suggesting that after a stroke VEGF may be involved in angiogenesis. However, whether administration of rhVEGF165 to the ischemic brain has the potential to promote angiogenesis and thereby improve functional neurological outcome has not been tested. In addition, VEGF is a potent vascular permeability factor that increases microvascular permeability to blood plasma proteins within minutes after exposure (18). Administration of rhVEGF165 after focal cerebral ischemia may exacerbate blood-brain barrier (BBB) permeability. After a stroke, disruption of the BBB occurs acutely whereas regeneration of cerebral microvessels develops relatively late in ischemic brain (6, 19–24). Using a model of focal embolic cerebral ischemia in rats, we therefore tested the hypotheses that administration of VEGF at a late stage of stroke improves cerebral microvascular perfusion in the ischemic penumbra and that administration of VEGF early after a stroke increases BBB leakage.
Methods
All experimental procedures have been approved by the Care of Experimental Animals Committee of Henry Ford Health Sciences Center.
General experimental procedures.
Male Wistar rats weighing 320–400 g were used in the experiments. Animals were anesthetized with 0.5–3.5% halothane in 70% N2O and 30% O2 during the surgical procedure and magnetic resonance imaging (MRI) measurements. Rectal temperature was maintained at 37 ± 0.5°C throughout the experiments (25, 26). The right femoral artery and vein were cannulated with a PE-50 catheter for continuous monitoring of blood pressure, measurement of blood gases, and for drug administration, respectively.
Animal model.
The MCA was occluded by placement of a single, intact, fibrin-rich, 24-hour-old, homologous clot (∼1 μl) at the origin of the MCA (25). This model is most relevant to human stroke (25, 26).
Administration of rhVEGF165.
One hour or 48 hours after embolization, using a Harvard pump (Harvard Apparatus; South Natick, Massachusetts, USA), rhVEGF165 (Genentech Inc., San Francisco, California, USA) was infused intravenously to rats at a dose of 1 mg/kg over a 4-hour interval (5 μl/min) to reduce the hemodynamic side effects of VEGF (27, 28). The dose of rhVEGF165 was chosen based on previous studies in the same species (27, 28). An equal volume of 0.9% saline was administered to rats as a control group.
MRI measurements.
MRI measurements were performed using a 3.0 T whole-body magnet (Magnex Scientific, Abingdon, United Kingdom) equipped with actively shielded gradients and a SMIS (Surrey, United Kingdom) console. MRI images were recorded using a birdcage radial frequency coil.
The apparent diffusion coefficient of water (ADCw) was measured using the method described by LeBihan et al. (29). The diffusion-weighting gradient was increased, in a nonlinear manner, from 0 to approximately 2.8 gauss/cm to obtain a series of five images with gradient b values of 0, 200, 400, 600, 800 s/mm2.
Perfusion estimates employed methods of Williams et al. that were further refined in our laboratory (30, 31). This technique is based on the selective inversion of blood-water protons at the level of the carotid arteries before 1H MRI measurement in the brain (31). Two images were obtained for perfusion measurement with parameters: repetition time (TR) = 1.055 seconds; echo time (TE) = 30 milliseconds, 64 × 64 image matrix, 2-mm slice thickness, and a 40-mm field of view (FOV). The duration of the inversion pulse was 1 second and 0.3 kHz in amplitude.
A gadolinium-chelate (Gd-DTPA) contrast material (Omniscan Gadodiamide; Nycomed Inc., Princeton, New Jersey, USA; 0.1 mmol/kg) was injected (intravenous bolus) at the end of the infusion of rhVEGF165 or saline to measure the BBB leakage. Contrast-enhanced MRIs were acquired in a multislice mode (nine slices) with a 2-mm slice thickness, a 40-mm FOV, and a 256 × 256 image matrix. The contrast-enhanced MRI images were obtained using multislice T1-weighted images with spin-echo sequences (TR = 500 milliseconds, TE = 30 milliseconds).
MRI analyses of ADCw and cerebral blood flow (CBF) maps were performed using Eigentool software (32). ADCw maps were produced on a pixel-by-pixel basis using a least-squares fit to a straight-line plot of the natural logarithm of normalized image intensity versus gradient b value (ADCw). CBF maps were calculated according to the method published previously (31).
Accelerating rotarod test.
An accelerating rotarod test was employed to measure motor function of rats (33, 34). Each rat received a test trial on the accelerating rotarod at 2, 7, 14, and 28 days after embolic MCA occlusion, and the time the animal remained on the device was recorded. Data are presented as the time the rat remained on the rotating spindle normalized to the preischemia duration.
Adhesive removal test.
The adhesive removal test, which measures somatosensory deficit, was performed (35). Briefly, two small pieces of adhesive-backed paper dots (of equal size, 113.1 mm2) were used as bilateral tactile stimuli occupying the distal-radial region on the wrist of each forelimb. The time required to remove the stimuli from each limb was recorded. Each animal received five trials at 2, 7, 14, and 28 days after embolic MCA occlusion, and the mean time required to remove both stimuli from limbs was recorded.
Tissue preparation.
For paraffin sections, brains were fixed in 10% buffered formalin and embedded in paraffin. Six-micrometer-thick coronal sections were cut and stained with hematoxylin and eosin (H&E) for histopathological evaluation. For vibratome sections, brains were fixed in 4% paraformaldehyde at 4°C for 48 hours. Coronal sections (100 μm) were cut on a vibratome.
Immunohistochemistry.
A mouse mAb against microtubule-associated protein 2 (MAP2; Serotec Ltd., Oxford, United Kingdom), the neuronal cytoskeletal protein, was used as a marker for early ischemic neuronal damage (36). For immunofluorescence, 100-μm thick vibratome sections were incubated with primary mAb’s against MAP2 (1:200) and with Cy 5–conjugated secondary Ab’s.
Three-dimensional image acquisition.
The vibratome sections were analyzed with a Bio-Rad MRC 1024 (argon and krypton) laser-scanning confocal imaging system (Bio-Rad, Cambridge, Massachusetts, USA). With the Evans blue– and FITC-perfused tissue samples, vibratome sections corresponding to the MRI coronal sections (interaural 6.20 mm to interaural 1.00 mm) (37) were scanned throughout the entire coronal section in 512 × 512 pixel (1042.4 × 1042.4 μm) format in the x-y direction using a 4× frame-scan average, and 16 optical sections along the z-axis with a 5-μm step-size under a ×10 objective were acquired with two separate photomultiplier tubes. Each image was assembled into a coronal section using Adobe Photoshop software (Adobe Systems Inc., Mountain View, California, USA). For FITC-dextran–perfused tissue stained with anti-MAP2 Ab (Cy5), green (FITC-dextran–perfused microvessels), and far red (Cy5, MAP2 immunoreactivity) fluorochromes on the sections were excited by a laser beam at 488 nm and 647 nm, respectively, and emissions were acquired simultaneously with two separate photomultiplier tubes through 522-nm and 680-nm emission filters, respectively.
Three-dimensional image analysis.
Measurements of vascular diameters are limited in two dimensions and are based on the assumption that vessel contours are round (38, 39). However, this is not the case in vivo in which cerebral microangioarchitecture has a highly irregular pattern of tortuous, anastomosing capillaries. To overcome most limitations of previous methods, we developed a new computerized system, MIRAGE, to examine cerebral microvessels perfused in vivo by FITC-dextran (K. Davies et al., manuscript submitted for publication). The methodology used by MIRAGE for measurement of blood vessel diameter involves a linearly applied spherical inflation technique. For each vessel under analysis, the system follows the center of the vessel and determines the long-axis path of flow. Vessel diameter is continuously measured as the center of the vessel is traced by using the length of 24 different divergent vectors, each perpendicular to this axis of flow. In a preliminary study, we measured 8,930 microvascular diameters in a nonischemic rat cortex, and the average microvessel diameter was 4.15 ± 1.24 μm (3–8 μm), which is comparable to cortical microvessel diameters of 4.4 ± 1 μm (2.5–8.7 μm) obtained from intravital microscopy in vivo (39), 4.26 ± 0.07 μm from manually measured fixed coronal sections stained with toluidine blue (38), and 3–8 μm measured by scanning electron microscopy on corrosion casts (40). We therefore employed this program to analyze cerebral microvessels in the present study. Images obtained under a ×40 objective from a laser-scanning confocal microscope (LSCM) had a resolution of 0.5 μm/pixel in the x and y dimensions and 1 μm/pixel along the z axis. All quantitative measurements were performed blindly.
Statistical analysis.
Repeated measures analysis was conducted using a generalized estimating equation to test the treatment effect on the rotarod and the adhesive removal tests, respectively. Student’s t tests were performed to compare cerebral microvascular plasma perfusion and vascular diameters in control and VEGF-treated groups. A paired t test was used to analyze the effects of VEGF and saline on measurement of perfusion- and diffusion-weighted MRIs. All data are presented as mean plus or minus SE, and a P value of 0.05 was considered to be statistically significant.
Results
The effects of administration of rhVEGF165 on cerebral microvascular perfusion and neurological outcome.
To determine whether administration of rhVEGF165 enhances cerebral microvascular perfusion, embolic ischemic rats were treated with rhVEGF165 at a dose of 1 mg/kg 48 hours after a stroke to avoid the peak of brain edema in this stroke model and were sacrificed at 9 days after ischemia. The time point of 9 days after ischemia was chosen based on data from a previous study using a cranial window: newly formed vessels arising from mouse brain were first visible at 4 days and peaked at 12 days after VEGF administration (41). Cerebral microvascular plasma perfusion and vascular diameters were examined in three dimensions by LSCM and analyzed using the MIRAGE program. Compared with the saline-treated rats (n = 6, Figure 1, a and g), treatment with rhVEGF165 significantly (P < 0.05, n = 5) increased numbers of voxels of plasma-perfused vessels in the penumbra of the cortex (Figure 1b and 1g). However, treatment with rhVEGF165 did not alter cerebral microvascular plasma perfusion in the contralateral hemisphere (Figure 1d) compared with the contralateral hemisphere from the saline-treated rats (Figure 1c). We measured in three dimensions the diameters of plasma-perfused cerebral microvessels in the penumbra (Figure 1e) and in the contralateral homologous tissue (Figure 1f). Eighty-five percent of plasma-perfused vessels in the penumbra had diameters within 3 μm, whereas the majority of vessels in the contralateral tissue had diameters of 4 and 5 μm (Figure 1h), suggesting that increased plasma-perfused vessels in the penumbra are newly grown vessels.
Figure 1.
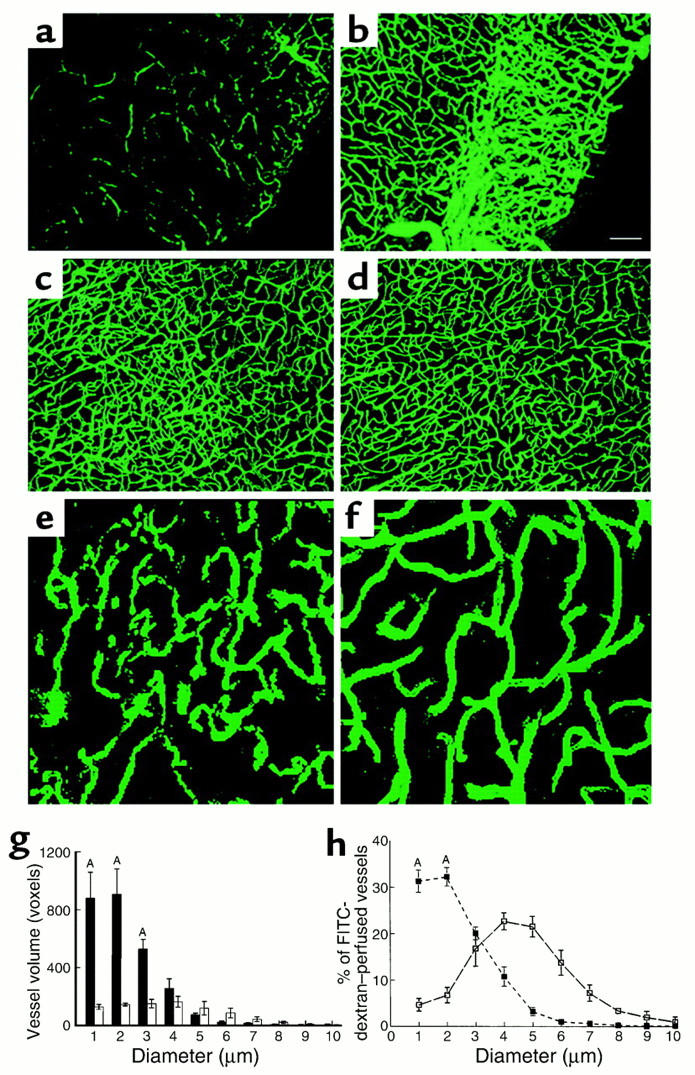
Composite images (through stacks of 14 optical sections of 5-μm thickness) of cerebral microvascular plasma perfusion obtained from the ischemic penumbra of the cortex 9 days after MCA occlusion. Late treatment with rhVEGF165 enhances cerebral microvascular plasma perfusion in the penumbra (b) compared with the control rat (a). Treatment with rhVEGF165 did not alter cerebral microvascular plasma perfusion in the contralateral hemisphere (d), compared with the contralateral hemisphere in the control rat (c). Bar, 100 μm. Quantitative data analyzed by the MIRAGE program (g) show a significant increase of plasma perfusion in vessels within 3 μm of diameters in the rhVEGF165–treated group (filled bars; n = 5) compared with the saline-treated group (open bars; n = 6; AP < 0.05). Higher-magnification images show that cerebral microvessels in the ipsilateral hemisphere had an irregular pattern of tortuosity (e) compared with the vessels in the contralateral hemisphere (f). Dimension of the composite image is 260.6 × 260.6 × 20 μm3. (h) The percentage of vessels with different diameters in the entire vessels perfused by FITC-dextran. The number of vessels with diameters within 2 μm is significantly (P < 0.01) higher in the ipsilateral hemisphere (filled squares; n = 5) than in the contralateral hemisphere (open squares; n = 5).
To determine whether late administration of rhVEGF165 improves functional neurological recovery, embolic ischemic rats were treated with rhVEGF165 at a dose of 1 mg/kg (n = 7) or saline (n = 10) at 48 hours after a stroke, and the rotarod and adhesive removal tests were performed on these rats at 2, 7, 14, and 28 days after stroke. To avert potential effects of rhVEGF165 on the rotarod test due to the likelihood of hindlimb ischemia caused by cannulated femoral arteries and veins, rhVEGF165 or saline was administered in a tail vein, and mean artery blood pressure was not measured during infusion of rhVEGF165 or saline in these groups. Rats in the rhVEGF165 or the saline-treated groups showed marked motor and somatosensory impairments 2 days after a stroke. Partial recovery was detected in the rhVEGF165 and saline-treated groups at 7, 14, and 28 days after a stroke (Figure 2a). However, the ischemic rats treated with rhVEGF165 exhibited significant (P < 0.05) improvements on the rotarod test at 7, 14, and 28 days after the stroke compared with the saline-treated rats (Figure 2a). The adhesive removal test did not reveal significant differences between the rhVEGF165-treated and the saline-treated groups at 2 days (116 ± 3.6 vs. 115 ± 3.5 seconds), 7 days (66 ± 16.5 vs. 65.2 ± 11.1 seconds), and 14 days (40 ± 1.4 vs. 34.8 ± 3.2 seconds). However, a significant (P < 0.05) difference was detected at 28 days after stroke (11 ± 1.2 vs. 15 ± 1.1 seconds). In addition, brain tissue obtained at 28 days after ischemia exhibited more small vessels in the ipsilateral ischemic border zone (Figure 2, e and f) in rats treated with rhVEGF165 when compared with the vehicle-treated rats (Figure 2, b and c). An increase in microvessels in the ischemic border zone was confirmed by detecting more FITC-dextran–perfused microvessels (Figure 2g) in the rats treated with rhVEGF165 when compared with FITC-dextran–perfused microvessels in the penumbra from the saline-treated rats (Figure 2d).
Figure 2.
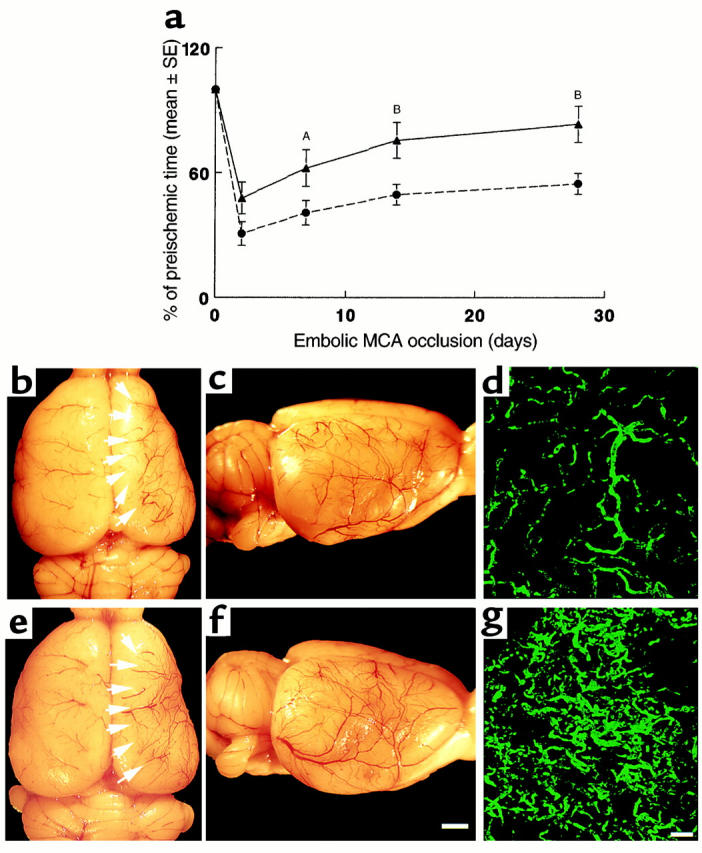
(a) The percentage of time that rats persisted on the rotarod after ischemia compared with the preischemic value as a function of time of stroke. Rotarod test was performed at 2, 7, 14, and 28 days after stroke on the ischemic rats treated with rhVEGF165 or saline initiated at 48 hours after ischemia. Treatment with rhVEGF 165 (triangles; n = 7) significantly (AP < 0.05 or BP < 0.01) increases time spent on the rotarod compared with the saline-treated group (circles; n = 10). Cerebral vessels and FITC-dextran–perfused cerebral microvessels from the saline-treated rat (b–d) and the rhVEGF165-treated rat (e–g) 28 days after the right MCA occlusion. Dorsal view of the right and left hemisphere vessels (b and e), a lateral view of the right hemisphere vessels (c and f), and FITC-dextran–perfused cerebral microvessels in the penumbra of the cortex (d and g). More vessels in the ipsilateral ischemic border zone (e, arrows, and f) and more FITC-dextran–perfused vessels (g) were observed in the rhVEGF165-treated rat compared with that in the saline-treated rat (b–d). Bar = 2 mm in f for b, c, e, and f; bar = 200 μm in g for d and g. Rats were decapitated one minute after injection of FITC-dextran and their brains were rapidly removed from the severed heads and digitized at 8× magnification (for panels b, c, e, and f) using an MCID image analysis system (Imaging Research, St. Catherines, Canada). After digitization the brains were placed in 4% paraformaldehyde at 4°C for 48 hours (for panels d and g).
The effects of rhVEGF165 administration on BBB leakage.
To examine early effects of rhVEGF165 administration initiated 1 hour after MCA occlusion on BBB leakage, the MRI contrast agent Gd-DTPA, along with 1 ml 2% of Evans blue, were administered to the ischemic rats at the end of the 4-hour rhVEGF165 (n = 5) or saline (n = 4) infusion, and FITC dextran (42) was administered intravenously at the end of the MRI measurements. These rats were sacrificed 1 minute after injection of FITC-dextran. Contrast agent Gd-DTPA within the tissue causes hyperintensity on T2-weighted MRI and reflects a breakdown of the BBB (43, 44). Coadministration of Evans blue, which binds to albumin in plasma with Gd-DTPA contrast agent, permits comparison of changes in BBB leakage measured by MRI with disruption of BBB detected by LSCM; FITC-dextran, which remains dissolved and free in plasma, allows analysis of microvascular plasma perfusion (42). In the saline-treated ischemic rats, the hyperintense area localized to the ipsilateral MCA supplied the subcortex (Figure 3e). Administration of rhVEGF165 resulted in the hyperintense areas localized to the ipsilateral MCA-supplied cortex and subcortex (Figure 3b). The hyperintense areas on the MRI in VEGF-treated rats were significantly (P < 0.05) larger than the areas in the control animals (Figure 3i). LSCM images of coronal sections that matched MRI sections from the same animal revealed extensive Evans blue and FITC-dextran extravasation in rats treated with rhVEGF165 (Figure 3c), compared with the control animals (Figure 3f). Areas with leakage of fluorescent dyes localized only to the ipsilateral area supplied by the MCA (Figure 3c) and corresponded closely to hyperintense areas detected on Gd-DTPA MRI (Figure 3b). High-magnification images of LSCM show that the extravasation of Evans blue and FITC-dextran occurred in capillaries and in the lesion boundary areas with nonplasma and low-plasma perfusion (Figure 3g), and extravasation of fluorescent dyes was not detected in cerebral vessels in the contralateral homologous tissue (Figure 3h). These data demonstrate that administration of rhVEGF165 exacerbates BBB leakage. Disruption of the BBB was not detected in nonischemic rats treated with rhVEGF165 (n = 3, data not shown).
Figure 3.
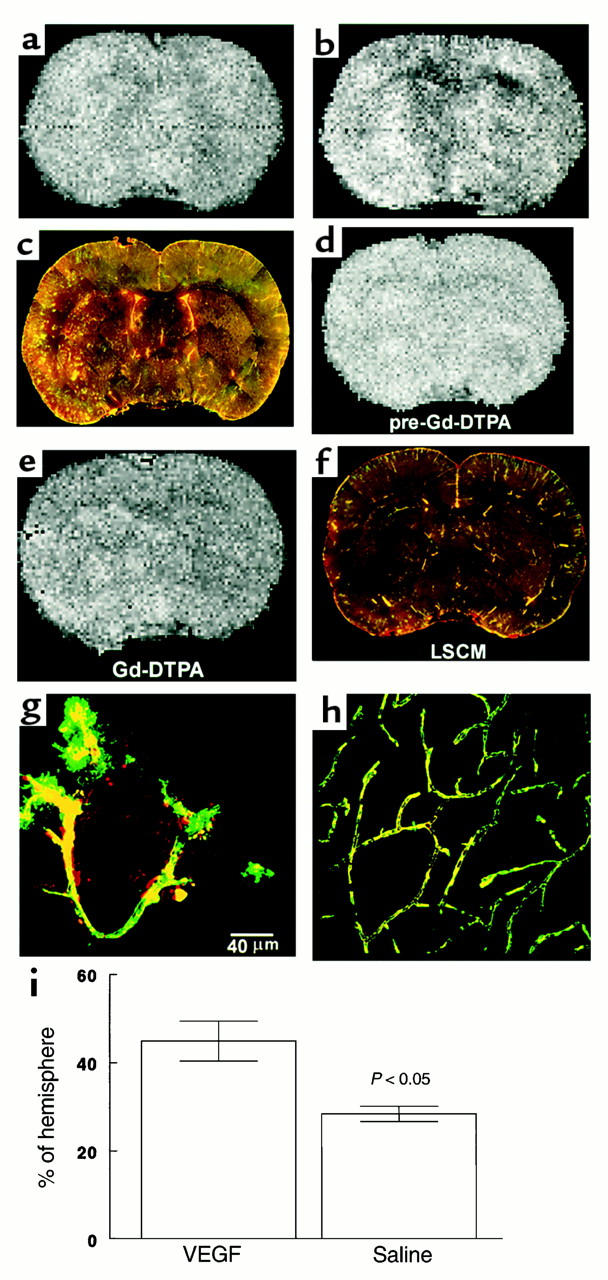
Gd-DTPA contrast-enhanced MRI images of ischemic brains treated with rhVEGF165 at 1 hour after MCA occlusion (a and b) or 0.9% saline (d and e) and corresponding images of LSCM (c and f). Postcontrast images obtained at 30 minutes after injection of Gd-DTPA agent show the hyperintense areas in the ipsilateral MCA-supplied cortex and subcortex of a rat treated with rhVEGF165 (b) and in the ipsilateral MCA-supplied subcortex of a rat treated with 0.9% saline (e), which are not seen on the precontrast images (a and d). Composite images of LSCM (80 μm thick) from the same locations exhibit extravasation of Evans blue and FITC-dextran in the cortex and subcortex from the rat treated with rhVEGF165 (c) and extravasation of Evans blue in the subcortex from the rat treated with saline (f) and sacrificed 6 hours after MCA occlusion. High-magnification images of LSCM from the rat treated with rhVEGF165 show that the extravasation of Evans blue (g, red) and FITC-dextran (g, green) was present in capillaries with nonplasma and low-plasma perfusion in the ipsilateral hemisphere (g) and that cerebral microvessels in the contralateral hemisphere were well perfused with both dyes and did not exhibit any extravasation of fluorescent dyes (h). Bar, 40 μm for g and h. Hyperintense areas on Gd-DTPA-enhanced MRI are significantly (P < 0.05) larger in rhVEGF165–treated rats (n = 5) than the saline-treated rats (n = 4) (i). Hyperintense areas were measured on Gd-DTPA–enhanced images obtained at 30 minutes after injection of Gd-DTPA agent, which was injected after 4 hours of infusion of rhVEGF165 or saline.
To examine the effect of exogenous VEGF on BBB leakage and the incidence of hemorrhagic transformation, rats were sacrificed at 24 hours after a stroke, since in this embolic stroke model hemorrhagic transformation primarily appears during this time period (25, 45). Administration of rhVEGF165 at the onset of 1 hour of MCA occlusion (n = 6) resulted in a significant (P < 0.05) increase in the incidence of gross hemorrhage (four out of six rats had gross hemorrhage, 80%) compared with non-rhVEGF165–treated rats (two out of ten, 20%; n = 10). Areas with gross hemorrhage detected on the coronal sections at 24 hours after a stroke (Figure 4, c and d) match the anatomic hyperintense areas detected on Gd-DTPA–enhanced MRI measurements at 5 hours after a stroke (Figure 4b), suggesting that the early increase in the BBB leakage to large molecules may be related to the incidence of hemorrhage in this model.
Figure 4.

Increase in the BBB leakage and hemorrhagic transformation. Large hyperintense areas on Gd-DTPA–enhanced MRI were observed 30 minutes after injection of Gd-DTPA in a rat treated with rhVEGF165 (b), compared with the precontrast image (a). A coronal section corresponding to the MRI section obtained at 24 hours after a stroke shows extensive Evans blue leakage and hemorrhage (c and d). Bar, 1 mm in d.
To assess the effects of rhVEGF165 given 1 hour after onset of ischemia on the evolution of the tissue damage and ultimate lesion volume, we performed diffusion-weighted MRIs to measure the evolution of the ischemic lesion in vivo, MAP2 immunohistochemistry to examine neuronal damage, and H&E staining to measure ischemic areas. The ischemic areas defined as the hyperintensity on the diffusion-weighted images (DWI) were relatively similar at 1 hour of ischemia for rats treated with saline (Figure 5a, 1 hour) or with rhVEGF165 (Figure 5b, 1 hour), suggesting the same basal level of ischemic severity for rats in both groups. However, areas of hyperintensity in rhVEGF165-treated rats increased substantially during and after infusion of rhVEGF165 (Figure 5b, 6 hours and 24 hours), compared with the vehicle-treated ischemic rats at the same time points (Figure 5a, 6 hours and 24 hours). Brain tissue obtained from the same animal immediately after the last MRI measurements at 24 hours after ischemia, shows a larger ischemic lesion on H&E-stained coronal section in the rhVEGF165-treated rat (Figure 5b, H&E) than the ischemic lesion in the vehicle-treated rat (Figure 5a, H&E). Quantitative measurements of the ischemic areas defined as the hyperintensity on the DWI as a percentage of hemisphere reveal that the ischemic areas increased by 78% at 6 hours and 151% at 24 hours after ischemia compared with the ischemic areas at 1 hour after ischemia for rats treated with rhVEGF165 (n = 6, Figure 5c). In contrast, the ischemic areas increased only by 32% at 6 hours and 114% at 24 hours of ischemia for rats treated with the vehicle (n = 7, Figure 5c). The differences of the increased ischemic areas between two groups at 6 hours and 24 hours of ischemia are statistically significant (P < 0.05; Figure 5c). Neurons in areas with extravasation of FITC-dextran (green, Figure 5d) exhibited much less MAP2 immunoreactivity (red, Figure 5d) at 6 hours of ischemia compared with homologous tissue in the contralateral hemisphere (Figure 5e) for rats treated with rhVEGF165, indicating acute ischemic neuronal damage. Taken together, these data suggest that exogenous VEGF accelerates ischemic damage.
Figure 5.

Evolution of the ischemic lesion from 1 hour to 24 hours after MCA occlusion in a rat treated with 0.9% saline (a) or rhVEGF165 (b). Although areas of hyperintensity on the MRI were relatively similar at 1 hour of MCA occlusion in saline-treated (a) or rhVEGF165–treated (b) rat, an area of hyperintensity on MRI was larger at 6 hours and 24 hours of ischemia in the rhVEGF-treated (b) rat compared with an area of hyperintensity at the same time points in the saline-treated rat (a). Differences of ischemic lesions observed on MRI between these two rats are confirmed on H&E-stained coronal sections obtained at 24 hours of ischemia (H&E) (a and b). Quantitative analysis of the ischemic area as a percentage of hemisphere measured on DWI at 6 hours and 24 hours of MCA occlusion (c) shows that ischemic areas are significantly (AP < 0.05) larger in rats treated with rhVEGF165 (solid line, n = 6) than in the control rats (dotted line, n = 7) at 6 hours and 24 hours of embolic MCA occlusion. Composite images (135 × 135 × 20 μm3) of microvessels (FITC-dextran, green) and MAP2 immunoreactivity (Cy5, red) show much less MAP2 immunoreactivity (red) in low plasma-perfused areas with extravasation of FITC-dextran (green) in the ipsilateral hemisphere (d) compared with the homologous tissue in the contralateral hemisphere (e) in a rat treated with rhVEGF165 (1 hour) and sacrificed at 6 hours after MCA occlusion. Bar, 20 μm for d and e.
To examine whether administration of rhVEGF165 48 hours after ischemia increased leakage of Gd-DTPA across the BBB, Gd-DTPA–enhanced MRI measurements were made on a subset of rats (n = 6) immediately after infusion of rhVEGF165. These rats were sacrificed after the last MRI measurements, and cerebral microvascular leakage and plasma perfusion were measured by LSCM on these brains. Late administration of rhVEGF165 did not result in a significant increase in the BBB leakage measured on Gd-DTPA–enhanced MRIs (Figure 6, a and b) and on LSCM (Figure 6c).
Figure 6.

Late (48 hours) administration of rhVEGF165 to ischemic rats did not result in increases in BBB leakage. Postcontrast images (b) obtained at 30 minutes after injection of the Gd-DTPA agent did not show an increase in areas of hyperintensity compared with the precontrast image (a) in a rat treated with rhVEGF165 at 48 hours of MCA occlusion. Non- and low cerebral microvascular plasma perfusion was detected in the ipsilateral MCA-supplied territory on a composite image of LSCM (80 μm thickness) (c). However, extravasation of Evans blue and FITC-dextran was not detected (c). Images of LSCM were obtained immediately after the last Gd-DTPA–enhanced MRI measurements.
The effects of rhVEGF165 administration on CBF.
To examine the effects of early administration of rhVEGF165 on CBF, we performed perfusion-weighted MRI measurements using an arterial spin-tagging technique (31). CBF increased in the ischemic hemisphere during the 1–3 hour infusion of rhVEGF165 (Figure 7a, 2 hours) and returned to the preinfusion level at 4 hours of infusion of rhVEGF165 (Figure 7a, 4 hours). Transient increases in CBF were not detected in the ischemic hemisphere in the control rats (Figure 7b), and the differences of changes in CBF between the VEGF (n = 6) and the control groups (n = 6) were statistically significant (P < 0.05; Figure 7d), suggesting that early administration of rhVEGF165 induces a transient increase of CBF in the ischemic brain.
Figure 7.
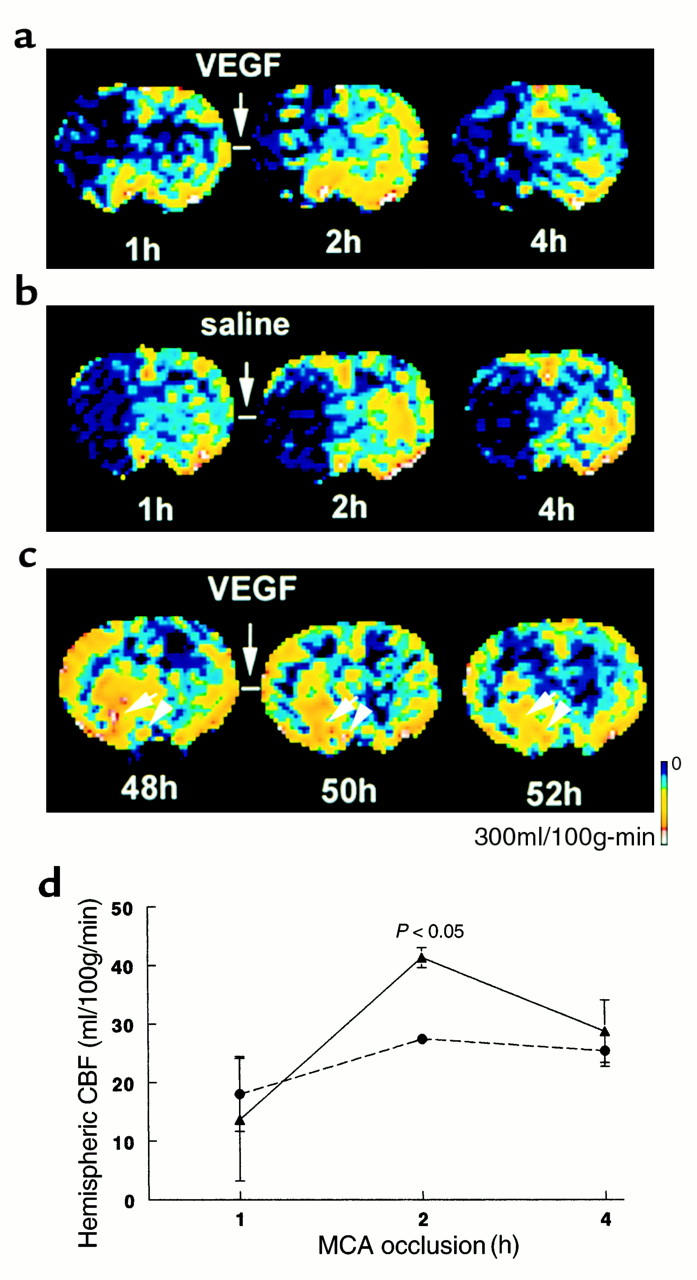
CBF maps of coronal sections measured by perfusion-weighted MRIs. Images of CBF were obtained before, during, and at the end of infusion of rhVEGF165 (a) and saline (b) initiated at 1 hour after embolization. Increase of CBF was observed in the ischemic lesion during infusion of rhVEGF165 (2 hours) (a). The increase in CBF in the rhVEGF165–treated rats (triangles; n = 6) (d) was significant (P < 0.05) compared with that in the saline-treated animals (circles; n = 6). Images of CBF were obtained before, during, and at the end of infusion of rhVEGF165 initiated at 48 hours after embolization (c). Hyperemia was evident in the ischemic lesion before infusion of rhVEGF165 (48 hours) (c). Infusion of rhVEGF165 decreases hyperemic areas in the ischemic lesion (50 hours and 52 hours) (c).
Hyperemia was detected by a perfusion-weighted MRI in the ischemic lesion at 48 hours after the stroke before infusion of rhVEGF165 (Figure 7c). Administration of rhVEGF165 at 48 hours after ischemia induced changes in the distribution of CBF within the ischemic lesion (Figure 7c). CBF was reduced in hyperemic areas during and after infusion of rhVEGF165 (Figure 7c), suggesting that exogenous VEGF at 48 hours after ischemia causes a redistribution of CBF within the ischemic lesion.
Discussion
The present study demonstrates that rhVEGF165 significantly enhances cerebral microvascular plasma perfusion and improves functional neurological recovery when rhVEGF165 is administered to ischemic rats at 48 hours after a stroke. However, administration of rhVEGF165 at 1 hour after stroke exacerbates BBB leakage, increases hemorrhagic transformation, and promotes ischemic cell damage. These findings provide the first evidence of a dual role of VEGF for enhancing cerebral microvascular perfusion and for increasing the BBB leakage in the ischemic brain.
Studies from human and experimental strokes indicate that angiogenesis is present in the focal ischemic brain (3, 6, 46). Development of angiogenesis in the ischemic brain is incompletely understood. The spatial and temporal profiles of VEGF and its receptor mRNA correspond closely with angiogenesis during embryonic development in the mouse brain, suggesting that VEGF and its receptors regulate brain angiogenesis (47, 48). Our data show that late treatment of ischemic rats with rhVEGF165 resulted in a significant increase of cerebral microvascular plasma perfusion in the penumbra of the cortex. A significant increase in microvascular plasma perfusion may be attributed to newly formed capillaries, based on the morphology in which diameters (≤3 μm) of plasma-perfused vessels in the penumbra are significantly smaller than diameters (5 μm) in the contralateral homologous areas (49). In addition, microvessels in the ipsilateral border of ischemia in the VEGF-treated rats exceeded that observed in the saline-treated rats at 28 days after ischemia (Figure 2, b–g). Taken together, our data indicate that late treatment with rhVEGF165 enhances angiogenesis in ischemic brain. Our observation is consistent with results obtained from a phase-1 trial, where administration of rhVEGF to patients with severe coronal artery disease significantly increased myocardial perfusion (10).
Our data also demonstrate that late administration of rhVEGF165 to ischemic rats significantly improves neurological function. The rotarod test is a sensitive index for assessing motor impairment after traumatic brain injury and focal cerebral ischemia (33, 34). In the present study, rats exhibited marked motor impairment at 2 days after a stroke, and partial recovery on the rotarod test was detected in the vehicle or the VEGF-treated groups at 7 and 28 days after a stroke, which is consistent with the fact that partial functional neurological recovery from a stroke occurs commonly (1, 2). However, VEGF-treated rats show a significant improvement on the rotarod test compared with vehicle-treated animals. An early significant improvement of the rotarod test (which primarily detects motor impairment in the cortex), but late modest improvement on the adhesive removal test (which detects somatosensory impairment in the cortex and the striatum that is the ischemic core), is consistent with a significant increase in plasma perfusion in the penumbra of the cortex, suggesting that exogenous VEGF may promote therapeutic angiogenesis in the ischemic rats (49). Our data are consistent with findings that functional imaging of stroke patients shows increased cerebral blood flow and metabolism in tissue surrounding focal brain infarcts (4, 5) and that stroke patients with a higher blood vessel density in the ischemic penumbra appear to have better progress and longer survival (3), indicting that restoration of cerebral microvascular circulation is important in the ischemic brain for functional recovery after a stroke. Therefore, treatment with VEGF to potentiate new vessel formation may be an important therapy for functional neurological recovery during the repair process after a stroke.
Mechanisms by which exogenously administered VEGF enhances cerebral microvascular perfusion and improves functional neurological outcome cannot be deduced from the present study. Increases in microvascular perfusion may be attributed to angiogenesis. Regeneration of cerebral microvessels requires a vascular basement membrane upon which endothelial cells migrate and proliferate to reform vascular channels after brain injury (50). VEGF is a specific mitogen for endothelial cells (8). VEGF may bind to the vascular basement membrane and cause proliferation and migration of endothelial cells (51, 52). Angiogenesis may promote ischemic brain plasticity and thereby improve functional neurological outcome (53). In addition to its effect on angiogenesis, VEGF may have direct effects on neuronal plasticity by its neurotrophic activity and by stimulating axonal outgrowth (54–56).
Based on the acute appearance of disruption of the BBB and early upregulation of VEGF in the ischemic core (our unpublished observation), we tested the hypothesis that an early increase of endogenous VEGF increases BBB leakage. We administered rhVEGF165 to the ischemic rats at 1 hour after onset of MCA occlusion and found a significant increase in BBB leakage in the ischemic lesion, but not in nonischemic areas, as measured by Gd-DTPA–enhanced MRI in the ischemic rats treated with rhVEGF165 compared with the saline treated ischemic rats. An increase in BBB leakage was further confirmed by LSCM. Furthermore, intravenous administration of rhVEGF165 did not increase BBB leakage in nonischemic rats. These findings are consistent with and extend previous studies (57–60). Intracarotid infusion of VEGF to nonischemic rats did not increase extravasation of FITC and 125I-BSA from normal brain microvessels (59). However, topical application of VEGF to the parietal cortex in nonischemic rats resulted in increases of BBB leakage to FITC-labeled dextran of molecular mass 10,000 d (58). Studies in vitro show that when it was applied to the abluminal plasma membrane of the endothelial cells, but not to luminal side, VEGF increased permeability of cultured brain microvessel endothelial cells (58, 60, 61). These data indicate that the receptors for VEGF are located on the abluminal side of vessels (58, 60, 61). However, our findings extend previous observations by demonstrating that intravenous administration of VEGF increases BBB leakage only in the ischemic lesion. An acute induction of VEGF receptors (Flt-1 and Flk-1) in the ischemic vessels and upregulation of neuropilin-1 (our unpublished observation) (62) may be responsible for the effect of exogenous VEGF on increasing BBB leakage only within the ischemic lesion. In contrast to early administration, late administration of VEGF does not increase the BBB leakage in the ischemic lesion, suggesting that there are some molecules that may inhibit VEGF-induced BBB leakage. Angiopoietin-1 has been demonstrated to counter peripheral vascular leakage induced by VEGF (63, 64). Expression of angiopoietin-1 is upregulated in this model (our unpublished observation). Future experiments to examine whether upregulation of angiopoietin-1 blocks ischemic cerebral vascular plasma leakage induced by VEGF are warranted.
Disruption of BBB was concomitant with increases in hemorrhagic transformation and ischemic lesions in rats treated with VEGF, suggesting that the early increase in the BBB leakage to large molecules causes subsequent hemorrhage and promotes ischemic cell damage. Overexpression of VEGF165 causes tumor-associated intracranial hemorrhage in mice (65). Furthermore, astrocytes play an important role in BBB integrity (66). Early upregulation of endogenous VEGF on astrocytes in the ischemic core may interact with receptors for VEGF on the ischemic vessels and contribute to the disruption of BBB leakage, thereby promoting hemorrhagic transformation and ischemic cell damage. Indeed, a recent study shows that administration of soluble VEGF-receptor chimeric protein Flt-(1–3)-IgG, inactivating VEGF to transient MCA-occluded mice, significantly reduced ischemic lesion volume measured by T2-weighted MRI (67). In contrast, local application of VEGF to the ischemic rats subjected to 90 minutes of transient MCA occlusion resulted in a reduction of infarct volume and brain edema (68).
Mechanisms that contribute to increase of leakage of the BBB in response to VEGF administration are unknown. Some effects of exogenous VEGF on endothelial cells in vitro are partially attributed to the generation of nitric oxide (NO) from these cells (69, 70). In vivo, NO increases CBF (71). In the present study, a transient augmentation and redistribution of CBF were observed in the ischemic lesion after early and late administration of VEGF, respectively, suggesting that exogenous VEGF generates NO in ischemic brain. Therefore, a significant increase in BBB leakage after administration of rhVEGF165 may result, in part, from action of NO, as others have demonstrated that exogenous VEGF administration increases permeability of the BBB through a NO synthase/cGMP–dependent pathway (57). In addition to generating NO, VEGF acutely induces microvascular hyperpermeability through functional activation of vesicular-vacuolar organelles in the cytoplasm of endothelial cells (18, 72). In contrast to many vesicular-vacuolar organelles in endothelial cells of peripheral tissues, endothelial cells in brain capillaries contain few vesicular-vacuolar organelles under physiological conditions (73, 74). However, cerebral ischemia and brain trauma increase endothelial vesicles and vacuoles in cerebral capillaries (73, 74), which may contribute to increased leakage of the BBB in response to VEGF.
In summary, our data demonstrate that late administration of VEGF enhances angiogenesis in the ischemic brain, improving neurological recovery, and that early administration of VEGF exacerbates BBB leakage. This suggests a potentially effective therapeutic strategy aimed at administration of exogenous VEGF to promote therapeutic angiogenesis during the repair process after a stroke and inhibition of VEGF at the acute stage of stroke to reduce the BBB permeability and the risk of hemorrhagic transformation after cerebral ischemia.
Acknowledgments
The authors wish to thank Denice Bliesath for manuscript preparation, Cynthia Roberts for technical assistance, and Mei Lu for statistical analysis. This work was supported by National Institute of Neurological Disorders and Stroke grants PO1 NS23393 and RO1 NS33627.
References
- 1.Kawamata, T.S.E., and Finklestein, S.P. 1997. The role of polypeptide growth factors in recovery from stroke. In Brain plasticity. H.J. Freund, B.A. Sabel, and O.W. Witte, editors. Lippincott-Raven. Philadelphia, Pennsylvania, USA. 377–382. [PubMed]
- 2.Pons TP. Reorganizing the brain. Nat Med. 1998;4:561–562. doi: 10.1038/nm0598-561. [DOI] [PubMed] [Google Scholar]
- 3.Krupinski J, Kaluza J, Kumar P, Kumar S, Wang JM. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke. 1994;25:1794–1798. doi: 10.1161/01.str.25.9.1794. [DOI] [PubMed] [Google Scholar]
- 4.Weiller C, Ramsay SC, Wise RJ, Friston KJ, Frackowiak RS. Individual patterns of functional reorganization in the human cerebral cortex after capsular infarction. Ann Neurol. 1993;33:181–189. doi: 10.1002/ana.410330208. [DOI] [PubMed] [Google Scholar]
- 5.Cramer SC, et al. A functional MRI study of subjects recovered from hemiparetic stroke. Stroke. 1997;28:2518–2527. doi: 10.1161/01.str.28.12.2518. [DOI] [PubMed] [Google Scholar]
- 6.Sbarbati A, et al. The microvascular system in ischemic cortical lesions. Acta Neuropathol (Berl) 1996;92:56–63. doi: 10.1007/s004010050489. [DOI] [PubMed] [Google Scholar]
- 7.Coyle P, Heistad DD. Blood flow through cerebral collateral vessels one month after middle cerebral artery occlusion. Stroke. 1987;18:407–411. doi: 10.1161/01.str.18.2.407. [DOI] [PubMed] [Google Scholar]
- 8.Ferrara N. The role of vascular endothelial growth factor in pathological angiogenesis. Breast Cancer Res Treat. 1995;36:127–137. doi: 10.1007/BF00666035. [DOI] [PubMed] [Google Scholar]
- 9.Carmeliet P, Collen D. Molecular analysis of blood vessel formation and disease. Am J Physiol. 1997;273:H2091–H2104. doi: 10.1152/ajpheart.1997.273.5.H2091. [DOI] [PubMed] [Google Scholar]
- 10.Hendel RC, et al. Effect of intracoronary recombinant human vascular endothelial growth factor on myocardial perfusion: evidence for a dose-dependent effect. Circulation. 2000;101:118–121. doi: 10.1161/01.cir.101.2.118. [DOI] [PubMed] [Google Scholar]
- 11.Takeshita S, et al. Therapeutic angiogenesis. A single intraarterial bolus of vascular endothelial growth factor augments revascularization in a rabbit ischemic hind limb model. J Clin Invest. 1994;93:662–670. doi: 10.1172/JCI117018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banai S, et al. Angiogenic-induced enhancement of collateral blood flow to ischemic myocardium by vascular endothelial growth factor in dogs. Circulation. 1994;89:2183–2189. doi: 10.1161/01.cir.89.5.2183. [DOI] [PubMed] [Google Scholar]
- 13.Aiello LP. Vascular endothelial growth factor. 20th-century mechanisms, 21st-century therapies. Invest Ophthalmol Vis Sci. 1997;38:1647–1652. [PubMed] [Google Scholar]
- 14.Bauters C, et al. Site-specific therapeutic angiogenesis after systemic administration of vascular endothelial growth factor. J Vasc Surg. 1995;21:314–324. doi: 10.1016/s0741-5214(95)70272-5. [DOI] [PubMed] [Google Scholar]
- 15.Kovacs Z, Ikezaki K, Samoto K, Inamura T, Fukui M. VEGF and flt. Expression time kinetics in rat brain infarct. Stroke. 1996;27:1865–1872. doi: 10.1161/01.str.27.10.1865. [DOI] [PubMed] [Google Scholar]
- 16.Hayashi T, Abe K, Suzuki H, Itoyama Y. Rapid induction of vascular endothelial growth factor gene expression after transient middle cerebral artery occlusion in rats. Stroke. 1997;28:2039–2044. doi: 10.1161/01.str.28.10.2039. [DOI] [PubMed] [Google Scholar]
- 17.Lennmyr F, Ata KA, Funa K, Olsson Y, Terent A. Expression of vascular endothelial growth factor (VEGF) and its receptors (Flt-1 and Flk-1) following permanent and transient occlusion of the middle cerebral artery in the rat. J Neuropathol Exp Neurol. 1998;57:874–882. doi: 10.1097/00005072-199809000-00009. [DOI] [PubMed] [Google Scholar]
- 18.Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol. 1995;146:1029–1039. [PMC free article] [PubMed] [Google Scholar]
- 19.Gotoh O, Asano T, Koide T, Takakura K. Ischemic brain edema following occlusion of the middle cerebral artery in the rat. I. The time courses of the brain water, sodium and potassium contents and blood-brain barrier permeability to 125I-albumin. Stroke. 1985;16:101–109. doi: 10.1161/01.str.16.1.101. [DOI] [PubMed] [Google Scholar]
- 20.Belayev L, Busto R, Zhao W, Ginsberg MD. Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 1996;739:88–96. doi: 10.1016/s0006-8993(96)00815-3. [DOI] [PubMed] [Google Scholar]
- 21.Hatashita S, Hoff JT. Role of blood-brain barrier permeability in focal ischemic brain edema. Adv Neurol. 1990;52:327–333. [PubMed] [Google Scholar]
- 22.Kuroiwa T, Shibutani M, Okeda R. Blood-brain barrier disruption and exacerbation of ischemic brain edema after restoration of blood flow in experimental focal cerebral ischemia. Acta Neuropathol (Berl) 1988;76:62–70. doi: 10.1007/BF00687681. [DOI] [PubMed] [Google Scholar]
- 23.Lyden PD, Zivin JA. Hemorrhagic transformation after cerebral ischemia: mechanisms and incidence. Cerebrovasc Brain Metab Rev. 1993;5:1–16. [PubMed] [Google Scholar]
- 24.Liu HM, Chen HH. Correlation between fibroblast growth factor expression and cell proliferation in experimental brain infarct: studied with proliferating cell nuclear antigen immunohistochemistry. J Neuropathol Exp Neurol. 1994;53:118–126. doi: 10.1097/00005072-199403000-00002. [DOI] [PubMed] [Google Scholar]
- 25.Zhang RL, Chopp M, Zhang ZG, Jiang Q, Ewing JR. A rat model of embolic focal cerebral ischemia. Brain Res. 1997;766:83–92. doi: 10.1016/s0006-8993(97)00580-5. [DOI] [PubMed] [Google Scholar]
- 26.Jiang Q, et al. Diffusion-, T2-, and perfusion-weighted nuclear magnetic resonance imaging of middle cerebral artery embolic stroke and recombinant tissue plasminogen activator intervention in the rat. J Cereb Blood Flow Metab. 1998;18:758–767. doi: 10.1097/00004647-199807000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Yang R, et al. Substantially attenuated hemodynamic responses to Escherichia coli-derived vascular endothelial growth factor given by intravenous infusion compared with bolus injection. J Pharmacol Exp Ther. 1998;284:103–110. [PubMed] [Google Scholar]
- 28.Yang R, et al. Effects of vascular endothelial growth factor on hemodynamics and cardiac performance. J Cardiovasc Pharmacol. 1996;27:838–844. doi: 10.1097/00005344-199606000-00011. [DOI] [PubMed] [Google Scholar]
- 29.LeBihan D, et al. MR imaging of intravoxel incoherent motions: application to diffusion and perfusion in neurologic disorders. Radiology. 1986;161:401–407. doi: 10.1148/radiology.161.2.3763909. [DOI] [PubMed] [Google Scholar]
- 30.Jiang Q, et al. The temporal evolution of MRI tissue signatures after transient middle cerebral artery occlusion in rat. J Neurol Sci. 1997;145:15–23. doi: 10.1016/s0022-510x(96)00286-9. [DOI] [PubMed] [Google Scholar]
- 31.Williams D, Detre J, Leigh J, Koretsky A. Magnetic resonance imaging of perfusion using spin inversion of arterial water. Proc Natl Acad Sci USA. 1992;89:212–216. doi: 10.1073/pnas.89.1.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peck D, Spickler E, Knight R, Hearshen D, Windham J. Analysis of the evolution of focal cerebral ischemia in the rat using the Eigenimage filter. Magn Reson Med. 1992;26:259–273. doi: 10.1002/mrm.1910260207. [DOI] [PubMed] [Google Scholar]
- 33.Zhang L, Chen J, Li Y, Zhang ZG, Chopp M. Quantitative measurement of motor and somatosensory impairments after mild (30 min) and severe (2 h) transient middle cerebral artery occlusion in rats. J Neurol Sci. 2000;174:141–146. doi: 10.1016/s0022-510x(00)00268-9. [DOI] [PubMed] [Google Scholar]
- 34.Hamm RJ, Pike BR, O’Dell DM, Lyeth BG, Jenkins LW. The rotarod test: an evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. J Neurotrauma. 1994;11:187–196. doi: 10.1089/neu.1994.11.187. [DOI] [PubMed] [Google Scholar]
- 35.Schallert T, et al. Tactile extinction: distinguishing between sensorimotor and motor asymmetries in rats with unilateral nigrostriatal damage. Pharmacol Biochem Behav. 1982;16:455–462. doi: 10.1016/0091-3057(82)90452-x. [DOI] [PubMed] [Google Scholar]
- 36.Matesic DF, Lin RC. Microtubule-associated protein 2 as an early indicator of ischemia-induced neurodegeneration in the gerbil forebrain. J Neurochem. 1994;63:1012–1020. doi: 10.1046/j.1471-4159.1994.63031012.x. [DOI] [PubMed] [Google Scholar]
- 37.Paxinos, G., and Watson, C. 1986. The rat brain in sterotaxic coordinates. 2nd edition. Academic Press Inc. New York, New York, USA. 29–50.
- 38.Gesztelyi G, et al. Parenchymal microvascular systems and cerebral atrophy in spontaneously hypertensive rats. Brain Res. 1993;611:249–257. doi: 10.1016/0006-8993(93)90510-t. [DOI] [PubMed] [Google Scholar]
- 39.Hudetz AG, Greene AS, Feher G, Knuese DE, Cowley AW., Jr Imaging system for three-dimensional mapping of cerebrocortical capillary networks in vivo. Microvasc Res. 1993;46:293–309. doi: 10.1006/mvre.1993.1054. [DOI] [PubMed] [Google Scholar]
- 40.Miyoshi Y, Date I, Ohmoto T. Three-dimensional morphological study of microvascular regeneration in cavity wall of the rat cerebral cortex using the scanning electron microscope: implications for delayed neural grafting into brain cavities. Exp Neurol. 1995;131:69–82. doi: 10.1016/0014-4886(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 41.Dellian M, Witwer BP, Salehi HA, Yuan F, Jain RK. Quantitation and physiological characterization of angiogenic vessels in mice: effect of basic fibroblast growth factor, vascular endothelial growth factor/vascular permeability factor, and host microenvironment. Am J Pathol. 1996;149:59–71. [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang ZG, Davies K, Prostak J, Fenstermacher J, Chopp M. Quantitation of microvascular plasma perfusion and neuronal microtubule-associated protein in ischemic mouse brain by laser-scanning confocal microscopy. J Cereb Blood Flow Metab. 1999;19:68–78. doi: 10.1097/00004647-199901000-00008. [DOI] [PubMed] [Google Scholar]
- 43.Knight RA, et al. Prediction of impending hemorrhagic transformation in ischemic stroke using magnetic resonance imaging in rats. Stroke. 1998;29:144–151. doi: 10.1161/01.str.29.1.144. [DOI] [PubMed] [Google Scholar]
- 44.Mathews V, et al. Combined effects of magnetization transfer and gadolinium in cranial MR imaging and MR angiography. Am J Roentgenol. 1995;164:169–172. doi: 10.2214/ajr.164.1.7998532. [DOI] [PubMed] [Google Scholar]
- 45.Zhang RL, Zhang ZG, Chopp M. Increased therapeutic efficacy with rt-PA and anti-CD18 antibody treatment of stroke in the rat. Neurology. 1999;52:273–279. doi: 10.1212/wnl.52.2.273. [DOI] [PubMed] [Google Scholar]
- 46.Krupinski J, Kaluza J, Kumar P, Wang M, Kumar S. Prognostic value of blood vessel density in ischaemic stroke. Lancet. 1993;342:742. doi: 10.1016/0140-6736(93)91734-4. [DOI] [PubMed] [Google Scholar]
- 47.Breier G, Albrecht U, Sterrer S, Risau W. Expression of vascular endothelial growth factor during embryonic angiogenesis and endothelial cell differentiation. Development. 1992;114:521–532. doi: 10.1242/dev.114.2.521. [DOI] [PubMed] [Google Scholar]
- 48.Risau W. Development and differentiation of endothelium. Kidney Int Suppl. 1998;67:S3–S6. doi: 10.1046/j.1523-1755.1998.06701.x. [DOI] [PubMed] [Google Scholar]
- 49.Klagsbrun, M., and Folkman, J. 1990. Angiogenesis. In Peptide growth factors and their receptors II. M.B. Sporn and A.B. Roberts, editors. Springer-Verlag. New York, New York, USA. 459–586.
- 50.Cancilla PA, Frommes SP, Kahn LE, DeBault LE. Regeneration of cerebral microvessels: a morphologic and histochemical study after local freeze-injury. Lab Invest. 1979;40:74–82. [PubMed] [Google Scholar]
- 51.Rosenstein JM, Mani N, Silverman WF, Krum JM. Patterns of brain angiogenesis after vascular endothelial growth factor administration in vitro and in vivo. Proc Natl Acad Sci USA. 1998;95:7086–7091. doi: 10.1073/pnas.95.12.7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ingber DE, Madri JA, Folkman J. A possible mechanism for inhibition of angiogenesis by angiostatic steroids: induction of capillary basement membrane dissolution. Endocrinology. 1986;119:1768–1775. doi: 10.1210/endo-119-4-1768. [DOI] [PubMed] [Google Scholar]
- 53.Stroemer RP, Kent TA, Hulsebosch CE. Enhanced neocortical neural sprouting, synaptogenesis, and behavioral recovery with D-amphetamine therapy after neocortical infarction in rats. Stroke. 1998;29:2381–2393. doi: 10.1161/01.str.29.11.2381. [DOI] [PubMed] [Google Scholar]
- 54.Schratzberger P, et al. Favorable effect of VEGF gene transfer on ischemic peripheral neuropathy. Nat Med. 2000;6:405–413. doi: 10.1038/74664. [DOI] [PubMed] [Google Scholar]
- 55.Sondell M, Lundborg G, Kanje M. Vascular endothelial growth factor has neurotrophic activity and stimulates axonal outgrowth, enhancing cell survival and Schwann cell proliferation in the peripheral nervous system. J Neurosci. 1999;19:5731–5740. doi: 10.1523/JNEUROSCI.19-14-05731.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gale NW, Yancopoulos GD. Growth factors acting via endothelial cell-specific receptor tyrosine kinases: VEGFs, angiopoietins, and ephrins in vascular development. Genes Dev. 1999;13:1055–1066. doi: 10.1101/gad.13.9.1055. [DOI] [PubMed] [Google Scholar]
- 57.Mayhan WG. VEGF increases permeability of the blood-brain barrier via a nitric oxide synthase/cGMP-dependent pathway. Am J Physiol. 1999;276:C1148–C1153. doi: 10.1152/ajpcell.1999.276.5.C1148. [DOI] [PubMed] [Google Scholar]
- 58.Wang W, Merrill MJ, Borchardt RT. Vascular endothelial growth factor affects permeability of brain microvessel endothelial cells in vitro. Am J Physiol. 1996;271:C1973–C1980. doi: 10.1152/ajpcell.1996.271.6.C1973. [DOI] [PubMed] [Google Scholar]
- 59.Criscuolo GR, Merrill MJ, Oldfield EH. Characterization of a protein product of human malignant glial tumors that induces microvascular permeability. Adv Neurol. 1990;52:469–474. [PubMed] [Google Scholar]
- 60.Zhao L, Zhang MM, Ng KY. Effects of vascular permeability factor on the permeability of cultured endothelial cells from brain capillaries. J Cardiovasc Pharmacol. 1998;32:1–4. doi: 10.1097/00005344-199807000-00001. [DOI] [PubMed] [Google Scholar]
- 61.Qu H, Nagy JA, Senger DR, Dvorak HF, Dvorak AM. Ultrastructural localization of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) to the abluminal plasma membrane and vesiculovacuolar organelles of tumor microvascular endothelium. J Histochem Cytochem. 1995;43:381–389. doi: 10.1177/43.4.7534783. [DOI] [PubMed] [Google Scholar]
- 62.Kawasaki T, et al. A requirement for neuropilin-1 in embryonic vessel formation. Development. 1999;126:4895–4902. doi: 10.1242/dev.126.21.4895. [DOI] [PubMed] [Google Scholar]
- 63.Thurston G, et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286:2511–2514. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- 64.Thurston G, et al. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med. 2000;6:460–463. doi: 10.1038/74725. [DOI] [PubMed] [Google Scholar]
- 65.Cheng SY, Nagane M, Huang HS, Cavenee WK. Intracerebral tumor-associated hemorrhage caused by overexpression of the vascular endothelial growth factor isoforms VEGF121 and VEGF165 but not VEGF189. Proc Natl Acad Sci USA. 1997;94:12081–12087. doi: 10.1073/pnas.94.22.12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hatashita S, Hoff J. Brain edema and cerebrovascular ischemia in rats. Stroke. 1990;21:582–588. doi: 10.1161/01.str.21.4.582. [DOI] [PubMed] [Google Scholar]
- 67.van Bruggen N, et al. VEGF antagonism reduces edema formation and tissue damage following ischemic reperfusion injury in the mouse brain. J Clin Invest. 1999;104:1613–1620. doi: 10.1172/JCI8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hayashi T, Abe K, Itoyama Y. Reduction of ischemic damage by application of vascular endothelial growth factor in rat brain after transient ischemia. J Cereb Blood Flow Metab. 1998;18:887–895. doi: 10.1097/00004647-199808000-00009. [DOI] [PubMed] [Google Scholar]
- 69.Levin ER, et al. Role of vascular endothelial cell growth factor in Ovarian Hyperstimulation Syndrome. J Clin Invest. 1998;102:1978–1985. doi: 10.1172/JCI4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu HM, Huang Q, Yuan Y, Granger HJ. VEGF induces NO-dependent hyperpermeability in coronary venules. Am J Physiol. 1996;271:H2735–H2739. doi: 10.1152/ajpheart.1996.271.6.H2735. [DOI] [PubMed] [Google Scholar]
- 71.Iadecola C. Regulation of the cerebral microcirculation during neural activity: is nitric oxide the missing link? Trends Neurosci. 1993;16:206–214. doi: 10.1016/0166-2236(93)90156-g. [DOI] [PubMed] [Google Scholar]
- 72.Feng D, Nagy JA, Hipp J, Dvorak HF, Dvorak AM. Vesiculo-vacuolar organelles and the regulation of venule permeability to macromolecules by vascular permeability factor, histamine, and serotonin. J Exp Med. 1996;183:1981–1986. doi: 10.1084/jem.183.5.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reese TS, Karnovsky MJ. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol. 1967;34:207–217. doi: 10.1083/jcb.34.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Simionescu M, Simionescu N, Palade GE. Morphometric data on the endothelium of blood capillaries. J Cell Biol. 1974;60:128–152. doi: 10.1083/jcb.60.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]