

Abstract
Hypoxic, hyperosmotic, and genotoxic stress slow mouse trophoblast stem cell (mTSC) proliferation, decrease potency/stemness, and increase differentiation. Previous reports suggest a period of reversibility in stress-induced mTSC differentiation. Here we show that hypoxic stress at 0.5% O2 decreased potency factor protein by ∼60%–90% and reduced growth to nil. Hypoxia caused a 35-fold increase in apoptosis at Day 3 and a 2-fold increase at Day 6 above baseline. The baseline apoptosis rate was only 0.3%. Total protein was never less than baseline during hypoxic treatment, suggesting 0.5% O2 is a robust, nonmorbid stressor. Hypoxic stress induced ∼50% of trophoblast giant cell (TGC) differentiation with a simultaneous 5- to 6-fold increase in the TGC product antiluteolytic prolactin family 3, subfamily d, member 1 (PRL3D1), despite the presence of fibroblast growth factor 4 (FGF4). Hypoxia-induced TGC differentiation was also supported by potency and differentiation mRNA marker analysis. FGF4 removal at 20% O2 committed cell fate towards irreversible differentiation at 2 days, with similar TGC percentages after an additional 3 days of culture under potency conditions when FGF4 was readded or under differentiation conditions without FGF4. However, hypoxic stress required 4 days to irreversibly differentiate cells. Runted stem cell growth, forced differentiation of fewer cells, and irreversible differentiation limit total available stem cell population. Were mTSCs to respond to stress in a similar mode in vivo, miscarriage might occur as a result, which should be tested in the future.
Keywords: differentiation, hypoxic stress, potency, trophoblast stem cells
INTRODUCTION
Mouse embryos grow exponentially to rapidly accumulate cell mass starting 1 day before implantation into the uterus and persisting for a week or more after implantation [1]. Necessary first differentiated lineages also arise during this rapid growth. Before implantation, trophoblast and embryonic stem cells (TSC and ESC lineages, respectively) initiate and allocate [2] in the embryo to further develop into extraembryonic and embryonic structures. Exponential growth starts first in the trophoblast lineage [1]. Rapid trophoblast cell growth produces PRL3D1 (aka placental lactogen 1; PL1) to maintain ovarian function and enable maternal recognition of pregnancy early after implantation [3]. This is similar to the function of human chorionic gonadotropin in early human pregnancy recognition and maintenance [4].
Hypoxia is commonly encountered during pregnancy. It can happen to pregnancies at high altitude [5] or in urban areas because of carbon monoxide (CO) pollution. CO has higher binding affinity to hemoglobin than O2 [6]. Increased CO exposure during pregnancy could reduce the amount of O2 delivered to the developing fetus by as much as 10% [7]. Cigarette smoking also increases maternal blood CO levels [8], which may further compromise O2 delivery to the fetus. Other conditions, such as maternal hypertension, anemia, and pulmonary disease, also contribute to fetal hypoxia [9]. Chronic hypoxia has been associated with intrauterine growth restriction and low birth weight, as well as increased cardiovascular diseases in adults [10, 11]. It has been reported that embryos derived from females exposed to malnutrition and cortisol during only the preimplantation period show slowed growth and negative prenatal and postnatal outcomes [12, 13]. The negative impact of stress on early trophoblast cells is likely to play a role in that process, because aberrations in trophoblast proliferation and differentiation in the early pregnancy or peri-implantation period are associated with adverse pregnancy outcome [14, 15]. Here we used mouse TSCs (mTSCs) to model the effect of hypoxia during the peri-implantation period, which is also the period when the majority of pregnancy loss happens [16]. Notice that all the external stimuli that cause hypoxia in vivo may initiate stress responses in a more complex systemic way, which can modify the effect of hypoxia on TSCs. Here we only study the single variable hypoxia in a reductionist approach that reveals the response of mTSCs under hypoxic stress.
Mouse TSCs have been successfully isolated from polar trophectoderm or extraembryonic ectoderm of mouse embryos; their potency and proliferation can be maintained in vitro with FGF4 [17]. In vivo differentiation of mTSCs occurs when the cells grow away from their FGF4 source [18]. In vitro differentiation happens when FGF4 is removed [17]. However, even 1 day of hypoxic stress has been shown to decrease the mRNA level of potency factors and increase that of differentiation markers despite the presence of FGF4 and without an overt differentiated phenotype [19]. Other types of stress such as hyperosmotic sorbitol and genotoxic benzopyrene can also force potency loss and increased mTSC differentiation despite the presence of FGF4 [20–22]. Hyperosmotic stress induces global mRNA changes of mTSCs by 24 h that emulate normal first-lineage trophoblast giant cell (TGC) differentiation caused by FGF4 removal [23]. However, hyperosmotic stress-forced differentiation occurs largely in the absence of later lineages that would have been induced by normal differentiation with FGF4 removal. Stress-activated protein kinase (SAPK) mediates hyperosmolar stress-induced heart and neural crest derivatives-expressed protein 1 (HAND1) transcription factor protein increase [20], which leads to TGC differentiation and enables PRL3D1 production [24]. Hypoxic stress at 0.5% O2 also causes SAPK-dependent increase in Hand1 mRNA [25]. We hypothesize that long-term hypoxic stress diminishes mTSC growth and potency and forces TGC differentiation and antiluteolytic PRL3D1 production.
There are several subtypes of TGC identified in mouse placenta, and not all produce PRL3D1. Parietal TGCs are characterized as the main subtype expressing PRL3D1, whereas mature spiral artery TGCs (Spa-TGCs) and canal TGCs (C-TGCs) do not [26]. It is possible that earlier Spa-TGCs and C-TGCs also express PRL3D1 [27]. In support of this, it was shown that TGCs isolated from early placenta at Days 7 and 9 of pregnancy went through successive stages of PRL3D1+, then PRL3D1+/PRL3B1+, and finally PRL3D1−/PRL3B1+ expression [28]. There is emerging evidence showing stress forces mTSC and murine ESC to differentiate primarily toward the earliest lineages [23, 29]. Hypothetically, hypoxic stress-forced differentiation may also include a large portion of PRL3D1+ TGC subtypes.
Although terminally differentiated TGC do not revert to being stem cells, there is evidence suggesting some aspects of stress-forced differentiation can be reversed. Hyperosmotic stress produced a reversible ∼50% inhibitor of differentiation 2 (ID2) protein loss [21], whereas Id2 mRNA was preserved during the same period [23]. ID2 is a potency factor that can block the normal differentiation of human placental stem cells when overexpressed [30], and related ID1 blocks differentiation and Prl3d1 transcription in rat choriocarcinoma cells (Rcho)1 [31]. The signature response of stressed somatic cells is to disassemble ribosomes but save mRNA into stress granules from which the mRNAs are freed and translated once stress subsides [32]. Loss of ID2 protein while preserving Id2 mRNA may enable some reversibility in stress-induced mTSC differentiation. We hypothesize there is a period of reversibility in stress-induced mTSC differentiation, and it would be longer than normal differentiation with FGF4 removal. This would potentially enable the stem cell reserve to replenish the placenta during rebound growth after stress.
MATERIALS AND METHODS
Reagents
Fetal bovine serum, RPMI1640 (Cat. No. 21870), and FGF4 (Cat. No. PHG0154) were from Gibco. Heparin (Cat. No. H3149) was purchased from Sigma Chemical Co. Primary and secondary antibodies used were purchased from the following sources: CDX2 (CDX2-88; Biogenex), ID2 (SC489; Santa Cruz Biotechnology), cleaved caspase-3 (CS9664; Cell Signaling), B-actin (CS4970; Cell Signaling), tubulin (T9026; Sigma), anti-rabbit horseradish peroxidase (HRP)-linked antibody (CS7074; Cell Signaling), anti-mouse HRP-linked antibody (CS7076; Cell Signaling), and anti-rabbit IgG-TR (SC2780; Santa Cruz Biotechnology). PRL3D1 antiserum was a generous gift of Dr. Soares from the University of Kansas Medical Center, and it was characterized in [33]. All reagents (cell cryopreservation buffer, nucleus isolation solution, RNase, and propidium iodide) used for flow cytometry DNA content analysis were contained in the kit purchased from BD Bioscience (Cat. No. 340242).
Cell Lines and Culture Conditions
The mTSC isolate was gratefully received from Dr. Rossant (Lunenfeld Research Institute, Ontario, Canada) [17]. The mTSCs were cultured as described previously [34]. Briefly, RPMI-1640 medium supplemented with 20% fetal bovine serum, 70% mouse embryonic fibroblast conditioned medium (MEFCM), and 25 ng/ml FGF4 was used for routine mTSC culture at 20% O2. When the mTSCs reached 70%–80% confluence, cells were trypsinized and passaged into new dishes 24 h before the start of each experiment. The starting cell confluence was ∼10%. The time immediately before the start of experiment was designated as time 0 (Tzero). Then cells were moved to pre-equilibrated medium at 0.5% O2 or normal differentiation medium without FGF4 and MEFCM at 20% O2. The 20% O2 culture was carried out in a conventional CO2 incubator. Hypoxic 0.5% O2 culture was done in a gas chamber equilibrated with commercially premixed gas containing 0.5% O2:5% CO2:94.5% N2.
There were two periods of cell culture before flow cytometry. The first period was called the initial treatment, when 0.5% O2 plus FGF4 or FGF4 and MEFCM removal was applied at 20% O2. The second period was called fate determination, which was another 3 days of cell culture after the initial treatment. Fate determination was conducted under either potency (20% O2 plus FGF4 and MEFCM) or differentiation conditions (20% O2 minus FGF4 and MEFCM). Initial treatment was 2–5 days for 0.5% O2 and 1–4 days for normal differentiation. After each initial treatment day, one plate of cells was trypsinized, resuspended in citrate-dimethyl sulfoxide buffer, and snap frozen on dry ice. At the same time, two other plates of cells were put into fate determination under either potency or differentiation conditions. After the fate determination period, cells were also trypsinized and frozen.
Nuclear Staining and Nuclear Size Quantification by ImageJ
At the end of each 0.5% O2 treatment day, cells were stained with Hoechst 33342 (H1399; Molecular Probes) at 5 μg/ml for 30 min in a CO2 incubator. Afterwards, images were taken with a DM-IRE2 fluorescence microscope (Leica). The Analyze Particles function of ImageJ can quantify the size of the blue-stained nucleus in each image. All images were taken at 10× magnification. Nucleus size of ∼2000 nuclei from normally maintained mTSCs were measured. There is variation in the nucleus size of the normal undifferentiated mTSCs. The size range of mean ± 2 SD was calculated. Nuclei with values above or below the mean ± 2 SD range were individually checked against their appearance in the fluorescence image in order to make sure the low values truly represented an intact nucleus instead of debris or cells in the middle of mitosis where their nuclei appeared as two small separated groups of blue staining. Furthermore, nuclei with a large size were inspected to confirm they were from individual spontaneously differentiated giant cells and not several nuclei mistakenly counted as one. Based on that, we arbitrarily set the high cutoff value and low cutoff value. Nuclei with size measurement above the high cutoff value were considered to represent a giant cell. Blue stains with size below the low cutoff value were considered too small to be an intact nucleus and excluded in the final giant cell percentage calculation. The same standard was used for all experimental groups, and at least 2500 nuclei were measured to generate the giant cell percentage.
Western Blot
Cells were washed twice with ice-cold PBS (SH30256; Fisher Scientific) and lysed with RIPA buffer (PI89901; ThermoScientific). Next, 15–30 μg of whole-cell extracts was separated on a 4%–20% SDS-PAGE gel (Cat. No. 4561094; Bio-Rad) using Bio-Rad Mini Format 1-D Electrophoresis Systems and transferred to nitrocellulose membrane using Bio-Rad Mini Trans-Blot Electrophoretic Transfer Cell. The sizes of the probed proteins were 38 kDa for CDX2, 15 kDa for ID2, 30–35 kDa for PRL3D1, 17 kDa for cleaved caspase-3, 45 kDa for B-actin, and 52 kDa for tubulin. Every blot carrying transferred proteins was cut into multiple pieces containing each probed protein (B-actin was reprobed after stripping off CDX2 using the same piece of blot). The location of each protein on the blot was estimated based on its size relative to protein ladder (Cat. No. LC5800; ThermoFisher) and amido black staining showing band shape. Afterwards, the blots were blocked at room temperature (RT) for 1 h with 5% fat-free milk (Cat. No.1705016; Bio-Rad) and incubated with CDX2 (1:1500), ID2 (1:400), PRL3D1 (1:500), cleaved caspase-3 (1:500), B-actin (1:1200), or tubulin (1:10 000) antibodies overnight at 4°C. The next morning, the blots were washed and incubated in HRP-conjugated secondary antibody (1:10 000) at RT for 90 min. Primary and secondary antibodies were diluted in 2% fat-free milk/TBST. The protein bands were visualized using enhanced chemiluminescence (Amersham). ImageJ was used to quantify the intensity of the bands from proteins of interest and normalized to loading control. Value for Tzero was arbitrarily set as 1 to show fold changes due to treatment.
Immunofluorescence
Cell culture was done on sterile coverslips. At each end point, the coverslips were washed with PBS and fixed with 3% paraformaldehyde for 25 min, quenched with 0.1 M glycine, permeabilized with 0.25% Triton X-100 for 12 min, and blocked with 3% (w/v) bovine serum albumin for 45 min at RT. Incubation with monoclonal mouse PRL3D1 (SC376436; Santa Cruz) antibody at 1:100 dilution or cleaved caspase-3 (1:200) was carried out at 4°C overnight. Then the coverslips were washed and incubated with anti-mouse IgM-TR (SC2983; Santa Cruz) or anti-rabbit IgG-FITC (554020; BD Pharmingen) at 1:400 dilution for 90 min at RT. Images were taken with a DM-IRE2 fluorescence microscope (Leica) using Simple PCI image acquisition software (Hamamatsu).
RNA Isolation and Quantitative RT-PCR
Total RNA was isolated using Rneasy Mini Kit (Qiagen) and treated with DNase. The cDNA was prepared using QuantiTect Reverse Transcription Kit iScript (Qiagen), and assayed using SYBR Green by 7500 fast instrument (Applied Biosystems). Each independent biological experiment was performed four times and all genes were normalized to Rn18S rRNA. Relative mRNA expression levels were determined by the ΔΔCT method. Fold change of individual genes was determined by comparison to expression in cells cultured at 20% O2 potency conditions. Primers used are shown in Table 1. All primer pairs were checked for specificity using BLAST analysis and thermal dissociation curves to ensure amplification of a single product.
TABLE 1.
List of primers for potency and differentiation markers.

F, forward; R, reverse.
Flow Cytometry Analysis
On the day of flow cytometry analysis, cells were quickly thawed at RT. Cell nucleus isolation and staining were done following the manufacturer's instructions [35]. Flow cytometry was carried out using a BD LSR II flow cytometer (BD Biosciences) and the FACSDiva 6.0 software (BD Biosciences).
Statistical Analysis
All experiments were performed in at least three replications. Data were analyzed using SPSS version 22.0. Independent t-test was used for the comparison between potency and differentiation culture group in fate determination experiments. One-way ANOVA was used for the comparisons among treatment days. Data were logarithm transformed to meet the assumptions of one-way ANOVA when such assumptions were violated. Dunnett or Tukey post hoc tests were performed following significant one-way ANOVA to further investigate the differences between treatments and Tzero control or among different treatment days, respectively. Values are presented as means ± SEM. P < 0.05 indicates statistical significance.
RESULTS
To test the hypothesis that hypoxic stress diminishes mTSC growth, we compared cell mass accumulation at 20% and 0.5% O2 with FGF4 present. Normal stem cell culture at 20% O2 + FGF4 produced a 4-fold increase in total protein after 2 days (P < 0.05), whereas hypoxic mTSC showed near zero cell mass increase after 6 days (Fig. 1A). Although 1–2 days of 0.5% O2 treatment increased total protein amount compared with Day 0, there was significantly more cell growth in the 20% O2 condition. Normal stem cell culture was ended at 2 days because by that time cells had already become confluent. There was significant increase in apoptosis starting from Day 2 of 0.5% O2 treatment, peaking at Day 3 with a ∼35-fold increase as indicated by the level of cleaved caspase-3 analyzed by Western blot (Fig. 1B). On Day 6, the level of apoptosis was ∼2-fold over background. We next studied the fraction of apoptotic cells at baseline and Day 3 or 6 of 0.5% O2 treatment using cleaved caspase-3 immunofluorescence staining (Fig. 1C). The baseline level of apoptosis was 0.3%. The fraction of apoptotic cells was 14.6% at Day 3 and 5.7% at Day 6 (P < 0.05, ANOVA followed by Dunnett post hoc test).
FIG. 1.

Treatment with 0.5% O2 restricted mTSCs cell growth assayed by protein and induced apoptotic response assayed by cleaved caspase-3. A) Comparison of cell growth at 0.5% O2 with 20% O2. B) The level of apoptosis over 1–6 days of 0.5% O2 culture compared with Day 0 baseline. C) The quantification of the fraction of apoptotic cells at the indicated days by immunofluorescence. Blue, Hoechst; green: cleaved caspase-3. Bar = 200 μm. Asterisk (*) indicates statistical difference was found in treatment groups compared with 0-day control (P < 0.05), and letter a indicates significant difference in cell growth between 20% and 0.5% O2 at Days 1 and 2 of stimulation.
To test the hypothesis that hypoxia forces differentiation despite the presence of FGF4, we next stained the cells cultured at 0.5% O2 with nuclear staining dye Hoechst 33342 to observe the formation of TGC. The fraction of TGC during 6 days of 0.5% O2 culture was quantified and compared with starting Day 0. There was a significant increase in TGC percentage starting from 2 days of 0.5% O2 exposure (P < 0.05). TGC percentage increased, then plateaued at ∼50% by Days 4–6 (Fig. 2). There was no statistical difference in TGC percentage among 4, 5, and 6 days of 0.5% O2 culture.
FIG. 2.

Treatment with 0.5% O2 forced ∼50% of TGC differentiation from mTSCs by Day 6. A) The quantification of TGC percentage over 6 days of 0.5% O2 culture. B–D) Examples of the nucleus-staining image taken at Days 0 (B), 3 (C), and 6 (D). Bar in B = 200 μm. Asterisk (*) indicates where statistical significance was found compared with 0-day control (P < 0.05), and letter a indicates significantly higher TGC percentage at 4–6 days of 0.5% O2 exposure compared with 2–3 days.
Consistent with the observation of increased TGC percentage, caudal type homeobox 2 (CDX2) and ID2 potency proteins were significantly decreased by Day 2 of 0.5% O2 treatment (Fig. 3A). At Day 6, CDX2 and ID2 were decreased by ∼90% and ∼60% respectively compared with unstressed mTSCs at Day 0. PRL3D1 increased 5–6-fold at Days 5 and 6 of 0.5% O2 culture compared with Day 0 (P < 0.05) (Fig. 3B). O2 at 0.5% induced comparable levels of PRL3D1 at 6 days of culture as normal differentiation with FGF4 removal (Fig. 3C). Both normal and hypoxic stress-induced differentiation produced PRL3D1-expressing cells and TGC formation (Fig. 4). However, 0.5% O2-induced giant cells appeared to be smaller and to express lower levels of PRL3D1 per cell compared with those PRL3D1-positive cells in normal differentiation.
FIG. 3.

Treatment with 0.5% O2 induced 60%–90% of mTSCs potency loss and 5–6-fold gain of TGC differentiation marker PRL3D1 that plateaued at Days 5–6. The levels of CDX2, ID2, and PRL3D1 were normalized to B-actin. Change in the levels of potency factors CDX2 and ID2 (A) and TGC differentiation marker PRL3D1 (B) over 6 days of 0.5% O2 culture compared with Tzero. C) PRL3D1 expression in 0.5% O2-forced differentiation and normal differentiation with FGF4 removal for 6 days. Asterisk (*) indicates where statistical significance was found compared with 0-day control (P < 0.05).
FIG. 4.
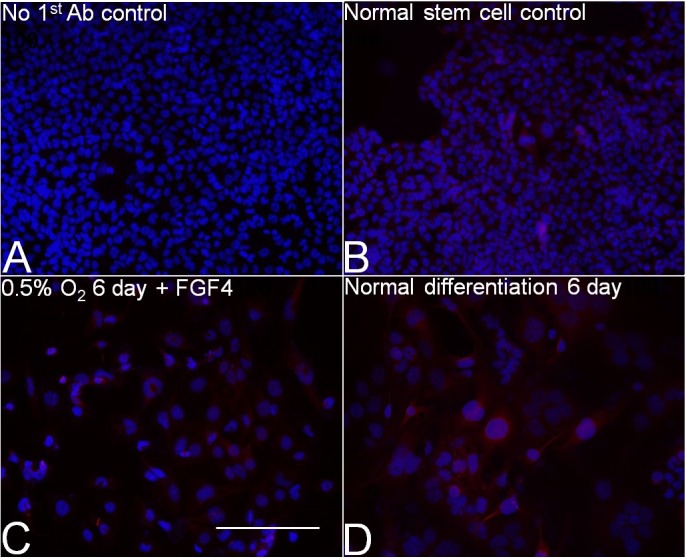
Six days of 0.5% O2 forced giant cell formation and PRL3D1 expression. Blue, Hoechst; red, PRL3D1. A) Normal mTSCs with no first antibody. B) Normal mTSCs with the same staining procedure as 0.5% O2. C) Treatment with 0.5% O2 for 6 days. D) Normal differentiation for 6 days (i.e. 20% O2 with FGF4 removal). Bar = 200 μm.
Next, we analyzed the mRNA expression of marker genes indicating potency and differentiation (Fig. 5). The hypothesis was that 0.5% O2 treatment for 6 days would cause loss of potency factor mRNA and gain of differentiation marker genes despite FGF4, similar to normal differentiation by FGF4 removal. Cells were cultured for 6 days under normal stem cell conditions (20% O2 + FGF4), hypoxic stress (0.5% O2 + FGF4), or normal differentiation conditions (20% O2 − FGF4). The result showed that normal differentiation led to significant 5–14-fold decrease in all four mRNA markers indicating potency (Cdx2, Fgfr2, Id2, and Elf5) compared with normal stem cell control. In contrast, hypoxic stress led to significant loss of Elf5 and Id2 mRNA, but not of Cdx2 and Fgfr2. Marker genes indicating differentiation, which include Hand1, Syna, Prl3d1, Prl2c2, Ctsq, and Tpbpa, were also significantly increased in normal differentiation. There was at least a trend to a significant increase in all of these genes in hypoxic stress-forced differentiation, with close to statistical significance for Tpbpa (P = 0.072). Hand1 and Gcm1 were increased significantly at 6 days of 0.5% O2 treatment and were even higher than under normal differentiation. There was a 600-fold increase in Prl3b1 under normal differentiation, but Prl3b1 did not increase at 0.5% O2 culture. Overall, the decrease in the mRNA expression of potency markers and the increase in the expression of differentiation markers support that 0.5% O2 induced TSC differentiation, despite the presence of FGF4.
FIG. 5.

Six days of 0.5% O2 treatment forced differentiation, and it was different from normal differentiation in marker mRNA expression. The relative expression level of each gene is presented as histogram bars. For gene names, see Table 1. Black bar indicates normal stem cell culture at 6 days, which was normalized to 1. Gray bar and slashed bar indicate the fold change of each individual gene against normal stem cell control at 0.5% O2 treatment and normal differentiation respectively. A) The four potency marker genes. B) The eight differentiation marker genes. Asterisk (*) indicates there was statistical difference with normal stem cell control. # indicates marginal P value compared with stem cell control (P = 0.052 for Hand1, P = 0.072 for Tpbpa). Letter a indicates there was significant difference between 0.5% O2-induced and normal differentiation.
We next tested the hypothesis that stress-induced differentiation has a longer period of reversibility than normal differentiation with FGF4 removal. Figure 6A shows the experimental design. As TGC was the major differentiated lineage at 0.5% O2 or normal in vitro differentiation, we focused on quantifying TGC formation. It takes 40–50 h for mTSCs or rat trophoblast cells to double their ploidy during TGC differentiation [17, 36]. The 3-day fate determination period was chosen to allow one to two cycles of DNA endoreduplication for TGC detection. TGC percentage after potency or differentiation fate determination conditions was compared. The day of irreversible differentiation was defined as the day of initial treatment after which, when cells are moved to fate determination culture, the fraction of TGC is comparable between potency and differentiation conditions. The rationale is that after the irreversible differentiation day, cells have lost their ability to maintain stemness by responding to FGF4, and TGC commitment will not be affected by the further presence or absence of FGF4. We found that with 2 days of FGF4 removal and 3 days of fate determination afterwards, TGC percentage was significantly higher than Day 0 baseline in both potency and differentiation conditions, but the conditions were not significantly different from each other (P = 0.26; Fig. 6B). Thus, 2 days was considered to be the day of irreversible differentiation for normal differentiation. In contrast, mTSCs did not commit irreversible differentiation until 4 days of hypoxic treatment (Fig. 6C). After 2 or 3 days of initial 0.5% O2 treatment, there was a higher TGC percentage after fate determination in differentiation conditions compared with potency conditions, suggesting that there were still stem cell reserves after 2 or 3 days of 0.5% O2 treatment, which responded to FGF4 in potency conditions and did not commit to TGC differentiation.
FIG. 6.

Irreversible differentiation happened at 4 days of 0.5% O2 exposure and at 1–2 days in normal differentiation with FGF4 removal. A) Diagram of experimental design. Time in the x-axis indicates the duration of initial normal differentiation (B) or 0.5% O2 (C) treatment. The Y-axis indicates percentage of cells with DNA > 4 N. N represents ploidy. >4 N means cells accumulated more DNA than tetraploidy, thus representing TGC. For each treatment day, there are three columns. The black column on the left represents percentage of cell with DNA > 4 N after initial treatment; the gray column in the middle represents initial treatment plus 3-day fate determination in differentiation conditions; the lighter gray column on the right represents initial treatment plus 3-day fate determination in potency conditions. The first comparison was made between each treatment group with the 0-day control using asterisk (*) to indicate statistical difference (P < 0.05). After every initial treatment day, a second comparison was made between fate determination in potency and differentiation conditions, with letter a indicating statistical difference.
To further test the day of irreversible differentiation, we next examined whether irreversibility was also reflected in the markers indicating potency (e.g., CDX2, ID2) or TGC differentiation (e.g., PRL3D1). The experimental design was the same as Figure 6, except instead of using flow cytometry to detect cells with DNA > 4 N, CDX2, ID2, and PRL3D1 proteins were measured. If irreversible differentiation has not happened yet, fate determination at potency conditions should promote higher CDX2 and ID2 and lower PRL3D1 compared with differentiation conditions. If irreversible differentiation has occurred after the initial treatment, the level of potency protein and PRL3D1 expression should not differ between potency or differentiation fate determination conditions.
Two initial treatment days, the irreversible day and 1 day before, were chosen for protein marker analysis. They were Days 3 and 4 of 0.5% O2 treatment (Fig. 7, A and C) and Days 1 and 2 of normal differentiation (Fig. 7, B and D). Each initial treatment plus two subsequent fate determination conditions together form a subgroup. After the day of irreversible differentiation (4 days of 0.5% O2 or 2 days of normal differentiation), fate determination in potency or differentiation conditions generated similar levels of CDX2, as shown in the second subgroup of Figure 7, A and B. In contrast, before the irreversible day (i.e., 3 days of 0.5% O2 treatment or 1 day of normal differentiation) potency conditions promoted higher CDX2 protein levels than differentiation conditions during fate determination, as shown in the first subgroup of each figure (Fig. 7, A and B). Another potency factor, ID2, did not show the same pattern of change as CDX2. For both 0.5% O2-induced and normal differentiation, there were no differences in average PRL3D1 expression after fate determination between potency and differentiation conditions on either day studied (Fig. 7, C and D).
FIG. 7.

Changes in the level of potency (CDX2, ID2) and differentiation (PRL3D1) protein markers after 3 or 4 days of 0.5% O2 treatment or 1 or 2 days of normal differentiation. The first histogram bars in A, B, C, and D show baseline protein level at Day 0. In A, the next three histogram bars following Day 0 form a subgroup showing 3 days of initial 0.5% O2 treatment, 3 days of initial 0.5% O2 plus 3 days of culture in differentiation conditions, and 3 days of initial 0.5% O2 plus 3 days in potency conditions, respectively. The last three histogram bars form another subgroup showing results after 4 days of 0.5% O2 treatment. C) The same experimental design as A, but shows PRL3D1 level. B and D follow the same structure as A and C. The only difference was that the initial treatment was FGF4 removal for 1 or 2 days. The first comparison was made between each treatment group and 0-day baseline using an asterisk (*) to indicate statistical difference (P < 0.05). The second comparison was made between fate determination in potency and differentiation conditions within each subgroup, with letter a indicating statistical difference in CDX2 level, a′ indicating statistical difference in ID2 level. # in C indicates marginal significant PRL3D1 increase compared with 0 days (P = 0.058).
DISCUSSION
The effect of hypoxic stress on mTSCs with FGF4 present was studied. We found that 0.5% O2 decreased growth and forced differentiation, but the durations of reversibility in 0.5% O2-induced differentiation and normal differentiation were not the same. Hypoxia decreased the mRNA expression of potency markers and increased the expression of differentiation markers in mTSCs despite the presence of FGF4. We showed for the first time that 0.5% O2-induced differentiation has a longer reversible period, but ultimately irreversible differentiation happens despite the presence of FGF4.
Hypoxia-reduced mTSC growth was reflected in the virtually nil net accumulation of protein. However, TGC differentiation and the associated larger cell size may mean protein amount might not correspond exactly to cell number. Prolonged 0.5% O2 exposure and TGC differentiation caused cells to become fragile to pipetting. Trypsinization for cell counts may lead to disproportionately more cell loss with longer 0.5% O2 exposure. So lysing cell in situ for protein measurement was adopted as a trade-off to avoid this problem. Apoptosis was analyzed at 0.5% O2 treatment to gain a better understanding of the nature of the stress hypoxia imposed on mTSC. Standard culture conditions at Day 0 created only 0.3% apoptosis by immunofluorescence for cleaved caspase-3, which echoes the nearly invisible cleaved caspase-3 protein band at Day 0 in Western blot analysis. In light of this, the ∼35-fold increase of cleaved caspase-3 by immunoblot at Day 3 would still represent a fairly low level of involved cells (i.e., 35-fold × 0.3% = 10.5%), and this is corroborated by the slightly higher estimate by immunofluorescence at Day 3 (14.6%). Similarly, immunofluorescence reports more involved cells than immunoblots at Day 6, ∼5.7% versus 0.6% (baseline × ∼2-fold), respectively. It is not clear what the reasons are for the higher estimates of involved apoptotic cells assayed by immunofluorescence than immunoblot for Days 3 and 6. Either estimate, confirmed by direct observation at the microscope, suggests that 0.5% O2 provides a TSC culture model that is not highly morbid at Day 6, when many final tests of differentiation were performed.
Differentiation was reflected in the formation of TGC, the loss of potency factors, and the gain of differentiation marker PRL3D1. Because spontaneous TGC differentiation and PRL3D1 expression can happen in normal stem cell maintenance, it is possible the observations might be in part due to the artifact of extended culture. However, we think stress-induced differentiation is more likely to be the reason. The cells started as stem cells at Day 0. During passages prior to the start of treatment, stem cells were enriched because giant cells, being more adhesive to the culture dish, tend to get lost during short trypsinization. This was reflected in the low PRL3D1 level at Day 0 baseline, so PRL3D1 expression and TGC formation was due to the effect of hypoxia on the cells. We previously estimated the average nuclear ploidy of cells after 7 days of in vitro culture based on morphology, and found that 20% O2 + FGF4 for 7 days produced average nuclear ploidy of 2.3 N [25]. However, after 7 days of normal differentiation, the average ploidy was 29.1 N, and 0.5% O2 treatment for 7 days (without FGF4 in that case) produced an average ploidy of 12.4 N. Thus, even with prolonged culture, stem cells are the dominant population in normal potency conditions. The finding of ∼90% loss of CDX2 and ∼60% loss of ID2 after 6 days of 0.5% O2 culture is more likely due to differentiation instead of artifact, as for PRL3D1 expression, especially when we know, based on morphology, that ∼50% of cells were giant cells after 6 days of 0.5% O2 treatment. In addition, the levels of PRL3D1 after 6 days of 0.5% O2 treatment or normal differentiation were comparable.
To assess the effects of normal or hypoxic stress-induced differentiation, mRNA marker analysis after 6 days of 0.5% O2 treatment or normal differentiation was compared with normal mTSC maintenance culture after the same period of time. The result further supports the contention that it is the hypoxic stress that induced the differentiation. The loss of Id2 and Elf5 under 0.5% O2 stress was comparable to normal differentiation. Elf5 was identified as a reliable marker for undifferentiated TSC [37]. However, unlike the uniform decrease of all four potency markers in normal differentiation, Cdx2 and Fgfr2 mRNA were preserved during TSC culture at 0.5% O2. There was only 10% CDX2 protein remaining after 6 days of 0.5% O2. Stress maintained high levels of vestigial mRNA for FGF4 signaling. We detected the Fgfr2c isoform of Fgfr2, the mRNA splice form that specifically recognizes FGF4 [38]. Different types of stress, such as heat shock, oxidative stress, ischemia, and viral infection, trigger sudden translational arrest but preserve mRNA in stress granules [39, 40]. Hypothetically, when stress subsides, the multiple processes involved in mRNA biogenesis will not be needed to reestablish cell identity, which may contribute to the reversibility in stress-induced differentiation. There is a possibility that cells at 0.5% O2 are still responsive to FGF4, but the signaling pathway used for maintaining potency after FGF4 binding to FGFR2 is inhibited.
Markers indicating TGC and syncytiotrophoblast differentiation (Prl3d1, Prl3b1, Prl2c2, Ctsq, Syna) were upregulated in both normal and stress-forced differentiation. However, the levels of those markers were much higher in normal differentiation conditions than in stressed cells. This suggests that 0.5% O2 forces differentiation but cannot fully sustain it, as reported previously [25]. Interestingly, Tpbpa, the marker of spongiotrophoblast and glycogen trophoblast cell differentiation [41], was highly upregulated by both 0.5% O2-induced and normal differentiation, and its levels were comparable between the two conditions. Tpbpa-positive spongiotrophoblasts can act as precursors for secondary TGC and glycogen trophoblast differentiation [42]. Tpbpa upregulation under hypoxic conditions has also been reported elsewhere [43]. Hypoxia-inducible factor functions to enhance spongiotrophoblast differentiation and simultaneously inhibits secondary TGC formation from spongiotrophoblast progenitors [44], which may explain the approximately 200-fold increase in Tpbpa seen under 0.5% O2. HAND1 mediates the differentiation of all TGC subtypes [26], and its mRNA was significantly upregulated by hypoxia-forced differentiation, higher than normal differentiation. GCM1 mediates the differentiation of syncytiotrophoblasts, and its mRNA was significantly increased by 0.5% O2 treatment. For normal differentiation, Gcm1 was not high at 6 days of FGF4 removal, which agrees with previous reports that show Gcm1 has only a transient increase at around 2 days of FGF4 removal, and by 6 or 7 days its level goes down again [25, 45]. Accompanying that is the significant increase in mature syncytiotrophoblast marker Syna, which suggests that by 6 days of normal differentiation, cells have passed the intermediate stage and committed to terminal differentiation. The relatively high levels of Hand1, Gcm1, and Tpbpa, together with the low terminal differentiation markers Prl3d1, Prl3b1, Prl2c2, Ctsq, and Syna at 0.5% O2, suggest that hypoxia drives cells toward differentiation, but more cells are in the intermediate stage of differentiation than under normal differentiation at Day 6.
Spontaneous differentiation is a common phenomenon in normal TSC maintenance culture. Compared with stem cell control, the relative fold change of each differentiation marker in stress-induced or normal differentiation would be influenced by its level in the stem cell maintenance control. For example, the 10-fold increase in Prl3d1 compared with the approximately 300-fold increase in Tpbpa and over-600-fold increase in Prl3b1 under normal differentiation conditions may be the combined result of a higher induction of Prl3b1 and Tpbpa at 6 days of normal differentiation and a lower stem cell control baseline. Hypothetically, if there were 5% of cells expressing Prl3d1 in normal stem cell control at Day 6, it would not be possible for stress-induced or normal differentiation to produce a more-than-20-fold change above the 5% background. And in the stem cell maintenance condition, we did consistently observe lower Ct values for Prl3d1 than for Prl3b1 and Tpbpa during quantitative PCR, which suggests that it is possible there was higher expression of Prl3d1 mRNA than of Prl3b1 and Tpbpa in stem cell maintenance conditions.
The reversibility of mTSC differentiation was also studied on a molecular level. Fate determination in potency conditions did not promote higher levels of CDX2 compared with differentiation conditions after the irreversible differentiation day, in contrast to the higher CDX2 level in potency fate determination after the reversible day. CDX2 is essential for trophoblast lineage establishment in mouse blastocyst and in vitro mTSCs maintenance, because mTSCs cannot be isolated from Cdx2 mutant mouse embryos [46]. Cdx2 knockdown leads to upregulation of Hand1 [47], which positively regulates Prl3d1 promoter and is necessary [45] and sufficient [48] for first-lineage TGC differentiation. Thus, irreversible CDX2 loss suggests loss of stemness and corroborates the irreversible differentiation day characterized by flow cytometry. Both molecular and morphological markers define the reversibility of stress-induced or normal differentiation.
The level of PRL3D1's being similar at the end of the fate determination between potency and differentiation conditions after both Days 3 and 4 of 0.5% O2 treatment and Days 1 and 2 of normal differentiation is intriguing because before the irreversible day, there was a higher TGC percentage after fate determination in differentiation conditions. We suspect that FGF4 supported a higher level of PRL3D1 expression per cell in a smaller fraction of PRL3D1-secreting cells under potency fate determination conditions. FGF4 is required to maintain the stemness of mTSCs [17]. However, after differentiation, FGF2 has been shown to increase PRL3D1 expression through ERK and the p38MAPK signaling pathway [49] in Rcho-1 cells, a rat trophoblast model that can be maintained in proliferative state or induced to differentiate and express PRL3D1 [50]. Thus, FGF signaling may both maintain potency before differentiation and increase differentiation marker PRL3D1 expression after differentiation.
It should be noted that the day of irreversible differentiation does not mean that absolutely all the cells are committed to differentiation with no stem cells left on that day. However, the stem cell reserve after the irreversible differentiation day is not substantial, and after a 3-day fate determination period, the minimal stem cell reserve was not able to change the overall trend toward TGC differentiation and TGC percentage. Moreover, there was a ∼5-fold rise and plateau of PRL3D1 in mTSC lysates due to stress-induced differentiation. Zero cell growth and irreversible differentiation together with plateaued PRL3D1 are unlikely to sustain pregnancy if in vivo stress responses happen in a similar way. In vitro differentiation of TGC and increased PRL3D1 expression go through an ordered sequence similar to in vivo circumstances [51], suggesting that this reductionist approach to study how stresses affect in vitro mTSCs may resemble the effects of stresses on placental stem cells in vivo.
The preparation of cell suspension for flow cytometry analysis has an inherent tendency to underestimate the proportion of giant cells, especially giant cells with DNA content >64 N, because of nuclear fragmentation and increased adherence to culture surface that causes trypsin resistance [36]. We found that under stress conditions, cells were more vulnerable to handling and there was more nuclear fragmentation. This is why there was a discrepancy in TGC percentage as estimated by measuring nuclear size or by flow cytometry. Nevertheless, flow cytometry is fast, and the same sample preparation procedure and machine settings were used for all samples. This defined the day of irreversibility, and the results were further supported by the potency protein assay for CDX2, which showed irreversible loss after 4 days of hypoxic stress or 2 days of FGF4 removal.
Using nuclear size measurement to define TGC percentage circumvents the potential problem of fragmentation and loss of larger trypsin-resistant cells. However, it may also underestimate the level of differentiation, because there could be small differentiated cells expressing PRL3D1, which have been shown to exist under hyperosmotic stress [20]. In addition, there may also be nonstem cells with small nuclei, such as the 2-N nuclei of multinuclear syncytiotrophoblasts. The mRNA marker analysis provides insights into the lineages formed under hypoxia-forced differentiation. However, we are not able to infer the population size of each lineage, because the mRNA copy number per cell for each marker is unknown. The reversibility of differentiation was studied based on TGC differentiation, because it is the major differentiated lineage, and non-TGC-differentiated lineages were not accounted here.
Another limitation is the interpretation of in vitro-derived data. For the in vivo situation, hypoxia can be buffered to a certain degree by the endometrium and other distant maternal organ systems, which are able to integrate and mount adaptive responses to local hypoxia [52, 53]. Using this reductionist approach, useful insight has been gained concerning the responses of isolated mTSCs to hypoxia. The embryonic response to the same level of hypoxia may not be exactly the same between in vivo and in vitro. Findings here suggest the hypothesis that hypoxia or other stresses may slow growth and force irreversible differentiation in vivo, which can lead to miscarriage even without a high level of cell death. In vitro findings and the resulting hypotheses will need to be tested in vivo in the future.
ACKNOWLEDGMENT
Thanks to Dr. Soares for the kind gift of PRL3D1 antibody. We acknowledge Frank Urban and Drs. Husam Abu-Soud, Awoniyi Awonuga, and Alan Bolnick for analysis and comments on the manuscript.
Footnotes
This research was supported by grants to D.A.R. from NIH (1R03HD061431 02), support for G.C.P. from NIH (P30 ES020957), and, from the Office of the Vice President for Research at Wayne State University, the Kam Moghissi Endowed Chair (E.E.P.).
REFERENCES
- McLaren A, Snow ML. Embryogenesis in Mammals. Amsterdam: Elsevier; 1976. [Google Scholar]
- Niwa H, Toyooka Y, Shimosato D, Strumpf D, Takahashi K, Yagi R, Rossant J. Interaction between Oct3/4 and Cdx2 determines trophectoderm differentiation. Cell. 2005;123:917–929. doi: 10.1016/j.cell.2005.08.040. [DOI] [PubMed] [Google Scholar]
- Cross JC. Genes regulating embryonic and fetal survival. Theriogenology. 2001;55:193–207. doi: 10.1016/s0093-691x(00)00454-4. [DOI] [PubMed] [Google Scholar]
- Wilcox AJ, Weinberg CR, O'Connor JF, Baird DD, Schlatterer JP, Canfield RE, Armstrong EG, Nisula BC. Incidence of early loss of pregnancy. N Engl J Med. 1988;319:189–194. doi: 10.1056/NEJM198807283190401. [DOI] [PubMed] [Google Scholar]
- Parraguez VH, Mamani S, Cofre E, Castellaro G, Urquieta B, De Los Reyes M, Astiz S, Gonzalez-Bulnes A. Disturbances in maternal steroidogenesis and appearance of intrauterine growth retardation at high-altitude environments are established from early pregnancy. Effects of treatment with antioxidant vitamins. PLoS One. 2015;10:e0140902. doi: 10.1371/journal.pone.0140902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cera E, Doyle ML, Morgan MS, De Cristofaro R, Landolfi R, Bizzi B, Castagnola M, Gill SJ. Carbon monoxide and oxygen binding to human hemoglobin F0. Biochemistry. 1989;28:2631–2638. doi: 10.1021/bi00432a041. [DOI] [PubMed] [Google Scholar]
- Ford MD. Clinical Toxicology. Philadelphia: Saunders; 2001. [Google Scholar]
- Ovari L, Aranyosi J, Balla G. Acute effect of cigarette smoking on placental circulation—a study by carbon-monoxide measurement and Doppler assessment. Acta Physiol Hung. 2009;96:243–250. doi: 10.1556/APhysiol.96.2009.2.9. [DOI] [PubMed] [Google Scholar]
- Jensen GM, Moore LG. The effect of high altitude and other risk factors on birthweight: independent or interactive effects? Am J Public Health. 1997;87:1003–1007. doi: 10.2105/ajph.87.6.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. Prenatal hypoxia and cardiac programming. J Soc Gynecol Investig. 2005;12:2–13. doi: 10.1016/j.jsgi.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Barker DJ. The fetal origins of coronary heart disease. Eur Heart J. 1997;18:883–884. doi: 10.1093/oxfordjournals.eurheartj.a015368. [DOI] [PubMed] [Google Scholar]
- Kwong WY, Wild AE, Roberts P, Willis AC, Fleming TP. Maternal undernutrition during the preimplantation period of rat development causes blastocyst abnormalities and programming of postnatal hypertension. Development. 2000;127:4195–4202. doi: 10.1242/dev.127.19.4195. [DOI] [PubMed] [Google Scholar]
- Burkus J, Kacmarova M, Kubandova J, Kokosova N, Fabianova K, Fabian D, Koppel J, Cikos S. Stress exposure during the preimplantation period affects blastocyst lineages and offspring development. J Reprod Dev. 2015;61:325–331. doi: 10.1262/jrd.2015-012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose P, Kadyrov M, Goldin R, Hahn S, Backos M, Regan L, Huppertz B. Aberrations of early trophoblast differentiation predispose to pregnancy failure: lessons from the anti-phospholipid syndrome. Placenta. 2006;27:869–875. doi: 10.1016/j.placenta.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Huppertz B. Placental origins of preeclampsia—challenging the current hypothesis. Hypertension. 2008;51:970–975. doi: 10.1161/HYPERTENSIONAHA.107.107607. [DOI] [PubMed] [Google Scholar]
- Macklon NS, Geraedts JP, Fauser BC. Conception to ongoing pregnancy: the “black box” of early pregnancy loss. Hum Reprod Update. 2002;8:333–343. doi: 10.1093/humupd/8.4.333. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Kunath T, Hadjantonakis AK, Nagy A, Rossant J. Promotion of trophoblast stem cell proliferation by FGF4. Science. 1998;282:2072–2075. doi: 10.1126/science.282.5396.2072. [DOI] [PubMed] [Google Scholar]
- Corson LB, Yamanaka Y, Lai KM, Rossant J. Spatial and temporal patterns of ERK signaling during mouse embryogenesis. Development. 2003;130:4527–4537. doi: 10.1242/dev.00669. [DOI] [PubMed] [Google Scholar]
- Zhou S, Xie Y, Puscheck EE, Rappolee DA. Oxygen levels that optimize TSC culture are identified by maximizing growth rates and minimizing stress. Placenta. 2011;32:475–481. doi: 10.1016/j.placenta.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awonuga AO, Zhong W, Abdallah ME, Slater JA, Zhou SC, Xie YF, Puscheck EE, Rappolee DA. Eomesodermin, HAND1, and CSH1 proteins are induced by cellular stress in a stress-activated protein kinase-dependent manner. Mol Reprod Dev. 2011;78:519–528. doi: 10.1002/mrd.21342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Xie Y, Abdallah M, Awonuga AO, Slater JA, Sipahi L, Puscheck EE, Rappolee DA. Cellular stress causes reversible, PRKAA1/2-, and proteasome-dependent ID2 protein loss in trophoblast stem cells. Reproduction. 2010;140:921–930. doi: 10.1530/REP-10-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie YF, Abdallah M, Awonuga N, Slater J, Puscheck E, Rappolee D. Benzo(a)pyrene activates stress enzymes and regulates transcription factors that favor differentiation of trophoblast stem cells. Biol Reprod. 2009:106–106. [Google Scholar]
- Liu J, Xu W, Sun T, Wang F, Puscheck E, Brigstock D, Wang QT, Davis R, Rappolee DA. Hyperosmolar stress induces global mRNA responses in placental trophoblast stem cells that emulate early post-implantation differentiation. Placenta. 2009;30:66–73. doi: 10.1016/j.placenta.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross JC, Anson-Cartwright L, Scott IC. Transcription factors underlying the development and endocrine functions of the placenta. Recent Prog Horm Res. 2002;57:221–234. doi: 10.1210/rp.57.1.221. [DOI] [PubMed] [Google Scholar]
- Xie YF, Zhou SC, Jiang ZL, Dai J, Puscheck EE, Lee I, Parker G, Huttemann M, Rappole DA. Hypoxic stress induces, but cannot sustain trophoblast stem cell differentiation to labyrinthine placenta due to mitochondrial insufficiency. Stem Cell Res. 2014;13:478–491. doi: 10.1016/j.scr.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DG, Fortier AL, Cross JC. Diverse subtypes and developmental origins of trophoblast giant cells in the mouse placenta. Dev Biol. 2007;304:567–578. doi: 10.1016/j.ydbio.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Hemberger M, Nozaki T, Masutani M, Cross JC. Differential expression of angiogenic and vasodilatory factors by invasive trophoblast giant cells depending on depth of invasion. Dev Dyn. 2003;227:185–191. doi: 10.1002/dvdy.10291. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Ogren L, Endo H, Thordarson G, Bigsby RM, Talamantes F. Production of mouse placental lactogen-I and placental lactogen-II by the same giant cell. Endocrinology. 1992;131:1595–1602. doi: 10.1210/endo.131.4.1396305. [DOI] [PubMed] [Google Scholar]
- Slater JA, Zhou S, Puscheck EE, Rappolee DA. Stress-induced enzyme activation primes murine embryonic stem cells to differentiate toward the first extraembryonic lineage. Stem Cells Dev. 2014;23:3049–3064. doi: 10.1089/scd.2014.0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janatpour MJ, McMaster MT, Genbacev O, Zhou Y, Dong J, Cross JC, Israel MA, Fisher SJ. Id-2 regulates critical aspects of human cytotrophoblast differentiation, invasion and migration. Development. 2000;127:549–558. doi: 10.1242/dev.127.3.549. [DOI] [PubMed] [Google Scholar]
- Cross JC, Flannery ML, Blanar MA, Steingrimsson E, Jenkins NA, Copeland NG, Rutter WJ, Werb Z. Hxt encodes a basic helix-loop-helix transcription factor that regulates trophoblast cell development. Development. 1995;121:2513–2523. doi: 10.1242/dev.121.8.2513. [DOI] [PubMed] [Google Scholar]
- Kedersha NL, Gupta M, Li W, Miller I, Anderson P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol. 1999;147:1431–1442. doi: 10.1083/jcb.147.7.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlin GP, Lu XJ, Roby KF, Soares MJ. Recapitulation of the pathway for trophoblast giant cell differentiation in vitro: stage-specific expression of members of the prolactin gene family. Endocrinology. 1994;134:2390–2396. doi: 10.1210/endo.134.6.8194465. [DOI] [PubMed] [Google Scholar]
- Quinn J, Kunath T, Rossant J. Mouse trophoblast stem cells. Methods Mol Med. 2006;121:125–148. doi: 10.1385/1-59259-983-4:123. [DOI] [PubMed] [Google Scholar]
- Ullah Z, Kohn MJ, Yagi R, Vassilev LT, DePamphilis ML. Differentiation of trophoblast stem cells into giant cells is triggered by p57/Kip2 inhibition of CDK1 activity. Genes Dev. 2008;22:3024–3036. doi: 10.1101/gad.1718108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAuley A, Cross JC, Werb Z. Reprogramming the cell cycle for endoreduplication in rodent trophoblast cells. Mol Biol Cell. 1998;9:795–807. doi: 10.1091/mbc.9.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motomura K, Oikawa M, Hirose M, Honda A, Togayachi S, Miyoshi H, Ohinata Y, Sugimoto M, Abe K, Inoue K, Ogura A. Cellular dynamics of mouse trophoblast stem cells: identification of a persistent stem cell type. Biol Reprod. 2016;94:122. doi: 10.1095/biolreprod.115.137125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271:15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci. 2008;33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- McEwen E, Kedersha N, Song B, Scheuner D, Gilks N, Han A, Chen JJ, Anderson P, Kaufman RJ. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J Biol Chem. 2005;280:16925–16933. doi: 10.1074/jbc.M412882200. [DOI] [PubMed] [Google Scholar]
- Simmons DG, Rawn S, Davies A, Hughes M, Cross JC. Spatial and temporal expression of the 23 murine prolactin/placental lactogen-related genes is not associated with their position in the locus. BMC Genomics. 2008;9:352. doi: 10.1186/1471-2164-9-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Cross JC. Ablation of Tpbpa-positive trophoblast precursors leads to defects in maternal spiral artery remodeling in the mouse placenta. Dev Biol. 2011;358:231–239. doi: 10.1016/j.ydbio.2011.07.036. [DOI] [PubMed] [Google Scholar]
- Gultice AD, Selesniemi KL, Brown TL. Hypoxia inhibits differentiation of lineage-specific Rcho-1 trophoblast giant cells. Biol Reprod. 2006;74:1041–1050. doi: 10.1095/biolreprod.105.047845. [DOI] [PubMed] [Google Scholar]
- Cowden Dahl KD, Fryer BH, Mack FA, Compernolle V, Maltepe E, Adelman DM, Carmeliet P, Simon MC. Hypoxia-inducible factors 1alpha and 2alpha regulate trophoblast differentiation. Mol Cell Biol. 2005;25:10479–10491. doi: 10.1128/MCB.25.23.10479-10491.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes M, Dobric N, Scott IC, Su L, Starovic M, St-Pierre B, Egan SE, Kingdom JC, Cross JC. The Hand1, Stra13 and Gcm1 transcription factors override FGF signaling to promote terminal differentiation of trophoblast stem cells. Dev Biol. 2004;271:26–37. doi: 10.1016/j.ydbio.2004.03.029. [DOI] [PubMed] [Google Scholar]
- Strumpf D, Mao CA, Yamanaka Y, Ralston A, Chawengsaksophak K, Beck F, Rossant J. Cdx2 is required for correct cell fate specification and differentiation of trophectoderm in the mouse blastocyst. Development. 2005;132:2093–2102. doi: 10.1242/dev.01801. [DOI] [PubMed] [Google Scholar]
- Wu G, Gentile L, Fuchikami T, Sutter J, Psathaki K, Esteves TC, Arauzo-Bravo MJ, Ortmeier C, Verberk G, Abe K, Scholer HR. Initiation of trophectoderm lineage specification in mouse embryos is independent of Cdx2. Development. 2010;137:4159–4169. doi: 10.1242/dev.056630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley P, Anson-Cartwright L, Cross JC. The Hand1 bHLH transcription factor is essential for placentation and cardiac morphogenesis. Nat Genet. 1998;18:271–275. doi: 10.1038/ng0398-271. [DOI] [PubMed] [Google Scholar]
- Peters TJ, Chapman BM, Wolfe MW, Soares MJ. Placental lactogen-I gene activation in differentiating trophoblast cells: extrinsic and intrinsic regulation involving mitogen-activated protein kinase signaling pathways. J Endocrinol. 2000;165:443–456. doi: 10.1677/joe.0.1650443. [DOI] [PubMed] [Google Scholar]
- Faria TN, Soares MJ. Trophoblast cell differentiation: establishment, characterization, and modulation of a rat trophoblast cell line expressing members of the placental prolactin family. Endocrinology. 1991;129:2895–2906. doi: 10.1210/endo-129-6-2895. [DOI] [PubMed] [Google Scholar]
- Carney EW, Prideaux V, Lye SJ, Rossant J. Progressive expression of trophoblast-specific genes during formation of mouse trophoblast giant cells in vitro. Mol Reprod Dev. 1993;34:357–368. doi: 10.1002/mrd.1080340403. [DOI] [PubMed] [Google Scholar]
- Chakraborty D, Rumi MA, Soares MJ. NK cells, hypoxia and trophoblast cell differentiation. Cell Cycle. 2012;11:2427–2430. doi: 10.4161/cc.20542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho-Chen JK, Ain R, Alt AR, Wood JG, Gonzalez NC, Soares MJ. Hypobaric hypoxia as a tool to study pregnancy-dependent responses at the maternal-fetal interface. Methods Mol Med. 2006;122:427–434. doi: 10.1385/1-59259-989-3:427. [DOI] [PubMed] [Google Scholar]