

Abstract
Cardiac mitochondrial function is altered in a variety of inherited and acquired cardiovascular diseases. Recent studies have identified the transcriptional coactivator peroxisome proliferator–activated receptor γ coactivator-1 (PGC-1) as a regulator of mitochondrial function in tissues specialized for thermogenesis, such as brown adipose. We sought to determine whether PGC-1 controlled mitochondrial biogenesis and energy-producing capacity in the heart, a tissue specialized for high-capacity ATP production. We found that PGC-1 gene expression is induced in the mouse heart after birth and in response to short-term fasting, conditions known to increase cardiac mitochondrial energy production. Forced expression of PGC-1 in cardiac myocytes in culture induced the expression of nuclear and mitochondrial genes involved in multiple mitochondrial energy-transduction/energy-production pathways, increased cellular mitochondrial number, and stimulated coupled respiration. Cardiac-specific overexpression of PGC-1 in transgenic mice resulted in uncontrolled mitochondrial proliferation in cardiac myocytes leading to loss of sarcomeric structure and a dilated cardiomyopathy. These results identify PGC-1 as a critical regulatory molecule in the control of cardiac mitochondrial number and function in response to energy demands.
Introduction
The energy demands of the postnatal mammalian heart are met primarily by ATP produced in mitochondria through oxidative phosphorylation. Consistent with the high energy requirements of the heart, the differentiated cardiac myocyte contains numerous mitochondria and an impressive capacity for energy production. The chief mitochondrial energy substrate in the heart is fatty acids, which provide the greatest yield of ATP per mole compared with other substrates such as glucose or lactate. Fatty acids are catabolized in mitochondria via the fatty acid β-oxidation (FAO) pathway, generating reducing equivalents for the electron transport chain and acetyl-CoA, a substrate for further oxidation in the tricarboxylic acid cycle (TCA). The importance of high-capacity mitochondrial energy production for the maintenance of cardiac function is underscored by the severe phenotype of inherited and acquired mitochondrial diseases. Inborn errors in nuclear-encoded mitochondrial FAO-pathway enzymes are an important cause of childhood cardiomyopathy and sudden death (1). Mutations in mitochondrial DNA, which lead to abnormalities in oxidative phosphorylation, cause a variety of diseases, including cardiomyopathy, neuromuscular dysfunction, and diabetes mellitus (2). Abnormalities in mitochondrial function also occur with aging and in a variety of acquired pathophysiologic conditions such as ischemic heart disease and in the hypertrophied and failing heart (2–6).
Mitochondrial number and functional capacity are dynamically regulated in accordance with cardiac energy demands during developmental stages and in response to diverse physiologic conditions (7). For example, during the early postnatal period, as myocardial energy demands increase due to changes in ventricular loading conditions and cardiac output, the number of mitochondria and expression of mitochondrial proteins increase dramatically (7, 8). Dietary influences, such as short-term starvation, increase the capacity for mitochondrial energy production via the FAO pathway. Accordingly, mechanisms exist to transduce changes in cardiac energy requirements dictated by physiologic conditions to the coordinate control of nuclear and mitochondrial genes encoding mitochondrial proteins.
A series of elegant studies by Scarpulla and coworkers (9–11) have provided important insight into the regulatory mechanisms involved in the transcriptional control of nuclear and mitochondrial genes encoding several key mitochondrial proteins during mitochondrial biogenesis. These studies identified the nuclear respiratory factors-1 and -2 (NRF-1 and -2) as key transcriptional activators of nuclear genes encoding mitochondrial respiratory chain proteins. NRF-responsive regulatory elements were also identified in the promoter region of the gene encoding mitochondrial transcription factor A (mtTFA), a nuclear-encoded protein that stimulates mitochondrial DNA transcription (10). Collectively, these results delineated a regulatory circuit involved in the coordinate transcriptional control of nuclear and mitochondrial genes encoding mitochondrial proteins involved in electron transport and oxidative phosphorylation.
Despite significant progress in the elucidation of the molecular events involved in the coordinate control of nuclear and mitochondrial genes during mitochondrial biogenesis, several important questions regarding the regulation of mitochondrial functional capacity remain. First, how are physiologic signals transduced to activation of mitochondrial biogenesis in cell types such as the cardiac myocyte known to regulate dynamically mitochondrial number and energy-producing capacity? Second, what is the link between factors involved in the broad program of mitochondrial biogenesis and the control of specific energy substrate utilization pathways such as FAO? This latter question is particularly relevant given that we and others have not found a role for NRF-1 or NRF-2 in the transcriptional control of mitochondrial FAO pathway enzyme genes. Rather, the peroxisome proliferator–activated receptor α (PPARα) and other nuclear receptors have been identified as key transcriptional regulators of the FAO pathway (12, 13). The recent identification and characterization of the transcriptional coactivator peroxisome proliferator–activated receptor γ coactivator-1 (PGC-1) has provided important clues regarding these questions. PGC-1, a cold-inducible protein, was cloned based on its ability to interact with the adipose-enriched nuclear receptor, PPARγ (14). Forced expression of PGC-1 was shown to induce mitochondrial biogenesis in adipocyte and myogenic cell lines and to coactivate NRF-1 (15). Consistent with its cold inducibility and brown adipose tissue–enriched expression pattern, PGC-1 was shown to induce predominantly uncoupled mitochondrial respiration in adipogenic cell lines by the activation of uncoupling protein (UCP) expression. These results identified PGC-1 as a key component of the regulatory pathway involved in the control of uncoupled mitochondrial respiration and thermogenesis. Given that PGC-1 is also highly expressed in the heart, a tissue specialized for high-capacity production of ATP, we sought to determine whether this coactivator plays a role in the control of mitochondrial number and function in cardiac tissue. In support of this notion, we have shown recently that PGC-1 is capable of coactivating PPARα (16), a cardiac-enriched member of the PPAR family known to control mitochondrial FAO enzyme expression.
In this report we present data that identify PGC-1 as a regulator of mitochondrial function and number in the heart. PGC-1 is shown to induce mitochondrial FAO cycle enzyme expression and mitochondrial biogenesis in cardiac myocytes in culture and in the heart of transgenic mice. In contrast to its action in noncardiac cell types reported previously, PGC-1 induces coupled mitochondrial respiration in cardiac myocytes consistent with the biogenesis of mitochondria capable of ATP production.
Methods
Animal experiments.
Three- to five-month-old 129/SvJ mice were used for the fasting experiments. Comparisons were made with sex-matched littermates fed ad libitum. All mice were kept in separate cages with identical light/dark cycles and free access to water. The 24-hour fasting period began at 1700 hours.
For evaluation of gene expression in the developing mouse heart, total RNA was isolated from male and female C57B6 × SJL/J F1 mice. For embryonic days 16.5 and 18.5 and postnatal day 1, five to six hearts were pooled for each time point. For postnatal days 3 and 7, at least two hearts were pooled for each RNA sample.
All animal experiments conducted were approved by the Animal Studies Committee of Washington University School of Medicine.
Adenoviral expression system.
Ventricular myocytes were cultured from 1-day-old Sprague-Dawley rats as described (13). Recombinant adenoviruses were generated employing a system described previously (17). The parent plasmids (pAdEasy-1, pAdTrack, and pAdTrack-CMV) were obtained as a gift from T.-C. He (The Howard Hughes Medical Institute) and B. Vogelstein (The Johns Hopkins Oncology Center). Recombination between the pAdEasy-1 and pAdTrack vectors (17) resulted in adenovirus green fluorescent protein (Ad-GFP). For the construction of Ad-PGC-1, a HindIII fragment of PGC-1 cDNA (a gift from B. Spiegelman, Dana-Farber Cancer Institute and Harvard Medical School, Boston, Massachusetts, USA) encoding amino acids 1–793 of murine PGC-1 was inserted into pcDNA3.1/Myc-His (Invitrogen, Carlsbad, California, USA) and then cloned into pAdTrack-CMV before recombination with pAdEasy-1. The Ad-PGC-1 vector contains, in tandem, the GFP gene and the PGC-1 cDNA containing a carboxy-terminal myc epitope tag downstream of separate cytomegalovirus (CMV) promoters. Viruses were propagated in the 293 cell line with subsequent titering in cultured rat neonatal cardiac myocytes to obtain 95–100% infection efficiency, as determined by GFP expression 18–24 hours after infection. For all experiments using the adenoviral system, cardiac myocytes were infected with Ad-PGC-1 or Ad-GFP (vector backbone containing the GFP cDNA but lacking PGC-1 cDNA) at the time of transition to serum-free medium, approximately 24 hours after initial plating. Cells were harvested for isolation of RNA 48 hours after infection and for preparation of protein extracts 96 hours after infection.
RNA and protein-blotting studies.
The protocols for RNA isolation and Northern blotting have been described (18). Probes labeled with 32P were derived from the following cDNAs: human UCP II and rat UCP III (gifts from P. Muzzin, University of Geneva, Switzerland) (19), citrate synthase (20, 21), ATP synthase subunits c and β (American Type Culture Collection, Rockville, Maryland, USA), the coding region of human skeletal α-actin (22). The mouse PPARα, GAPDH, acyl-CoA oxidase, and muscle-type carnitine palmitoyltransferase I (M-CPT I) probes have been described (23).
Immunoblot analysis of whole-cell protein extracts was performed as described (20). Anti-murine PGC-1 Ab was prepared by immunizing rabbits with a GST-PGC-1 fusion protein containing the NH2-terminal 120 amino acids of murine PGC-1. Ab specificity for PGC-1 was confirmed by peptide competition assays (data not shown). Cytochrome c oxidase (COX) subunits were separated by SDS-PAGE (15% acrylamide wt/vol) and detected using a polyclonal COX Ab (a gift from L. Prochaska, Wright State University) (24). Cytochrome c was detected using a monoclonal mouse anti-cytochrome c Ab (PharMingen, San Diego, California, USA).
Mitochondrial quantification.
Cardiac myocyte pellets were prepared for electron microscopy 5 days after adenoviral infection, as described (25). In brief, pellets were fixed in 2% glutaraldehyde/osmium tetroxide, embedded with Spurr resin, and sectioned. Thin sections were obtained and viewed with a Philips transmission electron microscope. Cardiac myocyte cytoplasmic volume densities were determined in a blinded fashion using the principles of Weibel (26) and Steer (27). A 20 × 25-cm transparent grid was used that allowed approximately 75 points of reference per electron micrograph. Data were expressed as volume density (volume of mitochondria [μm3] per cytoplasmic volume [μm3]). A minimum of 12 cells from each treatment were assessed.
Mitochondrial-staining studies were performed 5 days after adenoviral infection. Cardiac myocytes were incubated for 45 minutes at 37°C in 3 μM MitoTracker (M-7513; Molecular Probes, Eugene, Oregon, USA) diluted in DMEM supplemented with 10% calf serum. The cells were then placed in fresh medium, washed with PBS, fixed with 4% paraformaldehyde in PBS with 0.1% Triton X-100, and mounted with anti-fade (SlowFade) solution (Molecular Probes) for subsequent fluorescence microscopy.
Production and characterization of transgenic mice.
The myc-PGC-1 cDNA was cloned downstream of the cardiac α myosin heavy-chain (MHC) promoter (clone 26; a gift from J. Robbins, The Children’s Hospital Research Foundation, Cincinnati, Ohio, USA) (28). Transgenic mice were produced by microinjection of the MHC-PGC-1 construct into fertilized one-cell C57B6 × CBA/J F1 or C57B6 × C3H/J F1 embryos. Transgenic founders were identified by Southern blot analysis as described (29) using a PGC-1 cDNA probe. Pairs of transgenic and wild-type mouse hearts from five independent lines (ages 1–5 weeks) were characterized. Echocardiographic studies were performed as described (30).
Mitochondrial function studies.
Mitochondrial respiratory function was measured in cultured cardiac myocytes 5 days after adenoviral infection in the absence or presence of saponin, which perforates the sarcolemma while leaving the mitochondria morphologically and functionally intact (31). Oxygen consumption was measured at 37°C in a closed, continuously stirred system with a Clark-type polarographic oxygen probe (Model 5300; Yellow Springs Instrument Co., Yellow Springs, Ohio, USA). In some samples, after initial oxygen consumption measurements, samples were treated with 2.5 μg/ml oligomycin. Saponin permeabilization was not used for the oligomycin experiments. For experiments involving saponin, oxygen-consumption rates were assessed at 37°C in a solution containing 20 μg/ml saponin, 100 μM ADP, 5 mM glutamic acid, 2 mM malic acid, 4 mM MgCl2, 100 mM 2-[N-Morpholino]ethanesulfonic acid, 20 mM taurine, 0.5 mM DTT, 3 mM KH2PO4, 20 mM imidazole, and 10 mM EGTA-Ca EGTA, pH 7.1.
Statistical analysis.
Statistical comparisons were made using Student’s t test, with a statistically significant difference defined as a P value less than 0.05. Error bars represent the SEM.
Results
PGC-1 gene expression parallels known changes in mitochondrial energy-producing capacity during perinatal development and in response to fasting.
As an initial step to determine whether the inducible transcriptional coactivator PGC-1 is involved in the physiologic control of mitochondrial function in the heart, the expression of its gene was delineated in a context in which cardiac fatty acid-utilization rates, cellular mitochondrial number, and respiratory functional capacity is increased: the perinatal developmental transition (32). Mouse heart PGC-1 mRNA levels were coordinately induced on the first day after birth and during later postnatal stages in parallel with the expression of its interacting partner PPARα; two PPARα target genes encoding mitochondrial FAO enzymes, M-CPT I and medium-chain acyl-CoA dehydrogenase (MCAD); and the mitochondrial ATP synthase β gene (Figure 1). As has been described (14), two PGC-1 transcripts of approximately 5 and 8 kb were observed. The nature of the two PGC-1 transcripts is unknown, although mRNAs of similar sizes are expressed by the human PGC-1 gene as a result of differential polyadenylation (33). The expression pattern of the gene encoding the glycolytic enzyme, GAPDH, was distinct from the other genes studied, exhibiting greatest expression during the prenatal period.
Figure 1.

Induction of PGC-1 gene expression during perinatal cardiac development. The expression of the genes encoding PGC-1, PPARα, and mitochondrial energy production–pathway enzymes during cardiac postnatal development. Representative autoradiograph of Northern blot analyses performed with total RNA (15 μg/lane) isolated from mouse hearts at several late embryonic (e), postnatal, and adult (A, 2 months old) time points using the cDNA probes denoted on the left. The ethidium bromide–stained 28S ribosomal subunit (28S) was used as a control for loading.
PGC-1 gene expression was also evaluated in response to short-term fasting, a physiologic condition known to rapidly increase cardiac mitochondrial fatty acid utilization rates (34) and FAO enzyme gene expression by PPARα-mediated transcriptional activation (23). PGC-1 mRNA levels were induced within 24 hours of the onset of fasting (Figure 2). As expected, PPARα target genes encoding mitochondrial (MCAD, M-CPT I) and peroxisomal (acyl-CoA oxidase [ACO]) FAO enzymes were also induced in the mouse heart during the fast (Figure 2). In contrast to the results observed in the heart, fasting did not induce PGC-1 gene expression in brown adipose, a mitochondrial-enriched tissue specialized for thermogenesis through uncoupled respiration (data not shown).
Figure 2.

Induction of cardiac PGC-1 gene expression during short-term starvation. Representative autoradiograph of Northern blot analyses performed with total RNA isolated from sex-matched, adult littermate 129SvJ mice after a 24-hour fast (F) or controls fed ad libitum (C). The expression of nuclear genes encoding mitochondrial (M-CPT I, MCAD) and peroxisomal (acyl-CoA oxidase [ACO]) FAO enzymes was also assessed. The results shown are representative of three independent experiments.
Forced expression of PGC-1 in neonatal cardiac myocytes induces the expression of nuclear and mitochondrial genes involved in multiple mitochondrial energy-transduction/energy-production pathways.
Gain-of-function studies were performed to determine whether PGC-1 could regulate cardiac mitochondrial oxidative capacity. Primary rat neonatal ventricular myocytes in culture were chosen for these studies because they exhibit a fetal energy metabolic phenotype, including low mitochondrial number, in contrast to the adult mammalian heart that has abundant mitochondria and a high capacity for oxidative energy production. In addition, although cultured neonatal cardiac myocytes express functional PPARα (13), PGC-1 transcripts and protein were not detectable by Northern and Western blot analyses, respectively (Figure 3a). Infection of the cardiac myocytes with an adenoviral vector designed to express a myc epitope-tagged PGC-1 (Ad-PGC-1) resulted in high-level expression of the myc-PGC-1 transcript and protein (Figure 3a). Expression of the endogenous PGC-1 gene was also increased in the Ad-PGC-1–infected cells (Figure 3a), suggesting the existence of an autoregulatory mechanism. Forced expression of PGC-1 markedly induced the expression of PPARα target genes encoding the mitochondrial FAO enzymes, MCAD and M-CPT I (top panels, Figure 3a). Immunodetectable MCAD protein levels were also significantly greater in the Ad-PGC-1–infected myocytes compared with control cells (bottom panels, Figure 3a).
Figure 3.

Forced expression of PGC-1 in cultured rat neonatal cardiac myocytes induces the expression of nuclear and mitochondrial genes involved in multiple mitochondrial energy-transduction/energy-production pathways. (a) The expression of mitochondrial FAO enzyme genes. The autoradiographs represent Northern (top panels) and Western (bottom panels) analyses, respectively. The panels depict the expression of the myc-tagged PGC-1 and endogenous FAO enzyme genes (MCAD and M-CPT I) in rat neonatal cardiac myocytes grown in serum-free culture conditions. Total RNA (15 μg/lane) or protein (10 μg/lane) was isolated from uninfected cells (U), or 48 hours after infection with control adenovirus expressing GFP alone (C), or an adenoviral vector expressing both GFP and PGC-1 (P). As determined by GFP fluorescence, 95–100% of the total cells plated were infected (data not shown). Adenoviral epitope-tagged PGC-1 mRNA is denoted myc-PGC-1 and endogenous PGC-1 transcripts are labeled endogenous PGC-1. The α-actin signal is shown as a control. PGC-1 protein was not detected in extracts from control adenovirus-infected cells by Western blot analysis (bottom panels), even with prolonged exposures. (b) The expression of genes and proteins involved in the TCA cycle, electron transport, and oxidative phosphorylation. The autoradiographs shown at the top represent Northern blot studies performed with total RNA isolated from cardiac myocytes under the conditions described in a. Western blot studies (bottom) were performed with cellular protein extracts prepared from myocytes 4 days after adenoviral infection. Bov. COX, bovine COX protein standards; Cyt C, cytochrome c. All data shown are representative of at least three independent experiments.
We next sought to explore a broader role for PGC-1 in the transcriptional control of genes involved in mitochondrial pathways critical for cardiac myocyte energy production. PGC-1 induced the expression of the nuclear genes encoding a TCA cycle enzyme (citrate synthase) and components of the oxidative phosphorylation complex (β and c subunits of F1–F0 ATP synthase; Figure 3b). PGC-1–expressing myocytes also exhibited increased protein levels of nuclear-encoded (COX subunits IV, Va, Vb, and cytochrome c) and mitochondrial-encoded (COX subunit I) components of the electron transport chain (Figure 3b). Taken together, these results indicate that PGC-1 coordinately activates expression of nuclear and mitochondrial genes encoding proteins involved in multiple mitochondrial energy-transducing/energy-producing pathways.
PGC-1 induces mitochondrial biogenesis in cardiac myocytes in culture.
The results of the gene and protein expression studies shown in Figure 3 are indicative of a mitochondrial biogenic response to PGC-1 overexpression. To determine whether PGC-1 promotes mitochondrial biogenesis in cardiac myocytes, a fluorescent, mitochondrion-selective dye (MitoTracker; see Methods) was used. MitoTracker staining was significantly greater in cells expressing PGC-1 compared with control cells infected with the viral backbone, consistent with increased mitochondrial capacity (Figure 4a). Electron microscopy was performed on histologic sections of pellets prepared from cardiac myocytes expressing PGC-1. Cells infected with Ad-PGC-1 exhibited increased cellular mitochondrial number compared with control myocytes (Figure 4b). Marked mitochondrial size heterogeneity was also noted in the Ad-PGC-1–infected myocytes, including the presence of very large mitochondria (Figure 4b, far right panel). Quantitative morphometric analysis demonstrated that the mean mitochondrial volume density (total mitochondrial area/total cytoplasmic area) in cardiac myocytes infected with Ad-PGC-1 was 57% higher relative to control virus-infected cells (0.36 ± 0.04, n = 15 cells, vs. 0.23 ± 0.04, n = 13 cells, μm3 per μm3 of cell cytoplasm; P < 0.05).
Figure 4.

PGC-1 promotes mitochondrial biogenesis in cardiac myocytes. (a) Fluorescence micrograph panels representing the use of the mitochondrion-selective dye MitoTracker to estimate mitochondrial capacity in cardiac myocytes 5 days after infection with either a GFP-expressing control adenovirus (Ad-GFP) or an adenovirus expressing both GFP and PGC-1 (Ad-PGC-1). MitoTracker, which is oxidized, sequestered, and conjugated in mitochondria, is identified by the orange-red color. GFP, which confirms adenoviral infection, is seen as a nuclear-localized green color. (b) Electron micrographs obtained from sections of cultured rat neonatal cardiac myocyte pellets representing cells infected with either Ad-GFP (Control) or Ad-PGC-1 (PGC-1). The far right panel is representative of regions containing markedly enlarged mitochondria observed in the PGC-1–expressing myocytes. Bar represents 1 μm, the size standard for all three panels. M, mitochondria; N, nucleus.
PGC-1 increases cardiac myocyte capacity for mitochondrial respiration.
To investigate whether forced expression of PGC-1 increased the functional capacity for mitochondrial respiration in cardiac myocytes in culture, oxygen consumption rates were determined. For these studies, saponin-permeabilized, cultured rat neonatal cardiac myocytes were studied under conditions in which mitochondrial substrate and ADP were not limiting, to mimic state 3 respiration. Relative to control cells, PGC-1–expressing cardiac myocytes consumed oxygen at a 2.8-fold greater rate, consistent with an increase in mitochondrial oxidative capacity and the observed increase in mitochondrial number (Figure 5a).
Figure 5.

Forced expression of PGC-1 in neonatal cardiac myocytes increases oxygen consumption and coupled respiration. (a) Oxygen-consumption rates assessed in saponin-permeabilized cultured rat neonatal cardiac myocytes under conditions in which mitochondrial substrate and ADP were not limiting. The bars represent mean (± SE) oxygen consumption rates (nanomole of oxygen per minute per milligram of protein) in cells infected with control adenovirus expressing GFP alone (C) or adenovirus expressing GFP and PGC-1 (P), corrected for total cellular protein content (AP < 0.05). The values represent four samples in two independent experiments. (b) Myocyte oxygen-consumption rates in serum-free growth medium (without saponin) at base line and after exposure to the ATP synthase inhibitor, oligomycin. The values represent the mean of at least three samples in two independent experiments (AP < 0.001). (c) Representative autoradiograph of Northern blot analyses performed with total RNA isolated from cultured rat neonatal cardiac myocytes after a 48-hour exposure to three different conditions: uninfected cells (U), cells infected with control adenovirus expressing GFP alone (C), and cells infected with adenovirus expressing both GFP and PGC-1 (P). The blot was hybridized to cDNA probes encoding UCP-2, UCP-3, and the ATP synthase β subunit. The ethidium bromide–stained 28S ribosomal subunit (28S) was used as a control for lane loading.
A recent report demonstrated that PGC-1 increased uncoupled mitochondrial respiration and the expression of UCPs in adipocyte and C2C12 cell lines in culture (15). We hypothesized that in contrast to these cell types, PGC-1 would induce coupled mitochondrial respiration in the primary cardiac myocyte, given the obligate high level of ATP production in the heart. To determine whether PGC-1 induced coupled or uncoupled respiration in cardiac myocytes, the ATP synthase inhibitor, oligomycin, was employed to inhibit oxidative phosphorylation before the measurement of oxygen consumption rates. If PGC-1 overexpression induces predominantly coupled respiration in the cardiac myocytes, oligomycin would reduce oxygen consumption rates to a similar degree in cells infected with Ad-PGC and control cells. If, however, PGC-1 induces uncoupled respiration, oligomycin would reduce oxygen-consumption rates to a greater degree in control cells compared with PGC-1–expressing cells. The percentage of reduction in oxygen consumption after oligomycin treatment was similar in cardiac myocytes infected with either control or Ad-PGC-1 vector (Figure 5b), indicating that the PGC-1–induced increase in mitochondrial oxygen consumption mainly was due to an augmentation in coupled respiration through oxidative phosphorylation. Inhibition of the adenine nucleotide translocase by atractyloside also resulted in a similar degree of reduction in total oxygen consumption in control and PGC-1 adenovirus–infected cardiac myocytes (data not shown).
The effect of PGC-1 on cardiac myocyte UCP expression was delineated. UCP-1 is a brown adipose-specific inner mitochondrial membrane protein that generates heat by dissipating the inner mitochondrial membrane proton gradient (35). Two homologous proteins, UCP-2 and UCP-3, are expressed in multiple tissues, including skeletal muscle and heart (19). PGC-1 overexpression decreased cardiac myocyte UCP-2 mRNA levels by approximately 60%, while increasing ATP synthase-β subunit mRNA levels threefold (Figure 5c). Although detectable in adult heart total RNA (data not shown), UCP-3 mRNA was not detected in RNA isolated from neonatal rat cardiac myocytes in culture under either condition. Thus, in contrast to the effect of PGC-1 in several noncardiac cell types (15), PGC-1 overexpression decreased rather than increased mitochondrial UCP-2 gene expression in the cardiac myocyte, coincident with an observed increase in mitochondrial number and function. Taken together with the results of the oxygen consumption studies, these data indicate that PGC-1 is capable of increasing the capacity for cardiac myocyte energy production through coupled mitochondrial respiration.
Cardiac overexpression of PGC-1 in vivo in transgenic mice induces uncontrolled mitochondrial biogenesis.
To evaluate the effect of forced PGC-1 expression in the intact postnatal heart in vivo, the cardiac α-MHC gene promoter (28) was used to produce transgenic mice with high-level postnatal, cardiac-specific expression of PGC-1 (MHC-PGC-1 mice). MHC-PGC-1 transgenic mice from three independent lines (ages 1–5 weeks) exhibited high-level cardiac-specific transgene expression and increased mitochondrial DNA content compared with nontransgenic littermate controls (data not shown). The transgenic mice of all three independent lines exhibited massive edema, increased heart size, and four-chamber enlargement consistent with a dilated cardiomyopathy (Figures 6, a and b). Echocardiographic analysis of MHC-PGC-1 mice demonstrated marked four-chamber cardiac enlargement and severely decreased global contractile function (Figure 6c). All of the MHC-PGC-1 mice died by 6 weeks of age. The postnatal phenotype of the MHC-PGC-1 mice is consistent with the temporal pattern of activation of the cardiac α-MHC gene promoter used to drive expression of the transgene.
Figure 6.

MHC-PGC-1 mice develop a severe dilated cardiomyopathy. (a) Representative photographs of a nontransgenic mouse (Control) and a littermate MHC-PGC-1–transgenic mouse (MHC-PGC-1) at 6 weeks of age. (b) Midventricular transverse histologic sections of a nontransgenic control mouse (Control) and littermate MHC-PGC-1–transgenic mouse heart (MHC-PGC-1) at 4 weeks of age. The tissue was fixed in 10% neutral-buffered formalin and stained with Masson’s trichrome. (c) M-mode echocardiograms at the midventricular level of nontransgenic (Control) and littermate MHC-PGC-1–transgenic (MHC-PGC-1) mice at 6 weeks of age.
Histologic studies performed on left-ventricular tissue from the MHC-PGC-1 mice before death revealed marked structural abnormalities, including numerous regions of sarcomeric disruption (Figure 7a). Trichrome staining of the histologic sections revealed a granular appearance in areas where the sarcomeric assembly was altered or absent (Figure 7a). Patchy areas of mild fibrosis were also seen. Electron microscopic analysis of the transgenic hearts revealed that the regions of granular densities represented massive expansion of enlarged mitochondria (Figure 7b). The mitochondria in the cardiac ventricles of transgenic mice were more numerous and often significantly larger than the littermate nontransgenic control mouse hearts. In some myocytes, the marked mitochondrial proliferation appeared to have replaced the sarcomeric assembly (lower-left panel, Figure 7b). Thus, as predicted by the cell-culture studies, overexpression of PGC-1 resulted in a striking mitochondrial proliferation in the hearts of the transgenic mice.
Figure 7.
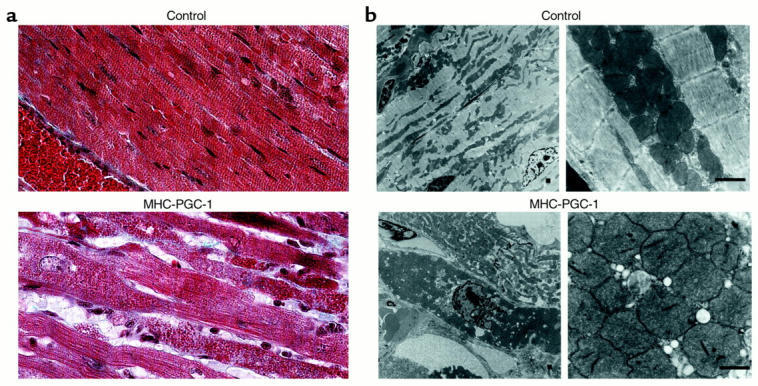
Induction of uncontrolled mitochondrial proliferation in the cardiac ventricle of MHC-PGC-1 mice. (a) Representative histologic sections of cardiac ventricles from a nontransgenic littermate control (Control) and a MHC-PGC-1–transgenic mouse heart (MHC-PGC-1) obtained at 3.5 weeks of age. The tissue was fixed in formalin and stained with Gomori’s trichrome. The red blood cells within a vessel present in the lower-left region of the Control panel serve as a relative size standard. Note the granular appearance of the myocytes and loss of sarcomeric structure in the section shown in the lower panel. (b) Low-power (left panels) and high-power (right panels) electron micrographs of histologic sections prepared from 3.5-week-old MHC-PGC-1 transgenic mouse ventricle (MHC-PGC-1) and littermate non-transgenic control (Control). Bar represents 1 μM.
To determine whether the cardiac phenotype of the MHC-PGC-1 mice was determined by the level of forced PGC-1 expression, additional lines with lower levels of transgene expression were characterized. Two independent MHC-PGC-1 lines with approximately fivefold lower PGC-1 mRNA expression compared with the three high-expressing lines described above did not develop cardiomyopathy (data not shown). However, a small number of homozygous offspring resulting from crosses of hemizygotes of the low-expressing MHC-PGC-1 lines developed cardiomyopathy associated with marked cardiac mitochondrial proliferation at 5–7 weeks of age. These data indicate that the level of transgene expression predicts the cardiac mitochondrial proliferative phenotype in the MHC-PGC-1 mice.
Discussion
The dynamic regulation of cardiac mitochondrial number and function has suggested the existence of a regulatory pathway involved in the physiologic control of mitochondrial biogenesis. Several lines of evidence presented here indicate that the transcriptional coactivator, PGC-1, is a key regulator of cardiac mitochondrial functional capacity and participates in the transduction of physiologic stimuli to energy production in the heart. First, the expression of the PGC-1 gene is upregulated after birth in the heart, before the known increase in mitochondrial biogenesis and switch from glucose to fatty acids as the chief energy substrate. Second, PGC-1 gene expression is activated by short-term fasting, a condition known to increase cardiac mitochondrial FAO rates. Third, forced expression of PGC-1 in cardiac myocytes induces mitochondrial biogenesis and coupled oxygen consumption, implicating this transcriptional coactivator in the regulation of mitochondrial respiration and, thus, ATP production in the heart.
Little is known about the mechanisms involved in the link between the broad program of mitochondrial biogenesis and the control of nuclear genes encoding enzymes of specific mitochondrial energy transduction pathways such as the FAO cycle. Recent studies indicate that PGC-1 coactivates NRF-1, a key transcriptional regulator of nuclear and mitochondrial genes encoding respiratory chain proteins (14, 15). Recently, we demonstrated that PGC-1 coactivates PPARα, a critical regulator of mitochondrial FAO enzyme gene expression (16). Collectively, these results suggested that PGC-1 may serve to link the actions of NRF-1 and other master regulators of mitochondrial biogenesis to the control of specific mitochondrial pathways such as the FAO cycle. The results shown here identify PGC-1 as a component of the regulatory communication between mitochondrial biogenesis and control of FAO and TCA cycle enzyme expression. Our observation that cardiac PGC-1 gene expression is induced by fasting, a physiologic condition known to activate FAO enzyme gene expression through PPARα (23) further supports a role for PGC-1 as a physiologically relevant coactivator of PPARα and mitochondrial fatty acid utilization in the heart. It will be of significant interest to determine whether PGC-1 coactivates other transcription factors involved in the transcriptional control of specific mitochondrial energy metabolic pathways.
The metabolic phenotype of mitochondria is cell and tissue specific. Mitochondria of the brown adipocyte are specialized for high-capacity, uncoupled respiration to generate heat. In contrast, cardiac mitochondria are efficient producers of ATP and exhibit significantly less respiratory uncoupling. Additionally, the contribution of specific mitochondrial energy metabolic pathways to the production of reducing equivalents for generation of ATP through oxidative phosphorylation varies among tissue types. For example, in contrast to the heart, which has a high capacity for the oxidation of fats, mitochondrial ATP production in the brain relies primarily on reducing equivalents generated by glucose oxidation. Consistent with the tissue-specific mitochondrial energy-substrate preferences, expression of FAO enzymes is markedly higher in heart mitochondria compared with that of the brain (18, 29). We propose that PGC-1 plays a role in determining the metabolic phenotype of mitochondria among specialized cell types. It is possible that the availability of PGC-1 partners in a given tissue, such as NRF-1, PPARα, PPARγ, and other, as yet unidentified, transcription factors dictate the level of expression of enzymes and proteins in specific mitochondrial pathways. This notion is supported by the observation that PGC-1 is capable of inducing either uncoupled (15) or coupled (shown here) respiration in a cell type–specific manner and the finding that prolonged cold exposure induces PGC-1 gene expression in brown adipose tissue and in skeletal muscle, but not in the heart (14). Taken together, these findings suggest that PGC-1 controls mitochondrial functional capacity in a tissue-specific manner.
Our data indicate that PGC-1 is capable of promoting biogenesis of mitochondria that support primarily coupled respiration in neonatal cardiac myocytes in culture. However, these results do not allow us to conclude that PGC-1 induces coupled respiration in the adult heart in vivo under all circumstances. It is possible that PGC-1 induces coupled or uncoupled mitochondrial respiration in vivo in the heart in different physiologic contexts. Unfortunately, given the cardiac dysfunction of the MHC-PGC-1 mice, functional studies of mitochondria from the transgenic mice would be difficult to interpret because of secondary effects related to heart failure. Future studies using inducible transgenic expression systems should allow the evaluation of the primary effects of PGC-1 on mitochondrial function in vivo.
Evidence is emerging that alterations in mitochondrial functional capacity are involved in the pathogenesis of a wide variety of inherited and acquired human diseases, many of which cause cardiomyopathy and heart failure (1–3). Inborn errors in mitochondrial FAO enzymes and mutations of the mitochondrial genome are now recognized as important causes of cardiomyopathy, metabolic disturbances, and skeletal myopathy in pediatric and adult populations. Interestingly, the clinical manifestations of mitochondrial diseases is often precipitated by environmental or physiologic “stressors” that increase the demand for cardiac or skeletal muscle mitochondrial energy production, such as exercise or fasting, suggesting that a mismatch between energy demand and supply precipitates organ dysfunction (1). Common acquired cardiac diseases are also associated with mitochondrial functional abnormalities. Cardiac hypertrophy due to pressure overload, such as that which occurs with hypertension, results in a downregulation of FAO enzyme expression and reduced capacity for mitochondrial oxidation of fats (22). Mitochondrial DNA deletions also occur with aging and in the ischemic heart (4–6). Future studies aimed at delineating the activity of PGC-1 in cardiac disease states associated with mitochondrial dysfunction may help define the potential role of this coactivator in the pathogenesis of specific cardiac diseases. Considering the wide range of pathophysiologic states in which cardiac mitochondrial functional capacity is altered, the PGC-1 regulatory pathway is a candidate target for the investigation of novel experimental and possibly therapeutic strategies aimed at the amelioration of energy metabolic dysfunction in a variety of cardiac diseases.
Acknowledgments
This work was supported by grants from the NIH (HL58493, DK45416, P30 DK56341, and P50 HL61006), and the American Heart Association (Established Investigator award 95001150). J. Lehman was supported by an institutional training grant from the NIH (T32 HL07873). We thank Kelly Hall for assistance with preparation of the manuscript. We are grateful to Bing Li and Clay Semenkovich for advice and assistance with the oxygen-consumption studies, Rick Vega and Zaza Khuchua for helpful discussions, and Teresa Leone for assistance with the fasting experiments.
References
- 1.Kelly DP, Strauss AW. Inherited cardiomyopathies. N Engl J Med. 1994;330:913–919. doi: 10.1056/NEJM199403313301308. [DOI] [PubMed] [Google Scholar]
- 2.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 3.Williams RS. Cardiac involvement in mitochondrial diseases, and vice versa. Circulation. 1995;91:1266–1268. doi: 10.1161/01.cir.91.4.1266. [DOI] [PubMed] [Google Scholar]
- 4.Corral-Debrinski M, et al. Hypoxia is associated with mitochondrial DNA damage and gene induction. Implications for cardiac disease. JAMA. 1991;266:1812–1816. [PubMed] [Google Scholar]
- 5.Corral-Debrinski M, Shoffner JM, Lott MT, Wallace DC. Association of mitochondrial DNA damage with aging and coronary atherosclerotic heart disease. Mutat Res. 1992;275:169–180. doi: 10.1016/0921-8734(92)90021-g. [DOI] [PubMed] [Google Scholar]
- 6.Cortopassi GA, Shibata D, Soong N-W, Arnheim N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc Natl Acad Sci USA. 1992;89:7370–7374. doi: 10.1073/pnas.89.16.7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Attardi G, Schatz G. Biogenesis of mitochondria. Annu Rev Cell Biol. 1988;4:289–333. doi: 10.1146/annurev.cb.04.110188.001445. [DOI] [PubMed] [Google Scholar]
- 8.Mayor F, Cuezva JM. Hormonal and metabolic changes in the perinatal period. Biol Neonate. 1985;48:185–196. doi: 10.1159/000242171. [DOI] [PubMed] [Google Scholar]
- 9.Evans MJ, Scarpulla RC. NRF-1: a trans-activator of nuclear-encoded respiratory genes in animal cells. Genes Dev. 1990;4:1023–1034. doi: 10.1101/gad.4.6.1023. [DOI] [PubMed] [Google Scholar]
- 10.Virbasius CA, Virbasius JV, Scarpulla RC. NRF-1, an activator involved in nuclear-mitochondrial interactions, utilizes a new DNA-binding domain conserved in a family of developmental regulators. Genes Dev. 1993;7:2431–2445. doi: 10.1101/gad.7.12a.2431. [DOI] [PubMed] [Google Scholar]
- 11.Virbasius JV, Scarpulla RC. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc Natl Acad Sci USA. 1994;91:1309–1313. doi: 10.1073/pnas.91.4.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gulick T, Cresci S, Caira T, Moore DD, Kelly DP. The peroxisome proliferator activated receptor regulates mitochondrial fatty acid oxidative enzyme gene expression. Proc Natl Acad Sci USA. 1994;91:11012–11016. doi: 10.1073/pnas.91.23.11012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brandt J, Djouadi F, Kelly DP. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor α. J Biol Chem. 1998;273:23786–23793. doi: 10.1074/jbc.273.37.23786. [DOI] [PubMed] [Google Scholar]
- 14.Puigserver P, et al. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 15.Wu Z, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 16.Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He TC, et al. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kelly DP, Gordon JI, Alpers R, Strauss AW. The tissue-specific expression and developmental regulation of the two nuclear genes encoding rat mitochondrial proteins: medium-chain acyl-CoA dehydrogenase and mitochondrial malate dehydrogenase. J Biol Chem. 1989;264:18921–18925. [PubMed] [Google Scholar]
- 19.Boss O, et al. Uncoupling protein-3: a new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett. 1997;408:39–42. doi: 10.1016/s0014-5793(97)00384-0. [DOI] [PubMed] [Google Scholar]
- 20.Cresci S, Wright LD, Spratt JA, Briggs FN, Kelly DP. Activation of a novel metabolic gene regulatory pathway by chronic stimulation of skeletal muscle. Am J Physiol. 1996;270:C1413–C1420. doi: 10.1152/ajpcell.1996.270.5.C1413. [DOI] [PubMed] [Google Scholar]
- 21.Annex BH, Kraus WE, Dohm GL, Williams RS. Mitochondrial biogenesis in striated muscles: rapid induction of citrate synthase mRNA by nerve stimulation. Am J Physiol. 1991;260:C614–C620. doi: 10.1152/ajpcell.1991.260.2.C266. [DOI] [PubMed] [Google Scholar]
- 22.Sack MN, Disch DL, Rockman HA, Kelly DP. A role for Sp and nuclear receptor transcription factors in a cardiac hypertrophic growth program. Proc Natl Acad Sci USA. 1997;94:6438–6443. doi: 10.1073/pnas.94.12.6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPARα) in the cellular fasting response: the PPARα-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci USA. 1999;96:7473–7478. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Estey LA, Lincoln AJ, Prochaska LJ. Chemical labeling studies on bovine heart mitochondrial cytochrome c oxidase dispersed in nonionic detergents. Biochemistry. 1990;29:9714–9720. doi: 10.1021/bi00493a029. [DOI] [PubMed] [Google Scholar]
- 25.Medeiros DM, Liao Z, Hamlin RL. Copper deficiency in a genetically hypertensive cardiomyopathic rat: electrocardiogram, functional and ultrastructural aspects. J Nutr. 1991;121:1026–1034. doi: 10.1093/jn/121.7.1026. [DOI] [PubMed] [Google Scholar]
- 26.Weibel, E.R. 1979. Stereological methods. Academic Press. London, United Kingdom. 755 pp.
- 27.Steer, M.W. 1981. Understanding cell structure. Cambridge University Press. Cambridge, United Kingdom. 126 pp.
- 28.Palermo J, Gulick J, Colbert M, Fewell J, Robbins J. Transgenic remodeling of the contractile apparatus in the mammalian heart. Circ Res. 1996;78:504–509. doi: 10.1161/01.res.78.3.504. [DOI] [PubMed] [Google Scholar]
- 29.Disch DL, et al. Transcriptional control of a nuclear gene encoding a mitochondrial fatty acid oxidation enzyme in transgenic mice: role for nuclear receptors in cardiac and brown adipose expression. Mol Cell Biol. 1996;16:4043–4051. doi: 10.1128/mcb.16.8.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rogers JH, et al. RGS4 causes increased mortality and reduced cardiac hypertrophy in response to pressure overload. J Clin Invest. 1999;104:567–576. doi: 10.1172/JCI6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veksler VI, Kuznetsov AV, Sharov VG, Kapelko VI, Saks VA. Mitochondrial respiratory parameters in cardiac tissue: a novel method of assessment by using saponin-skinned fibers. Biochim Biophys Acta. 1987;892:191–196. doi: 10.1016/0005-2728(87)90174-5. [DOI] [PubMed] [Google Scholar]
- 32.Wittels B, Bressler R. Lipid metabolism in the newborn heart. J Clin Invest. 1965;44:1639–1646. doi: 10.1172/JCI105270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esterbauer H, Oberkofler H, Krempler F, Patsch W. Human peroxisome proliferator activated receptor gamma coactivator 1 (PPARGC1) gene: cDNA sequence, genomic organization, chromosomal localization, and tissue expression. Genomics. 1999;62:98–102. doi: 10.1006/geno.1999.5977. [DOI] [PubMed] [Google Scholar]
- 34.Parvin R, Pande SV. Enhancement of mitochondrial carnitine and carnitine acylcarnitine translocase-mediated transport of fatty acids into liver mitochondria under ketogenic conditions. J Biol Chem. 1979;254:5423–5429. [PubMed] [Google Scholar]
- 35.Nicholls DG, Locke RM. Thermogenic mechanisms in brown fat. Physiol Rev. 1984;64:1–64. doi: 10.1152/physrev.1984.64.1.1. [DOI] [PubMed] [Google Scholar]