

Abstract
Despite prolonged treatment with highly active antiretroviral therapy (HAART), infectious HIV-1 continues to replicate and to reside latently in resting memory CD4+ T lymphocytes, creating a major obstacle to HIV-1 eradication. It is therefore not surprising to observe a prompt viral rebound after discontinuation of HAART. The nature of the rebounding virus, however, remains undefined. We now report on the genetic characterization of rebounding viruses in eight patients in whom plasma viremia was undetectable throughout about 3 years of HAART. Taking advantage of the extensive length polymorphism in HIV-1 env, we found that in five patients who did not show HIV-1 replication during treatment, the rebound virus was identical to those isolated from the latent reservoir. In three other patients, two of whom had been free of plasma viremia but had showed some residual viral replication, the rebound virus was genetically different from the latent reservoir virus, corresponding instead to minor viral variants detected during the course of treatment in lymphoid tissues. We conclude that in cases with apparent complete HIV-1 suppression by HAART, viral rebound after cessation of therapy could have originated from the activation of virus from the latent reservoir. In patients with incomplete suppression by chemotherapy, however, the viral rebound is likely triggered by ongoing, low-level replication of HIV-1, perhaps occurring in lymphoid tissues.
Introduction
In patients infected with HIV-1, early and aggressive treatment with highly active antiretroviral therapy (HAART) can achieve sustained suppression of plasma viremia below the limit of detection and partial repletion of CD4+ T lymphocytes (1–4). For the first time in this epidemic, the pathogenic course of HIV-1 infection has been dramatically attenuated, accounting for a substantial decrease in AIDS-related death in the developed world (5, 6). However, the presence of infectious HIV-1 residing latently in resting memory CD4+ T lymphocytes (7–11) and the persistence of residual viral replication despite prolonged treatment with HAART (12–18) represent major obstacles to HIV-1 eradication. Over the past few years, intensive efforts have been undertaken to characterize the decay of the latent reservoir on HAART. It has been estimated that its pool size is between 103 and 107 cells per patient (7, 19). In patients whose plasma viremia levels are particularly well suppressed by HAART, latent viruses were found to decay with a mean half-life of approximately 6 months (13, 16), a value similar to the observed turnover rate of memory lymphocytes in uninfected humans and monkeys (20–22). In contrast, in patients with episodes of intermittent viremia as a result of residual viral replication, the apparent decay rate of latent viruses could be as long as years (16, 23). It is therefore clear that viral persistence during prolonged treatment is due not only to the slow turnover of infected resting memory CD4+ T lymphocytes, but also to the inability of current antiretroviral regimens to completely suppress viral replication (12–18).
It is thus not surprising that viral rebound occurs in almost all patients after discontinuation of HAART (24–28). However, the nature of the rebounding viruses remains unknown. In the present study, using a newly developed genetic technique, we sought to delineate the genetic relationship between the rebounding viruses and those residing in the latent reservoir as well as in lymphoid tissues.
Methods
PCR amplification, length polymorphism, sequencing, and sequence analysis of HIV-1 gp120.
Viral RNA was extracted from plasma and culture supernatant, and cDNA was synthesized according to a previously described protocol (17, 29). Detection of unspliced (US) HIV-1 mRNA in PBMCs was carried out as described (17). Proviral DNA was extracted from PBMCs or tissue samples as previously described (13, 30). Our genetic analyses focused on the V1–V2 and V4–V5 domains of the viral envelope glycoprotein. These regions contain not only frequent base substitutions, but also extensive length polymorphisms. The env primers used in the PCR were as follows, with their positions in the HXB2 genome indicated in parentheses. For the V1–V2 region, the outer primers were V12–51 5′-GATGCATGAGGATATAATCAGTTTATGGG (+, 6533) and V12–52 5′-CCTAATTCCATGTGTACATTGTACTGT (–, 6954), while the inner primers were V12–50 5′-CCATGTGTAAAATTAACCCCACTCTGTGT (+, 6576) and V12–53 5′-TCAAAGGATACCTTTGGACAGGC (–, 6834). For the V3–V5 region, the outer primers were V3a 5′-CCAATTCCCATACATTATTG (+, 6858) and V3i 5′-GCGTTATTGACGCTGCGCCCAT (–, 7814), while the inner primers were V3e 5′-GTACAATGTACACATGGAAT (+, 6957) and V3h 5′-AATTCACTTCTCCAATTGTC (–, 7653). Each round of PCR consisted of 30 cycles, with the first five cycles at 94°C for 1 minute, 52°C for 1 minute, and 72°C for 1 minute, followed by 25 cycles at 94°C for 1 minute, 55°C for 1 minute, and 72°C for 1 minute. For length polymorphism studies, PCR amplifications were conducted on total viral cDNA in order to have an adequate representation of the viral population. One primer in the second round of PCR was labeled with a fluorophore, 6-carboxy-fluorescein, at the 5′ end (PE Biosystems, Foster City, California, USA). For the V1–V2 region, the labeled primer was V12–50, while for the V4–V5 region, it was V45–407 5′-GGGGAATTTTTCTACTGTAA (+, 7362). PCR-amplified fluorescently labeled products were purified using QIAquick PCR Purification Kit (QIAGEN Inc., Valencia, California, USA), separated on a 6% denaturing polyacrylamide gel using an automated sequencer (ABI PRISM 377; PE Biosystems) and analyzed by the GeneScan program (PE Biosystems). All RNA and DNA extractions and subsequent amplification reactions were carried out with appropriate negative controls in parallel to detect contamination at each step of the procedures. For nucleotide sequencing, single molecules of virus or provirus were amplified after limiting dilution and directly sequenced using an automated sequencer (ABI PRISM 377) to avoid errors introduced by PCR (30, 31). Obtained nucleotide sequences were aligned using the Clustal W program (32). Pairwise distances among sequences were estimated by the DNADIST program implemented in the PHYLIP package (33). Phylogenetic analysis of the nucleotide sequence data was carried out using the neighbor-joining method (34).
Isolation of replication-competent virus from resting memory CD4+ T cells.
To isolate infectious HIV-1 from the latent reservoir, limiting-dilution boosted cultures were performed as previously described (13, 16), based on the method of Finzi et al. (10).
Results
Patients.
Eight male patients, ages 31 to 39, were chosen for the study (Figure 1). They had a mean baseline CD4+ lymphocyte count of 498 cells/μl and an average initial plasma viral load of 199,872 RNA copies/ml (Amplicor HIV-1 Monitor Ultra Sensitive, Roche Molecular Systems, Branchburg, New Jersey, USA). All patients were enrolled into treatment protocols within the first 90 days of their acute HIV-1 infection. The antiviral regimens consisted of zidovudine (600 mg/d) and lamivudine (300 mg/d) along with either ritonavir (1200 mg/d) or indinavir (2400 mg/d). As shown in Figure 1, a precipitous decline in plasma viremia was seen in each case after initiation of therapy; levels below 50 RNA copies/ml were quickly reached and then sustained for 898–1200 days into therapy. After 90 days, plasma viremia was never again detected in any of the cases except on a single occasion in two patients (1306 and 1311) (Figure 1). An average increase of about 300 cells/μl was noted when comparing mean CD4+ lymphocyte counts for the last four measurements with baseline values. After stopping HAART, plasma viral rebound was found in all cases within a few weeks, reaching levels that are equivalent or higher than that during primary infection in six of eight patients (313-9, 1302, 1308, 1310, 1304, and 1311). In the other two patients (1306 and 1309), the viral rebound was delayed by 6 to 8 weeks and only reached low levels in patient 1309 despite his remaining off therapy (Figure 1). In general, there was a concomitant decline in the number of CD4+ T cells associated with the viral rebound (Figure 1). It is interesting to note that in a majority of the cases, the initial rebound in plasma viremia was spontaneously downmodulated without any further intervention. Correlating the changes in plasma viremia with fluctuations in HIV-1–specific immune responses will be the subject of a separate report.
Figure 1.

Changes in plasma viral load and CD4+ lymphocyte count during and after HAART. Shaded areas indicate the treatment periods. The drug regimens are as indicated: AZT, zidovudine; 3TC, lamivudine; RIT, ritonavir; and IND, indinavir. The open arrows indicate the days on which boosted cultures were undertaken to isolate latent viruses from resting memory CD4+ T cells. The solid arrows indicate the days when tissue biopsies were collected from the lymph node, tonsil, and/or rectum.
Length polymorphism of rebounding viruses and comparison with latent reservoir viruses during HAART and those at primary infection before treatment.
To compare the genetic relationship between different viral populations, we developed a fluorescent technique based on the known length polymorphism in V1–V2 and V4–V5 regions of HIV-1 gp120 (35). The new technique is sensitive enough to detect a single nucleotide deletion or insertion within PCR products, because of its fine resolution on a 6% denaturing polyacrylamide gel. We first tested this technique on plasma samples collected from eight acutely infected patients and eight chronically infected cases before treatment with HAART. As shown in Figure 2, single and uniform peaks of V1–V2 and V4–V5 products were found in acutely infected individuals, although the lengths varied from patient to patient. In contrast, multiple peaks with different lengths were identified for both V1–V2 and V4–V5 products in almost all chronically infected patients. This observation is consistent with previous findings that during acute infection, the viral population is relatively homogenous compared with that in the subsequent stages of infection (30, 36).
Figure 2.
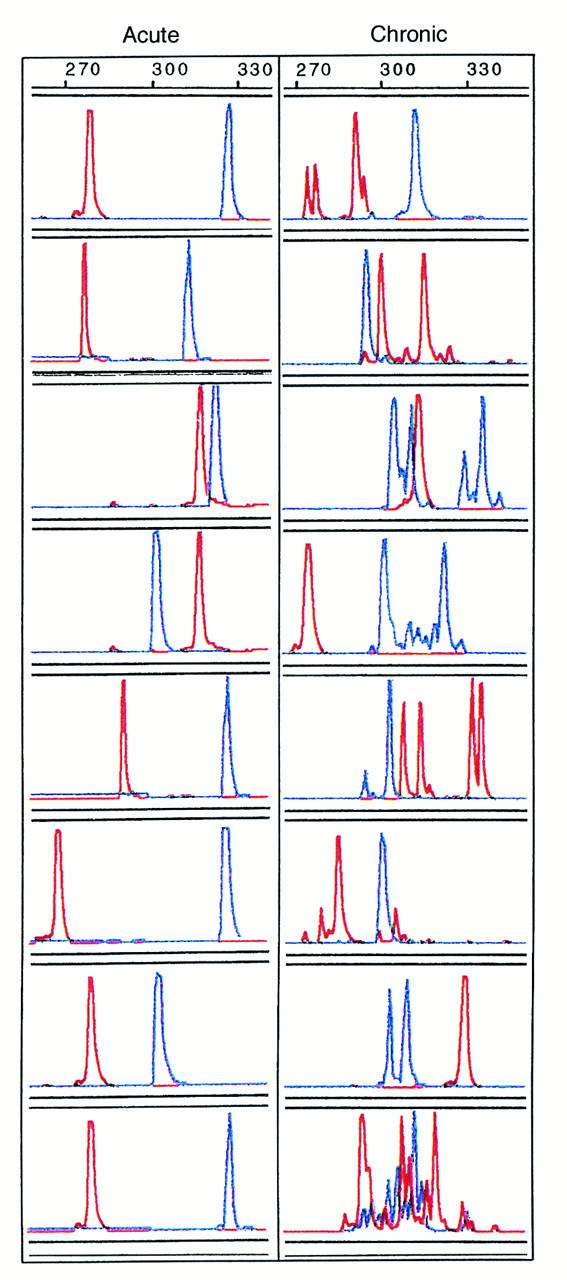
Length polymorphism in the V1–V2 (in red) and V4–V5 (in blue) regions of HIV-1 env found in the plasma of eight acutely infected (left) and eight chronically infected (right) patients. The length of each V1–V2 or V4–V5 region is indicated by the scale at the top of each panel.
To study the genetic relatedness between the rebounding viruses and those present during the primary infection and in the latent viral reservoir, viral RNA preparations derived from the plasma of the eight patients (Figure 1) before initiation of HAART and 14 to 280 days after stopping HAART were analyzed using the novel technique developed in this report. Viral RNA from the culture supernatant of resting memory CD4+ T cells was obtained at four different time points while on HAART (Figure 1) and was analyzed in a similar manner. As shown in Figure 3, replication-competent viruses recovered from the resting memory CD4+ T cells were identical in V1–V2 and V4–V5 lengths to those in the plasma during primary infection in each patient. In addition, in four independent samples collected up to 3 years after initiation of HAART, viruses residing in the resting memory CD4+ T cells remained unchanged in length in both V1–V2 and V4–V5 regions. Interestingly, when rebounding viruses in plasma were analyzed, it was found that in five of eight patients (1308, 1310, 1304, 1306, and 1311) they were identical in length to those recovered from latent reservoir while on HAART and from the plasma at primary infection. In three patients (313-9, 1302, and 1309), however, the initial rebounding viruses were different in length from those in latent viral reservoir and at primary infection. In patient 313-9, a 21-bp deletion was found in the V4–V5 region of the rebounding viruses, whereas the V1–V2 region stayed the same (Figure 3). In patient 1302, however, there was a 12-bp deletion in the V1–V2 region, whereas the V4–V5 region remained constant. In patient 1309, a 15-bp insertion was identified in the V4–V5 region of the rebounding virus. It is interesting to note that as patients 313-9 and 1309 remained off therapy, the initial rebounding viruses were gradually joined or overtaken by a strain similar in length to that found in the latent reservoir and at primary infection (Figure 3).
Figure 3.

Length polymorphism of the rebounding plasma virus in comparison with those isolated from the latent reservoir during therapy and those in the plasma before therapy. Multiple samples collected after cessation of HAART are separated into three time groups: 14–42 days, 43–140 days, and 141–280 days. The V1–V2 region is shown in red; the V4–V5 region is shown in blue. The size of each peak is indicated by the scale at the top of each panel.
To analyze the rebounding viruses in greater detail, extensive sequencing was conducted in four of eight patients (313-9, 1302, 1308, and 1310). A total of 295 V1–V2 and 312 V4–V5 sequences were obtained from the rebounding viruses, viruses at the primary infection, and those from the latent viral reservoir during treatment (GenBank accession numbers: AF290622–AF290675, AF290676–AF290722, AF290723–AF290778, AF290779–AF290843, AF292129–AF292236, AF292243–AF292306, and AF292327–AF292358). The most prominent result from the sequence analysis is that all of the length polymorphisms observed previously were confirmed by the actual sequences (data not shown). In patient 313-9, for example, an exact 21-bp deletion was identified in the V5 region of the initial rebounding virus, whereas in patient 1302, a 12-bp deletion in the V1–V2 region of the rebounding virus was noted, matching exactly findings based on length polymorphism studies. Importantly, the initial rebounding viruses in patients 1308 and 1310 were not only identical in length, but also identical in sequences to those found in the latent reservoir and at primary infections (data not shown). In addition, we also carried out an extensive phylogenetic analysis on the nucleotide sequences obtained from the four patients (313-9, 1302, 1308, and 1310). The most consistent result from this analysis was that nucleotide sequences from each individual patient clustered tightly together irrespectively of their source of origin, be it viruses from the primary infection, the latent viral reservoir, the rebounding population, or tissue biopsies (data not shown). The sequences on which the length polymorphism studies were carried out are therefore all specific to each individual without evidence of contamination.
Evidence of residual HIV-1 replication in two patients whose initial rebound viruses differ from latent viruses isolated during HAART and those at primary infection.
In patients whose rebounding viruses differ from the latent reservoir viruses, what could be the source? One likely possibility is the persistent, low-level HIV-1 replication during HAART. To examine this hypothesis, we measured the level of US HIV-1 RNA in PBMCs of each case approximately 3 weeks before stopping HAART. Among the five patients (1308, 1310, 1304, 1306, and 1311) whose rebounding viruses were identical in length to those recovered from the latent reservoir, no detectable US HIV-1 RNA was found (Table 1). Among the other patients (313-9, 1302, and 1309), however, two (313-9 and 1302) had detectable levels of US HIV-1 RNA in their PBMCs (Table 1). These results are consistent with our previous findings (summarized in Table 1) that residual HIV-1 replication occurred in the presence of HAART in patients 313-9 and 1302, as detected by continued sequence evolution in the viral envelope gene and expression of viral RNA in lymphoid tissues (13).
Table 1.
Detection of residual HIV-1 replication in blood and tissue biopsies

Further analysis revealed that sequences identical to the initial rebounding viruses in patients 313-9 and 1302 were in fact minor variants detected in biopsies of the lymph node, tonsil, or rectum during HAART. In patient 313-9, among a total of 40 proviral sequences obtained from the lymph node, tonsil, and rectum, four (one in the lymph node, one in the tonsil, and two in the rectum) were identical to the rebounding virus (data not shown). None of the proviral sequences in PBMCs during treatment, however, was identical to the rebounding virus. In patient 1302, among a total of 33 proviral sequences obtained from the tonsil and rectum, one sequence (rectum) was identical to the rebounding virus (data not shown).
Discussion
That HIV-1 rebounds promptly after cessation of HAART (24–28) should come as no surprise, given the persistence of infectious virus residing latently in resting memory CD4+ T cells (7–11) as well as the continuation of residual viral replication despite complete suppression of plasma viremia (12–17). Using a novel genetic technique together with nucleotide sequencing, we have studied eight acutely infected patients whose plasma viremia had been completely suppressed by HAART for about 3 years (Figure 1). Specifically, we compared the genetic relationship of viruses that rebound after stopping treatment with that present at primary infection, as well as with those in the latent reservoir during treatment. In five cases, the rebounding virus was identical in length to viruses found at primary infection and within the latent reservoir (Figure 3). This identity of viral populations was confirmed by additional nucleotide sequencing studies. Interestingly, these five patients had no detectable evidence of residual HIV-1 replication based on the absence of sequence evolution or mRNA expression in PBMCs and lymphoid tissues (Table 1) while on treatment. In contrast, in three other patients, the rebounding virus was distinct from the latent reservoir virus (Figure 3), but identical to minor viral variants found in lymphoid tissues (data not shown). Evidence of ongoing viral replication in blood and lymphoid tissues was noted in two of the latter cases (Table 1). Therefore, we now conclude that in patients with apparent complete HIV-1 suppression by HAART, the viral rebound could have resulted from the activation of the virus from the latent reservoir. In patients with incomplete suppression by chemotherapy, however, the viral spread upon cessation of treatment is probably triggered by ongoing, low-level replication of HIV-1, perhaps in tissue compartments such as lymph nodes, tonsils, or gut-associated lymphoid tissues. A similar study has recently been conducted by Chun et al. (37). Based on heteroduplex mobility and tracking assays, they found that in the majority of patients, the rebounding virus in plasma was genetically distinct from both the cell-associated viral RNA and the replication-competent virus in the resting memory CD4+ T cells. They concluded that the cause of viral rebound was a source other than the latent reservoir (37).
We believe that above conclusions, however, are subject to two important caveats. First, the identity of HIV-1 sequences cannot unequivocally pinpoint the latent reservoir as the source, since the viral rebound could have been triggered by a replicating viral population that happens to be the same as that in the latent reservoir. Second, nonidentity also cannot conclusively exclude the latent reservoir as the source, since the viral rebound could have originated from the reactivation of a divergent viral species in the latent reservoir that was not detected due to limited sampling. It is therefore imprudent to draw firm conclusions based on these preliminary genetic findings. Nevertheless, when placed in the context of our overall understanding, these new results do provide valuable insights into major obstacles on the road to HIV-1 eradication (19).
The cases in whom rebounding viruses match those in the latent pool (1308, 1310, 1304, 1306, and 1311) serve to emphasize the importance of this reservoir in retaining infectious HIV-1 during HAART. Particularly daunting is the observation that the viral reservoir established during primary infection persists for years on seemingly effective therapy. The intrinsic decay rate of the latent reservoir is likely to be slow, perhaps with half-life of approximately 6 months (13, 16). Purging this latent reservoir, therefore, remains a huge challenge in our therapeutic effort to eliminate HIV-1 (19).
On the other hand, the cases in whom rebounding viruses differ from those in the latent reservoir (313-9, 1302, and 1309) serve to illustrate the problem of residual HIV-1 replication during HAART even when plasma viremia is completely suppressed. Ongoing, low-level viral replication on treatment not only renders HIV-1 eradication infeasible but also exacerbates the persistence of infectious virus in the latent reservoir. Active viral replication could continually replenish the latent reservoir (16), thereby slowing its apparent decay to rates that are essentially equivalent to lifelong persistence (23). Thus, it is paramount that studies be undertaken to define the fundamental mechanism responsible for residual viral replication in the presence of several specific inhibitors, as this information will in turn inform the design of therapeutic regimens that have the potential to block HIV-1 replication completely.
Acknowledgments
We are indebted to our study patients for their participation; to Abbott Laboratories, Merck Inc., Hoffman-LaRoche Inc., and Glaxo Wellcome for sponsoring the clinical studies; to M. Yaman and M.R. Chaudhry for performing lymph node and tonsillar biopsies, and to A. Talal for performing rectal biopsies. This work was supported by NIH grants (AI-41534, AI-40387, AI-42848 [Center for AIDS Research], and AI-46964), the AIDS Clinical Trials Group, the General Clinical Research Center of The Rockefeller University (M01-RR00102), the Belotsky Foundation, the Bristol-Myers Squibb Foundation, the Irvin A. Hansen Memorial Foundation, and the Irene Diamond Fund.
References
- 1.Hammer SM, et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. N Engl J Med. 1997;337:725–733. doi: 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- 2.Gulick RM, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med. 1997;337:734–739. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- 3.Perelson AS, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 4.Markowitz M, et al. The effect of commencing combination antiretroviral therapy soon after human immunodeficiency virus type 1 infection on viral replication and antiviral immune responses. J Infect Dis. 1999;179:527–537. doi: 10.1086/314628. [DOI] [PubMed] [Google Scholar]
- 5.Palella FJ, Jr, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338:853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 6.Chiasson MA, et al. Declining HIV/AIDS mortality in New York City. J Acquir Immune Defic Syndr. 1999;21:59–64. doi: 10.1097/00126334-199905010-00008. [DOI] [PubMed] [Google Scholar]
- 7.Chun TW, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 8.Chun TW, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci USA. 1997;94:13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chun TW, Fauci AS. Latent reservoirs of HIV: obstacles to the eradication of virus. Proc Natl Acad Sci USA. 1999;96:10958–10961. doi: 10.1073/pnas.96.20.10958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finzi D, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 11.Wong JK, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- 12.Gunthard HF, et al. Evolution of envelope sequences of human immunodeficiency virus type 1 in cellular reservoirs in the setting of potent antiviral therapy. J Virol. 1999;73:9404–9412. doi: 10.1128/jvi.73.11.9404-9412.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L, et al. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. N Engl J Med. 1999;340:1605–1613. doi: 10.1056/NEJM199905273402101. [DOI] [PubMed] [Google Scholar]
- 14.Furtado MR, et al. Persistence of HIV-1 transcription in peripheral-blood mononuclear cells in patients receiving potent antiretroviral therapy. N Engl J Med. 1999;340:1614–1622. doi: 10.1056/NEJM199905273402102. [DOI] [PubMed] [Google Scholar]
- 15.Sharkey ME, et al. Persistence of episomal HIV-1 infection intermediates in patients on highly active anti-retroviral therapy. Nat Med. 2000;6:76–81. doi: 10.1038/71569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramratnam B, et al. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat Med. 2000;6:82–85. doi: 10.1038/71577. [DOI] [PubMed] [Google Scholar]
- 17.Lewin SR, et al. Use of real-time PCR and molecular beacons to detect virus replication in human immunodeficiency virus type 1-infected individuals on prolonged effective antiretroviral therapy. J Virol. 1999;73:6099–6103. doi: 10.1128/jvi.73.7.6099-6103.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dornadula G, et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA. 1999;282:1627–1632. doi: 10.1001/jama.282.17.1627. [DOI] [PubMed] [Google Scholar]
- 19.Ho DD. Toward HIV eradication or remission: the tasks ahead. Science. 1998;280:1866–1867. doi: 10.1126/science.280.5371.1866. [DOI] [PubMed] [Google Scholar]
- 20.Michie CA, McLean A, Alcock C, Beverley PC. Lifespan of human lymphocyte subsets defined by CD45 isoforms. Nature. 1992;360:264–265. doi: 10.1038/360264a0. [DOI] [PubMed] [Google Scholar]
- 21.Mohri H, Bonhoeffer S, Monard S, Perelson AS, Ho DD. Rapid turnover of T lymphocytes in SIV-infected rhesus macaques. Science. 1998;279:1223–1227. doi: 10.1126/science.279.5354.1223. [DOI] [PubMed] [Google Scholar]
- 22.Hellerstein M, et al. Directly measured kinetics of circulating T lymphocytes in normal and HIV-1-infected humans. Nat Med. 1999;5:83–89. doi: 10.1038/4772. [DOI] [PubMed] [Google Scholar]
- 23.Finzi D, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 24.Neumann AU, et al. HIV-1 rebound during interruption of highly active antiretroviral therapy has no deleterious effect on reinitiated treatment. Comet Study Group. AIDS. 1999;13:677–683. doi: 10.1097/00002030-199904160-00008. [DOI] [PubMed] [Google Scholar]
- 25.Harrigan PR, Whaley M, Montaner JS. Rate of HIV-1 RNA rebound upon stopping antiretroviral therapy. AIDS. 1999;13:F59–F62. doi: 10.1097/00002030-199905280-00001. [DOI] [PubMed] [Google Scholar]
- 26.Garcia F, et al. Dynamics of viral load rebound and immunological changes after stopping effective antiretroviral therapy. AIDS. 1999;13:F79–F86. doi: 10.1097/00002030-199907300-00002. [DOI] [PubMed] [Google Scholar]
- 27.Ortiz GM, et al. HIV-1-specific immune responses in subjects who temporarily contain virus replication after discontinuation of highly active antiretroviral therapy. J Clin Invest. 1999;104:R13–R18. doi: 10.1172/JCI7371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davey RT, Jr, et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci USA. 1999;96:15109–15114. doi: 10.1073/pnas.96.26.15109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vesanen M, Stevens CE, Taylor PE, Rubinstein P, Saksela K. Stability in controlling viral replication identifies long-term nonprogressors as a distinct subgroup among human immunodeficiency virus type 1-infected persons. J Virol. 1996;70:9035–9040. doi: 10.1128/jvi.70.12.9035-9040.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang LQ, et al. Selection for specific sequences in the external envelope protein of human immunodeficiency virus type 1 upon primary infection. J Virol. 1993;67:3345–3356. doi: 10.1128/jvi.67.6.3345-3356.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simmonds P, et al. Human immunodeficiency virus-infected individuals contain provirus in small numbers of peripheral mononuclear cells and at low copy numbers. J Virol. 1990;64:864–872. doi: 10.1128/jvi.64.2.864-872.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Higgins DG, Thompson JD, Gibson TJ. Using CLUSTAL for multiple sequence alignments. Methods Enzymol. 1996;266:383–402. doi: 10.1016/s0076-6879(96)66024-8. [DOI] [PubMed] [Google Scholar]
- 33.Felsenstein J. Phylogenies from molecular sequences: inference and reliability. Annu Rev Genet. 1988;22:521–565. doi: 10.1146/annurev.ge.22.120188.002513. [DOI] [PubMed] [Google Scholar]
- 34.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 35.Simmonds P, Balfe P, Ludlam CA, Bishop JO, Brown AJ. Analysis of sequence diversity in hypervariable regions of the external glycoprotein of human immunodeficiency virus type 1. J Virol. 1990;64:5840–5850. doi: 10.1128/jvi.64.12.5840-5850.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu T, et al. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science. 1993;261:1179–1181. doi: 10.1126/science.8356453. [DOI] [PubMed] [Google Scholar]
- 37.Chun TW, et al. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat Med. 2000;6:757–761. doi: 10.1038/77481. [DOI] [PubMed] [Google Scholar]