

Abstract
MMP activity with disruption of structural collagen has been implicated in the pathophysiology of dilated cardiomyopathy. To examine the role of this enzyme in cardiac function, a transgenic mouse was created that constitutively expressed human collagenase (MMP-1) in the heart. At 6 months of age, these animals demonstrated compensatory myocyte hypertrophy with an increase in the cardiac collagen concentration due to elevated transcription of type III collagen. Chronic myocardial expression of MMP-1 produced loss of cardiac interstitial collagen coincident with a marked deterioration of systolic and diastolic function at 12 months of age. This is the first animal model demonstrating that direct disruption of the extracellular matrix in the heart reproduces the changes observed in the progression of human heart failure.
Introduction
Congestive heart failure (CHF) is an important and rapidly growing cause of mortality and morbidity in the United States. Approximately 25,000 people die from CHF, with more than 875,000 hospitalizations annually in the United States alone (1). Although the primary underlying defect is an inability of the left ventricle to maintain adequate forward stroke volume, the etiology of this disease is complex and often undetermined (2). The progression of CHF is characterized by left ventricular remodeling coincident with altered neurohormonal signaling and ultimately cardiac muscle dysfunction (2). In addition to cardiac muscle dysfunction, the gradually progressive nature of CHF is associated with changes in the extracellular matrix (ECM) of the heart, leading to alterations in both the geometry and mechanical properties of this organ. This dynamic structural remodeling has a significant impact on overall cardiac function and is an important component of the progression of CHF (3).
Animal models have provided significant insight into the pathophysiology of CHF and in the treatment of this disease (4). In particular, the recent development of techniques specifically to alter gene expression in transgenic mice has added greatly to our understanding of heart failure (5). Because ventricular remodeling is a dynamic and slowly progressive process, the animal model is ideal for examining both the structural and functional changes that occur over time. The biologic response over time is not always uniform, and serial temporal analysis of transgenic animals with specifically targeted gene expression is one means toward understanding the evolutionary progression of CHF (5). Previous animal models of cardiomyopathy have studied alterations in the cardiac cytoskeleton and myocyte structural apparatus, ion channel function, neurohormonal activation, and cytokine/growth factor expression (4). However, none of these transgenic models has directly examined the role of the cardiac ECM in the development and progression of heart failure.
Although the cardiac myocytes are central to the contractile function of the myocardium, the cardiac interstitium and its fibrillar collagen matrix also play a critical role in determining cardiac performance (6). A network of collagen fibers serves a number of functions important to the overall function of the heart. Cardiac collagen provides supportive scaffolding for myocytes, myofibrils, and the vasculature critical to maintaining the structural integrity and overall geometry of the heart. The ECM tethers individual myocytes and myofibrils together in proper alignment, enabling optimal transduction of coordinated force generated by cardiac contraction. In addition, interstitial collagen provides tensile strength to the myocardium and is an important determinant of diastolic and systolic stiffness within the heart (3).
The remodeling of this matrix is critical in the progression of heart failure, yet very little direct evidence exists to demonstrate that disruption of this matrix leads to cardiac dysfunction. In the end stages of CHF, upregulation of MMP enzymatic activity and loss of collagen cross-linking can be observed (7). MMPs are a family of proteolytic enzymes capable of degrading a wide variety of the ECM (for review, see ref. 8). Fibrillar collagens are specifically degraded by interstitial collagenase, MMP-1, present in the heart in its enzymatically active form (9–11). Alterations in the balance of matrix synthesis and degradation through changes in protease activity and expression are postulated to be crucial to the process of ventricular remodeling and in the pathophysiology of chronic heart failure (12). Pharmacologic inhibition of MMP activity has been shown to attenuate the process of ventricular remodeling seen after experimental myocardial infarction in the mouse (13) and in the rapid cardiac pacing pig model of congestive heart failure (14). However, there is no direct evidence demonstrating that degradation of the ECM through increased activity of MMPs leads to the development of heart failure.
The alteration in the balance between synthesis and degradation of the ECM can be generated through the use of the transgenic mouse system. By specifically targeting expression of interstitial collagenase (MMP-1) to cardiac muscle, the change in cardiac remodeling over time can be examined. The present study provides the first demonstration to our knowledge that alterations in the balance of synthesis and degradation of interstitial collagen through the activity of collagenase and the disruption of the myocardial ECM lead to cardiac dysfunction.
Methods
Generation of α-MHC-collagenase.
The construct clone 20 containing the α-MHC promoter fragment was digested with Sal1, filled in with Klenow (15) and then digested with Not1. The 5.5-kb Not1/blunt end fragment was isolated (15). A Sma1/Sal1 9.3-kb genomic MMP-1 gene fragment was then ligated to the promoter at a Sma1 site that had been artificially introduced into the MMP-1 gene through in vitro mutagenesis (16) and immediately precedes the translational initiation site. The resulting plasmid, pJ1360, was confirmed by restriction enzyme analysis. In addition, the α-MHC-MMP-1 gene junction consisting of 356 bp of the 3′ end of the α-MHC and 222 bp of the 5′ end of the MMP-1 gene was sequenced. The translation start site and the reading frame of the MMP-1 gene were found to be intact, and Kozak’s rules for translational efficiency were maintained (17). Transcription initiation occurs within the α-MHC promoter.
DNA fragment preparation and microinjection.
To remove prokaryotic sequences that sometimes inhibit transgene expression (18), the 15.0-kb transgene was isolated from pJ1360 by Not1/Sal1 enzymatic digestion (12 bp and 17 bp of prokaryotic sequence remained, respectively), purified by CsCl centrifugation, and microinjected into fertilized mouse eggs (F1[C57BL/6 × CBA/J] × F1 [C57BL/6 × CBA/J]) (19).
Southern blot analysis of DNA.
Approximately 3 weeks after birth, DNA from the tails of pups was prepared and Southern blot analysis was performed (19). Briefly, 2 cm of clipped tail tip was digested overnight with 350 μg proteinase K in a buffered solution (50 mM Tris-HCl [pH 8.0], 100 mM EDTA, and 0.5% SDS) at 60°C. Protein and cellular debris were removed by serial phenol/sevag extractions. DNA was precipitated from the supernatant with 250 mM NaOAc (pH 6.0) and ethanol at room temperature. A total of 10 μg of DNA was cleaved with Hind III, separated on a 0.6% agarose gel, and transferred to 0.45 μm nitrocellulose (Trans-blot Transfer Membrane; Bio-Rad Life Science Research Group, Hercules, California, USA) (20). Blots were prehybridized with 50% formamide, 5× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), 10× Denhardt’s solution, 0.1 mg/ml sonicated denatured salmon sperm DNA. 0.2% SDS, 0.05 M sodium phosphate buffer (pH 6.8), and 1 mM EDTA for 2 hours at 42°C. Hybridization was performed for 16 hours at 42°C in 50% formamide, 5× SSC, 1× Denhardt’s solution, 0.02 M sodium phosphate buffer (pH 6.8), 0.1 mg/ml sonicated denatured salmon sperm DNA, 1 mM EDTA, 0.2% SDS, and 10% dextran sulfate with 5.0 × 108 cpm of a nick translated 32P-labeled probe (15) specific for the α-MHC-collagenase construct. Filters were washed at 65°C with 2× SSC containing 0.5% SDS for 3 hours followed by a wash in 0.2× SSC and 0.5% SDS for 40 minutes before exposure to x-ray film at –70°C.
Northern blot analysis of mRNA.
Total RNA was prepared from mouse tissue using the guanidine thiocyanate-cesium chloride method (21). Total RNA (10 μg) was resolved by electrophoresis on a 1.2% agarose gel containing 0.2% formaldehyde and blotted to nylon membranes (Hybond-N; Amersham Pharmacia Biotech, Buckinghamshire, United Kingdom). These membranes were hybridized with the respective probes. The transgene was used as a probe for MMP-1, the mouse cDNA probe for MMP-9 was a kind gift of Y. Okada (Keio University, Tokyo, Japan), and the probes for MMP-13 and MMP-3 were obtained by PCR amplification of mouse macrophages. The 5′ and 3′ primers used were 5′-CTCGAGCATGCTTCCTGATGATGACGTT-3′ and 5′-CTCGAGCCCCACCCCATACATCTGAAA-3′, respectively, for MMP-13 and 5′-GAATTCTGGGCTATACGAGGGCACGA-3′ and 5′-GAATTCGCACTTC CTTTCACAAAGACTC-3′, respectively for MMP-3. The resulting PCR products were subcloned into a pGEM-T Easy vector (Promega Corp., Madison, Wisconsin, USA) and inserts confirmed by sequencing. The cDNA probes corresponding to MMP-3 (432 bp) and MMP-13 (943 bp) were excised by digestion with EcoR1 and Not1, respectively. The membranes were prehybridized with 50% formamide, 5× SSC, 10× Denhardt’s solution, 0.1 mg/ml sonicated denatured salmon sperm DNA, 0.2% SDS, 0.05 M phosphate buffer (pH 6.8), and 0.001 M EDTA for 3 hours at 42°C. Hybridization was performed for 16 hours at 42°C in 50% formamide, 5× SSC, 1× Denhardt’s solution, 0.02 M phosphate buffer (pH 6.8), 10% dextran sulfate, 0.1 mg/ml sonicated denatured salmon sperm DNA, 0.001 M EDTA, and 0.2% SDS with 1 × 108 cpm of nick translated 32P-labeled probe specific for the target RNA. Membranes were washed at 65°C with 2× SSC containing 0.5% SDS for 1 hour before exposure to x-ray film at –70°C. Membranes were rehybridized with a complementary radiolabeled 24-mer oligonucleotide to mouse 28S rRNA (AAC GAT GAG AGT AGT GGT ATT TCA) to ensure that equal amounts of RNA were loaded into each lane (22).
In-situ hybridization for MMP-1 expression.
Cardiac tissue of both a wild-type mouse and a transgenic littermate was fixed in 4% paraformaldehyde and embedded in paraffin. Histological sections were probed with a sense and antisense MMP-1 probe. Linearized MMP-1 cDNAs were labeled with digoxigenin by in vitro transcription with T7 and T3 polymerase by using a DIG RNA Labeling Kit (Roche Molecular Biochemicals, Indianapolis, Indiana, USA). After hybridization, specimens were treated with AP-conjugated antidigoxigenin polyclonal IgG. The bound antibody was detected by adding NBT/BCIP as substrate.
Western blot analysis.
The hearts from transgenic and control mice were denuded of pericardium, and the lower two-thirds portion of the ventricles was excised. For each sample, tissue was placed directly into 1 ml of nonreducing PAGE sample buffer (2% SDS, 20% glycerol, 0.2 M Tris), homogenized, sonicated briefly, and then centrifuged at 2,400 g for 20 minutes. A 200-μl portion of the supernatant was added to 15 μl of 1 M DTT, boiled for 15 minutes, and then normalized with Bio-Rad Protein assay as per instructions (Bio-Rad Life Science Research Group). Equal amounts of protein were resolved by SDS-PAGE on a 10% gel and transferred to a nitrocellulose membrane (Micron Separations Inc., Westborough, Massachusetts, USA). The membrane was blocked with 5%(wt/vol) dry milk in PBT (0.1% [wt/vol] Tween in PBS) at room temperature for 4 hours, then probed with polyclonal antibody against MMP-1 at a 1:1,000 dilution for 1 hour at room temperature. The membrane was subjected to three 5-minute washes with PBT and then probed with Protein A conjugated Horseradish Peroxidase (Roche Molecular Biochemicals) at 1:20,000 dilution in PBT + 1% BSA for 1 hour at room temperature. The membrane was washed once again three times with PBT for 5 minutes each wash. Proteins were then visualized with ECL per manufacturer’s instructions (Pierce Chemical Co., Rockford, Illinois, USA).
Collagenase ELISA and activity analysis.
ELISA assays were performed on protein homogenate of cardiac tissue from both wild-type and littermate transgenic mice. Animals were sacrificed, and the heart was removed and homogenized in TNC buffer (50 mM Tris-HCl [pH 7.5], 0.15 M NaCl, 10 mM CaCl2, 0.05% Brij 35, and 0.02% NaN3) using a polytron homogenizer. The homogenate was centrifuged to sediment any particulate matter. The samples were concentrated using Amicon Centricon-30 columns (Millipore Corp., Bedford, Massachusetts, USA) to an approximate volume of 1–3 ml. The total protein concentration was determined using the standard BCA method (Pierce Chemical Co.). Total heart protein (20 μg) was used in an MMP-1 ELISA assay to determine the levels of MMP-1 protein using the Biotrak Human MMP-1 ELISA system (Amersham Life Sciences Inc., Arlington Heights, Illinois, USA). The assay was performed according to the manufacturer’s protocol. This assay is a “sandwich” format in which polyclonal and monoclonal MMP-1 antibodies are used with a horseradish peroxidase detection method. The lower limit of detection for the assay is 1 ng/ml.
Two lines (WH 1 and WH 11) were examined for the presence of MMP-1 activity in the heart. Protein homogenate was prepared as already described here. The MMP-1 assays were performed on the supernatant after the addition of 10 mM CaCl2. Because MMP-1 is secreted in a zymogen form, the supernatant was treated with 1.5 mM 4-aminophenylmecuric acetate for 5 hours at 37°C to activate the enzyme. The assay was performed as follows. Rat tail type I collagen (Collaborative Biomedical Products, Becton Dickinson, Bedford, Massachusetts, USA) was labeled using [14C] acetic anhydride (23) and then dialyzed against 0.1 M Tris-HCL (pH 7.5), 0.15 M NaCl, and 0.2% NaN3. The collagen gel assay was performed using 10 μl of each sample (20 μg) incubated at 27°C with 5 μl of the rat tail type I collagen (3 μg/μl) in a total volume of 20 μl in a buffer of TNC. The digestion products were analyzed by SDS-PAGE (9% total acrylamide). MMP-1 is extremely specific for fibrillar collagen and cleaves native type I collagen at residue numbers 775–776 of the α(I) chain to generate three-quarter– and one-quarter–size fragments (24) that are easily recognizable on polyacrylamide gel analysis.
L-HYDROXYPROLINE ASSAY. L-Hydroxyproline concentration in the hearts of the transgenic and wild-type mice was determined using a modified Woessner protocol (25, 26). The heart tissue was lyophilized for 12 hours, and the left ventricle was isolated by shaving away the great vessels, the right ventricle, and both atria. The left ventricle was then minced, weighed, and hydrolyzed with 4 ml 6N HCl at 125°C at 200 PSI in an autoclave for 3 hours. One milliliter of the hydrolysate was evaporated and reconstituted with 1 ml distilled H2O and then re-evaporated. Reconstitution was then done with 5 ml of distilled H2O. Hydroxyproline standard solutions of 0, 1, 2, 4, 6, 8, and 10 mg/ml were made. Sample solution (2 ml) was taken and oxidized with 1 ml of Chloramine-T (Sigma, St. Louis, Missouri, USA) solution for 20 minutes. The Chloramine-T was then destroyed with 1 ml of 3.15 M perchloric acid. After 5 minutes, 1 ml of p-dimethylaminobenzaldehyde solution was added. The sample was vortexed, incubated in a 60°C bath, and then cooled under tap water for 5 minutes. The absorbency of the solutions was determined at 557 mm. The hydroxyproline concentration was determined directly from the standard curve.
Histological analysis.
Hearts were arrested in diastole with PBS/20 mM KCl solution and pressure fixed at 20 mmHg with 4% paraformaldehyde or 10% neutral buffered formalin. Paraffin-embedded tissues were sectioned (8-μm thick) and stained with hematoxylin and eosin for light microscopy. Paraffin sections were also stained with picrosirius red and analyzed with a polarized light microscope to evaluate the distribution of myocardial collagen (27). Using an electron microscope, transgenic and wild-type littermates were anesthetized, and hearts at 3 and 6 months were pressure fixed in diastole with PBS/20 mM KCl solution. The hearts were perfused with 2.5% glutaraldehyde until the heart turned white. The hearts were minced and then placed in 2.5% glutaraldehyde for 24 hours, rinsed with PBS, and postfixed in 1% osmium solution. Sections from the left ventricular free wall were prepared for viewing with a transmission electron microscope.
Hemodynamic analysis.
In vivo intraventricular hemodynamic analysis was performed on transgenic and wild-type littermate mice at the 6- and 12-month time points. A total of 33 mice (6 months: ten wild-type and seven transgenic; 12-months: nine wild-type and seven transgenic) were anesthetized with 2.5% Avertin 0.015 ml/g body weight (19). A midline incision in the neck exposed the trachea, and the mouse was intubated intratracheally with a 22-gauge angiocatheter (Becton Dickinson) which was secured with a 3-0 silk suture (USSC). The mouse was mechanically ventilated with 0.5 cc ambient air tidal volume at 110 breaths per minute with a small animal respirator/ventilator (Columbus Instruments, Columbus, Ohio, USA). A median sternotomy was performed, and the heart was exposed. Digitized intraventricular hemodynamic measurements were obtained via a left ventricle apical puncture with a 26-gauge fluid-filled angiocatheter (Becton Dickinson) attached to a high-fidelity pressure transducer that was connected to an eight-channel chart recorder set at 1,000 Hz (MacLab 8s; ADInstruments, Mountain View, California, USA). The data were stored on a computer for subsequent analysis (PowerMac 5300C; Apple Computer Inc., Cupertino, California, USA).
Results
Generation of transgenic mice.
The α-MHC promoter has been shown previously to target gene expression within transgenic mice specifically to the myocyte in the heart (28). The α-MHC-collagenase (MMP-1) transgene was generated through removal of all sequences 5′ to the translation initiation codon of the full-length human interstitial collagenase gene (16) and placed under the control of the human α-MHC promoter fragment to dictate cardiac-specific expression of the transgene. Sixteen live mice were born of which four carried the transgene. All founder transgenic mice transmitted the transgene as a single integration, and lines were established.
The tissue specificity of transgene expression was investigated in hemizygous transgenic mice from the four lines harboring the α-MHC-collagenase transgene. RNA was isolated (21) and analyzed by Northern blot analysis (29). Specific expression of collagenase was observed only in the hearts from all four transgenic mouse lines (Figure 1). As expected, human collagenase MMP-1 was not expressed in the hearts of wild-type mice (data not shown).
Figure 1.

Cardiac-specific expression of MMP-1 transgene. Northern blot analysis was performed using 10 μg total RNA isolated from the indicated tissues of transgenic lines WH 6, 11, 1, and 15 and probed with the transgene. The lower panel shows the same membrane probed with ribosomal 28S oligonucleotide to ensure that equal amounts of RNA were loaded in each lane.
Cardiac myocyte–specific expression of interstitial collagenase by the transgene was demonstrated by in situ hybridization (Figure 2). Using a probe specific for human MMP-1, staining was seen in the cardiac myocytes of the transgenic hearts when hybridized with the antisense probe (Figure 2d). Staining was not seen when using the sense probe or in the wild-type heart with the antisense probe (Figure 2, a–c).
Figure 2.
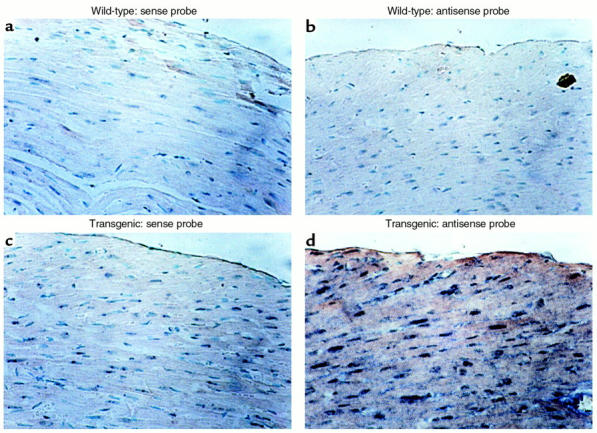
Localization of the MMP-1 transgene expression to cardiac myocytes by in situ hybridization. Photomicrographs (± 200) of transverse sections through the anterior wall of the heart of a wild-type mouse (a and b) and a transgenic littermate (c and d) from line WH 1, probed with the sense MMP-1 probe (a and c) and probed with the antisense MMP-1 probe (b and d). Staining can be seen only in the myocytes of the transgenic heart (d).
Once the transgene was shown to be transcriptionally active, the presence of MMP-1 protein was documented by ELISA assays performed on cardiac tissue homogenate from both wild-type and littermate transgenic mice. The total protein in the transgenic heart determined to be interstitial collagenase MMP-1 ranged from 30 ng/mg (line WH 11) to 300 ng/mg (line WH 1). The control littermates did not have any detectable MMP-1 collagenase. Collagenase activity was detected in the tissue homogenate from the transgenic hearts and not in the homogenate of hearts from the wild-type mice (Figure 3a). To determine the presence of active MMP-1, Western blot analysis was performed revealing the presence of inactive and active MMP-1 in the transgenic mouse heart (Figure 3b). The expression of the MMP-1 transgene did not appear to affect the endogenous expression of three MMPs known to be expressed in the heart, MMP-3, -9, and -13 (data not shown). In addition, gelatin zymography on heart protein extracts did not demonstrate any significant difference in protease activity between the transgenic and wild-type hearts (data not shown).
Figure 3.

(a) Presence of MMP-1 protein in transgenic mouse hearts. Western blot analysis was performed on ventricular extracts from 3-month-old transgenic mice (+) and control littermates (–) as described in Methods, and normalized for protein content. Equal amounts of protein were resolved by SDS-PAGE and subject to immunoblotting with anti–MMP-1 polyclonal antibodies. Arrows indicate the active and pro forms of MMP-1. (b) Collagenase activity in the homogenate from the heart of transgenic and control mice. Type I collagen was incubated with homogenate from the heart of a transgenic mouse treated with APMA (TG+) and without APMA (TG–) and the homogenate from the heart of a control littermate treated with APMA (WT+) and without APMA (WT–). As a control, 100 ng of active MMP-1 was incubated with type I collagen (MMP-1). The characteristic collagenase (one-quarter) degradation fragment can be seen only in the APMA-treated homogenate from the transgenic heart (TG+) and the positive control (MMP-1).
Phenotypic and histologic analysis of cardiac-collagenase mice.
The transgenic mice appeared normal at birth until weaning when compared with their wild-type littermates. However, the transgenic mice had a 20% increased mortality rate over 6 months when compared with their wild-type littermates. Grossly, the lungs and liver appeared normal with no pathological changes suggestive of pulmonary or hepatic congestion. At the six-month time point, the heart wet weight/body weight from the transgenic mice was increased (0.593 μg/g ± 0.006) when compared with age-matched control littermates (0.445 μg/g ± 0.002; P < 0.001). However, there was no statistically significant difference observed at the 1-year time point.
In the transgenic heart, hematoxylin and eosin staining revealed increased left ventricular wall thickness with a decrease in chamber size compared with the wild-type heart at the 3-month time point (Figure 4, a and b). Transgenic hearts contained enlarged hyperchromatic myocyte nuclei with increased myofibrillar width consistent with cardiac hypertrophy (Figure 4, c and d). There was no evidence of inflammatory infiltrate or myonecrosis in the transgenic hearts analyzed up to the 12-month time point. Morphometric analysis demonstrated that the myocyte cross-sectional area was increased in the transgenic mouse heart (199.6 ± 61.8 μm2) when compared with wild-type hearts (144.1 ± 35.5 μm2). The increase in the number of myocyte nuclei and the large irregular shaped myocyte nuclei in the transgenic heart (Figure 4d) is a well-described characteristic feature of myocyte hypertrophy (30, 31).
Figure 4.
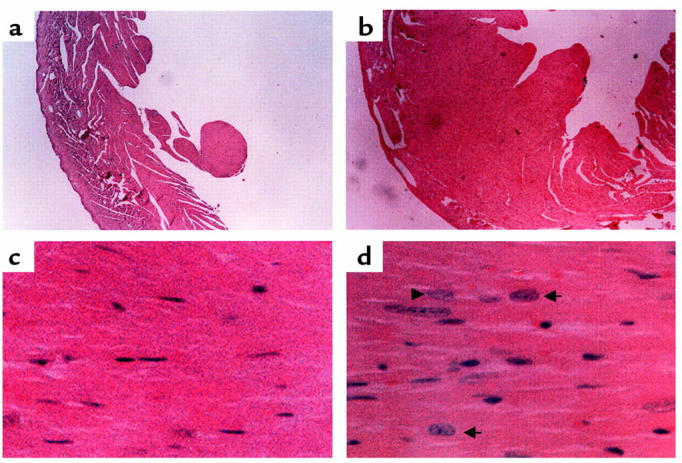
Histological analysis of mouse hearts from age-matched (3-month-old) littermates. Left ventricle from wild-type (a and c) and transgenic (b and d) hearts in cross-section. Note the increased thickness of the left ventricular free wall in the transgenic heart (b) compared with the wild-type (a). Enlarged hyperchromatic myocyte nuclei (arrows) with increased myofibrillar width consistent with cardiac hypertrophy is seen in the transgenic heart (d) compared with wild-type (c). Hematoxylin and eosin stains were used. a and b, ×40; c and d, ×400.
Ultrastructural analysis was performed on sections from transgenic and littermate wild-type mouse using transmission electron microscopy. Again, at the 6-month time point, transgenic hearts revealed widened myofibrils, thickened Z-bands with breakdown of normal Z-band registration, and myofibrillar disarray with disruption of sarcomeric architecture. Transgenic hearts contained numerous enlarged pleomorphic mitochondria distributed in a disorderly manner consistent with hypertrophy and reflective of the increased metabolic demands of this tissue (Figure 5).
Figure 5.

Representative transmission electron micrographs of wild-type and cardiac-collagenase transgenic mice. Ultrastructural analysis was performed on wild-type (a and c) and transgenic (b and d) hearts. The wild-type heart (a and c) shows an orderly row of myofibrils with neatly interposed mitochondria and sharp closely packed cristae. The transgenic heart (b and d) reveals changes consistent with pathological hypertrophy characterized by wider myofibrils, thicker Z-bands with breakdown of normal Z-band registration and myofibrillar disarray with disruption of the sarcomeric architecture. Note the numerous enlarged pleomorphic mitochondria distributed in a disorderly manner, all consistent with hypertrophy and reflective of the enormous metabolic demands of this tissue.
Collagen in the transgenic heart.
Picrosirius red staining was performed to evaluate the collagen content in the hearts of wild-type and littermate transgenic mice. Twelve-month-old transgenic hearts compared with littermate wild-type hearts revealed less birefringence staining under polarized light microscopic analysis consistent with decreased myocardial interstitial collagen in the transgenic mice (data not shown). Quantitative collagen analysis was performed using a hydroxyproline assay (25, 26). Despite constitutive interstitial collagenase expression in the transgenic heart, hydroxyproline concentration was greater in the transgenic heart compared with the wild-type littermate at the 6-month time point (Table 1). However, at the 12-month time point, the hydroxyproline concentration in the transgenic heart was significantly reduced compared with the 12-month-old wild-type heart and the transgenic heart at the 6-month time point (Table 1). There was no significant difference in the wild-type hearts comparing 6- and 12-month time points.
Table 1.
Hydroxyproline concentration (mg/g heart, dry wt) in transgenic and wild-type littermate hearts at 6 and 12 months

To determine whether the increased collagen concentration was the result of increased type I and/or type III collagen transcription, a Northern blot analysis was performed using cDNA probes specific for collagens type I and III on RNA obtained from 1.5- and 6-month– and 1-year–old transgenic and wild-type mice. At the 1.5-month time point, type III collagen expression was increased in the cardiac collagenase transgenic mice twofold when compared with wild-type littermates. This difference was no longer apparent at 6 months of age, with a slight increase in transcription of collagen III seen at 1 year of age (Figure 6). No differences in the expression of type I collagen were observed between transgenic and wild-type littermates at any of the time points examined (Figure 6).
Figure 6.

Upregulation of collagen type III but not collagen type I in cardiac-collagenase mice. Northern blot analysis was performed using 10 μg total RNA from heart tissue from transgenic (line WH 1) and wild-type littermate mice at 6-week, 6-month, and 12-month time points and probed with an xba I/Hind III fragment of the collagen type III cDNA (top). Increased collagen III message was detected in the transgenic mice compared with the wild-type littermates at the 6-week time point. No difference was noted by the 6- month or 12-month time point. The same membrane was probed with a Hind III fragment of the collagen type I cDNA (middle). No differences in type I collagen expression were observed between wild-type and transgenic mice at any time point. The lower panel shows the same membranes probed with ribosomal 28S oligonucleotide to ensure that equal amounts of RNA were loaded in each lane. WT, wild-type, TG, transgenic.
Functional evaluation of cardiac-collagenase transgenic mice.
To examine the functional consequences of constitutive collagenase expression in the heart, we performed in vivo intraventricular hemodynamic analysis on transgenic and wild-type littermate mice at the 6- and 9-month time points. Although the heart rates of transgenic mice were lower than those of wild-type mice at the 6-month time point, the left ventricular pressures generated in the transgenic mice were significantly increased by 16.3 mmHg at the 6- month time point compared with age-matched wild-type mice (Table 2) (Figure 7a). At the 12-month time point, however, the left ventricular pressure measured in the transgenic mice was 9.3 mmHg lower than in comparison age-matched wild-type mice, indicating a significant deterioration in contractile function. Consistent with the demonstration of severe temporal functional deterioration in the transgenic mice was the marked diminution in the rate of pressure development and relaxation observed in the transgenic mice comparing 6- and 12-month time points. The maximum first derivative of left ventricular pressure (dP/dT max) was reduced by 46.9%. The minimum first derivative of left ventricular pressure (dP/dT min) was reduced by 30.6% (Table 2) (Figure 7, b and c). There were no significant temporal changes in contractile or relaxation function indices in the wild-type mice.
Table 2.
Hemodynamic indices

Figure 7.

Hemodynamic measurements performed in wild-type and transgenic mice. Open chest measurements taken with a high-fidelity fluid-filled pressure transducer via a left ventricular apical puncture. (a) Increased peak left ventricular systolic pressure in transgenic mice from line WH 1 compared with controls at 6 months. (a–c) Significant temporal reduction in left ventricular systolic and diastolic function in transgenic mice from line WH 1 comparing 6- and 12-month time points. Control and transgenic mice were compared using unpaired Student’s t test. Values are means ± SEM.
Discussion
This study demonstrates that destruction of the collagen network in the myocardium of transgenic mice produces an initial adaptive response with myocardial hypertrophy and deposition of collagen. Prolonged expression of cardiac MMP-1 subsequently leads to loss of interstitial collagen with significant deterioration of contractile function. This animal model accurately mimics the human condition in that an initial adaptive response is seen, followed by progressive loss of function. In addition, this transgenic animal model for the first time provides direct evidence that MMA and disruption of the equilibrium of ECM synthesis and degradation can play a critical role in the process of cardiac remodeling and the pathophysiology of heart failure.
Although numerous transgenic mouse models have generated cardiac pathology through targeted perturbation of the myocyte cytoskeleton, myocardial metabolism and regulation, and the overexpression of cytokines (32, 33), this is the only transgenic animal directly demonstrating the role of the ECM in determining cardiac performance. The present model is unique in that it reproduces many of the evolutionary changes seen in the development of pressure-overload hypertrophy through the critical transition to cardiac dysfunction. This animal provides new insight into the pathophysiology of ventricular remodeling and heart failure and establishes the significance of the balance of ECM synthesis and degradation in the progression to cardiac dysfunction.
Multiple studies have now demonstrated that rodents do not possess the mouse homologue of the MMP-1 gene and MMP-13 is believed to provide the necessary collagenolytic activity in rodents (34). The enzyme MMP-1 expressed in this transgenic animal is identical immunologically and functionally to the collagenase demonstrated to be elevated in the heart tissue of patients with ischemic and dilated cardiomyopathy (35–37). In addition, other animal models of cardiac injury and dysfunction support the activity of MMP-1 in the remodeling process and its impact on function (38–42). Therefore, this heart failure model is strengthened as a homologue of the human disease, as it examines the effect of MMP-1 on the effectively null rodent background and mouse fibrillar collagen is susceptible to degradation by the human enzyme (43). The phenotype seen in these mice is not due to the upregulation of endogenous mouse MMP-3, -9, or -13, as expression levels in the transgenic hearts did not change for these proteases. It is possible that MMP-13 is solely responsible for normal matrix turnover and that MMP-1 is more important during the disease state. It is only through the generation of an MMP-13–overexpressing transgenic mouse that the differences in these two enzymes can be better defined.
The hearts from the cardiac-collagenase mice revealed myocardial hypertrophy at both the light and transmission electron microscope levels similar to changes seen in pressure-overload hypertrophy. However, myocyte hypertrophy was an unexpected finding in these mice, as increased collagenase expression would not have been expected to directly alter the myocyte. Myocardial hypertrophy is a well-known compensatory response of the left ventricle to excessive hemodynamic loads. Breakdown of the surrounding structural cardiac extracellular matrix in this transgenic mouse may produce added hemodynamic stress in the heart leading to the development of compensatory myocyte hypertrophy. Alternatively, disruption of the cardiac matrix may disrupt signaling between myocytes mimicking the hypertrophic state of the diseased heart.
These cardiac MMP-1 transgenic mice undergo a dynamic change in their collagen concentration over time due to the constitutive expression of MMP-1. First, there is a reactive increase in the hydroxyproline concentration at the 6-month time point followed by a decrease in hydroxyproline when the initial reactive phase has subsided. The initial increased production of collagen is consistent with the increase in transcription of type III collagen seen in the transgenic mouse. The elevated collagen concentration in the transgenic heart may represent a reactive fibrosis in response to collagen degradation from cardiac MMP-1 expression, which is consistent with the experiments of Weber and colleagues (44). Deposition of new collagen in response to upregulation of collagenolytic activity is an important part of the overall process of tissue remodeling (45). An increase in type III collagen expression can be seen during other remodeling states (46, 47) (44, 47–51) including ischemic cardiomyopathy (52). Stimulation of fibroblast collagen synthesis by exposure to collagen degradation products has also been demonstrated (53). Therefore, the generation of collagen degradation fragments in the heart by MMP-1 in the cardiac-MMP-1 transgenic mice may actually stimulate the enhanced collagen synthesis seen at the 6-month time point.
Although dynamic differences in collagen content are present in the heart of these transgenic mice these alterations alone do not necessarily explain the changes seen in cardiovascular function. Disruption of cell-ECM attachments through the activity of MMP-1 and the development of myocyte hypertrophy may activate intracellular signaling cascades affecting cell cycle and ECM molecule synthesis. Proteolytic modification and activation of cell surface proteins and receptors is postulated to liberate growth factors, such as TGF-α initiating downstream signaling pathways known to stimulate collagen synthesis (54, 55). Accordingly, the physical changes themselves induced by MMP-1 may play an important role in modulating ECM homeostasis. In addition, the phenotype seen in these mice may not be mediated through collagen degradation by MMP-1. It is possible that the disruption of a yet unidentified substrate of MMP-1 could generate the cardiac pathology seen in this transgenic animal. Although fibrillar collagen is clearly the major substrate of MMP-1 other proteins such as IL-1β have reportedly been targets of this enzyme (56). In addition, transgenic mice that express MMP-1 in the epidermis of the skin exhibit a distinct phenotype despite the lack of fibrillar collagen in the epidermis suggesting an alternative substrate for this enzyme (57).
Remarkably, functional hemodynamic evaluation of these animals correlated with the concentration of hydroxyproline in the heart. During the evolution of cardiac hypertrophy in response to hemodynamic stress, a several-fold increase in cardiac collagen content is seen in both animal models and humans (58). In the absence of myocyte necrosis, early adaptive ventricular hypertrophy and remodeling is associated with increased systolic stress-strain relations indicative of enhanced systolic force–generating capacity and performance. These findings are consistent with those reported in rat (59, 60), dog (61), and primate (44, 61) hypertrophy animal models. As hypertrophy progresses into the established phase; however, further deposition of reparative and replacement fibrosis occurs with the development of increased systolic and diastolic stiffness and dysfunction (62).
The results in this study are consistent with human studies that demonstrate the importance of the ECM and perturbation of the protease/antiprotease balance in the pathophysiology of cardiac dysfunction and congestive heart failure. Endomyocardial biopsies and explanted hearts from patients with idiopathic dilated cardiomyopathy reveal extensive changes in the collagenous framework of the heart (63). Although collagen content and concentration are increased in these hearts, reflecting significant reparative and replacement fibrosis, there is a remarkable decrease in the amount of stable mature pyridinoline cross-linking present. In addition, collagenolytic and gelatinolytic activity in these hearts is increased 30-fold (7). Other investigators have also reported similar findings in explanted heart samples from patients with idiopathic dilated cardiomyopathy and ischemic cardiomyopathy (35–37).
Although the role of matrix homeostasis and MMP activity in the acute and chronic phases of remodeling in myocardial infarction and the response to pressure-overload is not fully understood, this transgenic mouse with a perturbation in a pathway critical to maintaining ECM homeostasis provides important insight into the progression of compensated cardiac hypertrophy to decompensated cardiomyopathy. The dynamic functional changes in these animals are similar to those seen in humans with heart disease. In contrast to the acute animal models of cardiac dysfunction, this model more accurately represents the chronic progressive nature of the human disease. This transgenic animal model will help identify new avenues targeting ECM metabolism in the prevention and treatment of ischemic and dilated cardiomyopathy and provide an organismal model in which the action and efficacy of potentially novel therapeutic interventions can be tested.
Acknowledgments
This work was supported in part by an NIH RO1 grant AG16994-0 (J. D’Armiento) and an NIH National Research Service Award (H.E. Kim) through the Department of Nutrition, Columbia University. J. D’Armiento is a recipient of a Burroughs Wellcome Fund Career Award in the Biomedical Sciences. The authors acknowledge the critical reading of the manuscript provided by K. Chada and A. Marks.
References
- 1.O’Connell JB, Bristow MR. Economic impact of heart failure in the United States: time for a different approach. J Heart Lung Transplant. 1994;13:S107–S112. [PubMed] [Google Scholar]
- 2.Katz, A.M. 1992. Heart failure. Raven Press. New York, New York, USA. 200–280.
- 3.Weber KT. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol. 1989;13:1637–1652. doi: 10.1016/0735-1097(89)90360-4. [DOI] [PubMed] [Google Scholar]
- 4.Hasenfuss G. Animal models of human cardiovascular disease, heart failure and hypertrophy. Cardiovasc Res. 1998;39:60–76. doi: 10.1016/s0008-6363(98)00110-2. [DOI] [PubMed] [Google Scholar]
- 5.Franz WM, Mueller OJ, Hartong R, Frey N, Katus HA. Transgenic animal models: new avenues in cardiovascular physiology. J Mol Med. 1997;75:115–129. doi: 10.1007/s001090050096. [DOI] [PubMed] [Google Scholar]
- 6.Pelouch V, Dixon IMC, Golfman L, Beamish RE, Dhalla NS. Role of extracellular matrix proteins in heart function. Mol Cell Biochem. 1994;129:101–120. doi: 10.1007/BF00926359. [DOI] [PubMed] [Google Scholar]
- 7.Gunja-Smith Z, Morales AR, Romanelli R, Woessner JF. Remodeling of human myocardial collagen in idiopathic dilated cardiomyopathy. Role of metalloproteinases and pyridinoline cross-links. Am J Pathol. 1996;148:1639–1648. [PMC free article] [PubMed] [Google Scholar]
- 8.Birkedal-Hansen H, et al. Matrix metalloproteinases: a review. Crit Rev Oral Biol Med. 1993;4:197–250. doi: 10.1177/10454411930040020401. [DOI] [PubMed] [Google Scholar]
- 9.Caulfield JB, Wolkowicz P. Inducible collagenolytic activity in isolated perfused rat hearts. Am J Pathol. 1988;131:199–205. [PMC free article] [PubMed] [Google Scholar]
- 10.Chakraborty A, Eghbali M. Collagenase activity in the normal rat myocardium. An immunohistochemical method. Histochemistry. 1989;92:391–396. doi: 10.1007/BF00492496. [DOI] [PubMed] [Google Scholar]
- 11.Montfort I, Perez-Tamayo R. The distribution of collagenase in normal rat tissues. J Histochem Cytochem. 1975;23:910–920. doi: 10.1177/23.12.172556. [DOI] [PubMed] [Google Scholar]
- 12.Weber K, et al. Remodeling and reparation of the cardiovascular system. J Am Coll Cardiol. 1992;20:3–16. doi: 10.1016/0735-1097(92)90130-f. [DOI] [PubMed] [Google Scholar]
- 13.Rohde LE, et al. Matrix metalloproteinase inhibition attenuates early left ventricular enlargement after experimental myocardial infarction in mice. Circulation. 1999;99:3063–3070. doi: 10.1161/01.cir.99.23.3063. [DOI] [PubMed] [Google Scholar]
- 14.Spinale FG, et al. Matrix metalloproteinase inhibition during the development of congestive heart failure: effects on left ventricular dimensions and function. Circ Res. 1999;85:364–376. doi: 10.1161/01.res.85.4.364. [DOI] [PubMed] [Google Scholar]
- 15.Sambrook, J., Fritsch, E.F., and Maniatis, T. 1989. Molecular cloning. Volume 1. Cold Spring Harbor Laboratory Press. Cold Spring Harbor, New York, USA. Chapter 6.
- 16.D’Armiento J, Dalal SS, Okada Y, Berg RA, Chada K. Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell. 1992;71:955–961. doi: 10.1016/0092-8674(92)90391-o. [DOI] [PubMed] [Google Scholar]
- 17.Kozak M. Point mutations defines a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- 18.Chada K, et al. Specific expression of a foreign globin gene in erythroid cells of transgenic mice. Nature. 1985;314:377–380. doi: 10.1038/314377a0. [DOI] [PubMed] [Google Scholar]
- 19.Hogan, B., Costantini, F., and Lacy, E. 1986. Manipulating the mouse embryo: a laboratory manual. Cold Spring Harbor Laboratory Press. Cold Spring Harbor, New York, USA. 1–200.
- 20.Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975;98:503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- 21.Chirgwin J, Przybyla A, MacDonald R, Rutter WJ. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 22.Barbu V, Dautry F. Northern blot normalization with a 28S rRNA oligonucleotide probe. Nucleic Acids Res. 1989;17:7115. doi: 10.1093/nar/17.17.7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cawston TE, Barrett AJ. A rapid and reproducible assay for collagenase using (I-14C) acetylated collagen. Anal Biochem. 1979;99:340–345. doi: 10.1016/s0003-2697(79)80017-2. [DOI] [PubMed] [Google Scholar]
- 24.Berman, M.B. 1980. Collagenase and corneal ulceration. John Wiley & Sons. Chinchester, United Kingdom. 141–174.
- 25.Stegemann H, Stalder K. Determination of hydroxyproline. Clin Chim Acta. 1967;18:267–273. doi: 10.1016/0009-8981(67)90167-2. [DOI] [PubMed] [Google Scholar]
- 26.Woessner JF. The determination of hydroxyproline in tissue and protein samples containing small proportions of this amino acid. Arch Biochem Biophys. 1961;93:440–447. doi: 10.1016/0003-9861(61)90291-0. [DOI] [PubMed] [Google Scholar]
- 27.Dolber PC, Spach MS. Picrosirius red staining of cardiac muscle following phosphomolybdic acid treatment. Stain Technol. 1987;62:23–26. doi: 10.3109/10520298709107961. [DOI] [PubMed] [Google Scholar]
- 28.Subramaniam A, et al. Tissue-specific regulation of the α-myosin heavy chain gene promoter in transgenic mice. J Biol Chem. 1991;266:24613–24620. [PubMed] [Google Scholar]
- 29.D’Armiento J, Dalal SS, Chada K. Tissue, temporal and inducible expression pattern of haptoglobin in mice. Gene. 1997;195:19–27. doi: 10.1016/s0378-1119(97)00123-6. [DOI] [PubMed] [Google Scholar]
- 30.Olivetti G, Ricci R, Anversa P. Hyperplasia of myocyte nuclei in long-term cardiac hypertrophy in rats. J Clin Invest. 1987;80:1818–1821. doi: 10.1172/JCI113278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan SM, Finato N, Di Loreto C, Beltrami CA. Nuclear size of myocardial cells in end-stage cardiomyopathies. Anal Quant Cytol Histol. 1999;21:174–180. [PubMed] [Google Scholar]
- 32.Rossant J. Mouse mutants and development: new molecular insights into cardiogenesis. Circ Res. 1996;78:349–353. doi: 10.1161/01.res.78.3.349. [DOI] [PubMed] [Google Scholar]
- 33.Chien KR. Genes and physiology: molecular physiology in genetically engineered animals. J Clin Invest. 1996;97:901–909. doi: 10.1172/JCI118512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vincenti MP, et al. Cloning of the gene for interstitial collagenase-3 (matrix metalloproteinase-13) from rabbit synovial fibroblasts: differential expression with collagenase-1 (matrix metalloproteinase-1) Biochem J. 1998;331:341–346. doi: 10.1042/bj3310341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tyagi S, Campbell SE, Reddy HK, Tjahja E, Voelker DJ. Matrix metalloproteinase activity expression in infarcted, noninfarcted and dilated cardiomyopathic human hearts. Mol Cell Biochem. 1996;155:13–21. doi: 10.1007/BF00714328. [DOI] [PubMed] [Google Scholar]
- 36.Thomas CV, et al. Increased matrix metalloproteinase activity and selective upregulation in LV myocardium from patients with end-stage dilated cardiomyopathy. Circulation. 1998;97:1708–1715. doi: 10.1161/01.cir.97.17.1708. [DOI] [PubMed] [Google Scholar]
- 37.Li YY, Feldman AM, Sun Y, McTiernan CF. Differential expression of tissue inhibitors of metalloproteinases in the failing human heart. Circulation. 1998;98:1728–1734. doi: 10.1161/01.cir.98.17.1728. [DOI] [PubMed] [Google Scholar]
- 38.Charney RH, Takahashi S, Zhao M, Sonnenblick EH, Eng C. Collagen loss in the stunned myocardium. Circulation. 1992;85:1483–1490. doi: 10.1161/01.cir.85.4.1483. [DOI] [PubMed] [Google Scholar]
- 39.Zhao M, et al. Profound structural alterations of the extracellular collagen matrix in postischemic dysfunctional (“stunned”) but viable myocardium. J Am Coll Cardiol. 1987;10:1322–1334. doi: 10.1016/s0735-1097(87)80137-7. [DOI] [PubMed] [Google Scholar]
- 40.Spinale FG, Zellner JL, Johnson WS, Eble DM, Munyer PD. Cellular and extracellular remodeling with the development and recovery from tachycardia-induced cardiomyopathy: changes in fibrillar collagen, myocyte adhesion capacity and proteoglycans. J Mol Cell Cardiol. 1996;28:1591–1608. doi: 10.1006/jmcc.1996.0150. [DOI] [PubMed] [Google Scholar]
- 41.Spinale FG, et al. Time-dependent changes in matrix metalloproteinase activity and expression during the progression of congestive heart failure. Circ Res. 1998;82:482–495. doi: 10.1161/01.res.82.4.482. [DOI] [PubMed] [Google Scholar]
- 42.Weber KT, et al. Fibrillar collagen and remodeling of dilated canine left ventricle. Circulation. 1990;82:1387–1401. doi: 10.1161/01.cir.82.4.1387. [DOI] [PubMed] [Google Scholar]
- 43.Jeffrey, J.J. 1998. Interstitial collagenases. In Matrix metalloproteinases. W.C. Parks and R.P. Mecham, editors. Academic Press. San Diego, California, USA. 15–42.
- 44.Weber KT, et al. Collagen remodeling of the pressure-overloaded, hypertrophied nonhuman primate myocardium. Circ Res. 1988;62:757–765. doi: 10.1161/01.res.62.4.757. [DOI] [PubMed] [Google Scholar]
- 45.Cairns JA, Walls AF. Mast cell tryptase stimulates the synthesis of type I collagen in human lung fibroblasts. J Clin Invest. 1997;99:1313–1321. doi: 10.1172/JCI119290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clore JN, Cohen IK, Diegelmann RF. Quantitation of collagen types I and III during wound healing in rat skin. Proc Soc Exp Biol Med. 1979;161:337–340. doi: 10.3181/00379727-161-40548. [DOI] [PubMed] [Google Scholar]
- 47.Cleutjens JPM, Verluyten MJA, Smits JFM, Daeman MJAP. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–338. [PMC free article] [PubMed] [Google Scholar]
- 48.Eleftheriades EG, et al. Regulation of procollagen metabolism in the pressure-overloaded rat heart. J Clin Invest. 1993;91:1113–1122. doi: 10.1172/JCI116270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Medugorac I, Jacob R. Characterization of left ventricular collagen in the rat. Cardiovasc Res. 1983;17:15–21. doi: 10.1093/cvr/17.1.15. [DOI] [PubMed] [Google Scholar]
- 50.Dawson R, Milne G, Williams RB. Changes in the collagen of rat heart in copper deficiency-induced cardiac hypertrophy. Cardiovasc Res. 1982;16:559–565. doi: 10.1093/cvr/16.10.559. [DOI] [PubMed] [Google Scholar]
- 51.Okada H, et al. Alteration of extracellular matrix in dilated cardiomyopathic hamster heart. Mol Cell Biochem. 1996;156:9–15. doi: 10.1007/BF00239313. [DOI] [PubMed] [Google Scholar]
- 52.Mukherjee D, Sen S. Alteration of collagen phenotypes in ischemic cardiomyopathy. J Clin Invest. 1991;88:1141–1146. doi: 10.1172/JCI115414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gardi C, et al. Collagen breakdown products and lung collagen metabolism: an in vitro study on fibroblast cultures. Thorax. 1994;49:312–318. doi: 10.1136/thx.49.4.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Slack JL, Liska DJ, Bornstein P. Regulation of the expression of the type I collagen genes. Am J Med Genet. 1993;45:140–151. doi: 10.1002/ajmg.1320450203. [DOI] [PubMed] [Google Scholar]
- 55.Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 1997;91:439–442. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- 56.Ito A, et al. Degradation of interleukin 1 b by matrix metalloproteinases. J Biol Chem. 1996;271:14657–14660. doi: 10.1074/jbc.271.25.14657. [DOI] [PubMed] [Google Scholar]
- 57.D’Armiento J, et al. Collagenase expression in transgenic mouse skin causes hyperkeratosis and acanthosis and increases susceptibility to tumorigenesis. Mol Cell Biol. 1995;15:5732–5739. doi: 10.1128/mcb.15.10.5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium: fibrosis and renin-angiotensin-aldosterone system. Circulation. 1991;83:1849–1865. doi: 10.1161/01.cir.83.6.1849. [DOI] [PubMed] [Google Scholar]
- 59.Narayan S, Janicki JS, Shroff SG, Pick R, Weber KT. Myocardial collagen and mechanics after preventing hypertrophy in hypertensive rats. Am J Hypertens. 1989;2:675–682. doi: 10.1093/ajh/2.9.675. [DOI] [PubMed] [Google Scholar]
- 60.Jalil JE, et al. Fibrillar collagen and myocardial stiffness in the intact hypertrophied rat left ventricle. Circ Res. 1989;64:1041–1050. doi: 10.1161/01.res.64.6.1041. [DOI] [PubMed] [Google Scholar]
- 61.Sasayama S, Franklin D, Ross J., Jr Hyperfunction with normal inotropic state of the hypertrophied left ventricle. Am J Physiol. 1977;232:H418–H425. doi: 10.1152/ajpheart.1977.232.4.H418. [DOI] [PubMed] [Google Scholar]
- 62.Eghbali M, Weber KT. Collagen and the myocardium: fibrillar structure, biosynthesis and degradation in relation to hypertrophy and its regression. Mol Cell Biochem. 1990;96:1–14. doi: 10.1007/BF00228448. [DOI] [PubMed] [Google Scholar]
- 63.Weber K, Pick R, Janicki JS, Gadodia G, Lakier J. Inadequate collagen tethers in dilated cardiopathy. Am Heart J. 1988;116:1641–1646. doi: 10.1016/0002-8703(88)90763-6. [DOI] [PubMed] [Google Scholar]