

Abstract
The connexin carboxyl-terminal (CxCT) domain plays a role in the trafficking, localization, and turnover of gap junction channels, as well as the level of gap junction intercellular communication via numerous post-translational modifications and protein–protein interactions. As a key player in the regulation of gap junctions, the CT presents itself as a target for manipulation intended to modify function. Specific to intrinsically disordered proteins, identifying residues whose secondary structure can be manipulated will be critical toward unlocking the therapeutic potential of the CxCT domain. To accomplish this goal, we used biophysical methods to characterize CxCT domains attached to their fourth transmembrane domain (TM4). Circular dichroism and nuclear magnetic resonance were complementary in demonstrating the connexin isoforms that form the greatest amount of α-helical structure in their CT domain (Cx45 > Cx43 > Cx32 > Cx50 > Cx37 ≈ Cx40 ≈ Cx26). Studies compared the influence of 2,2,2-trifluoroethanol, pH, phosphorylation, and mutations (Cx32, X-linked Charcot-Marie Tooth disease; Cx26, hearing loss) on the TM4-CxCT structure. While pH modestly influences the CT structure, a major structural change was associated with phosphomimetic substitutions. Since most connexin CT domains are phosphorylated throughout their life cycle, studies of phospho-TM4-CxCT isoforms will be critical toward understanding the role that structure plays in regulating gap junction function.
Keywords: gap junctions, intrinsically disordered, circular dichroism, nuclear magnetic resonance
INTRODUCTION
Gap junctions allow the exchange of ions, second messengers, and small metabolites between adjacent cells. This enables individual cell events to synchronize into the functional response of an entire organ.1,2 Dysfunctional intercellular communication via gap junctions has been implicated as a causative factor in many human diseases.3 Gap junction channels are formed by the apposition of two connexins from adjacent cells. Each connexin consists of six connexin proteins, which are named based on their molecular mass (e.g., 43 kDa isoform, Connexin43 (Cx43)). The 21 known human connexin isoforms share a highly conserved N-terminus, two extracellular loops (EL1 and EL2), and four transmembrane domains. In contrast, the cytoplasmic loop and carboxyl-terminal (CT) domains are divergent and variable in length and sequence.
The CT plays a role in the gating, trafficking, and localization of gap junction channels, as well as the level of gap junction intercellular communication.4–11 The CT domain is involved in regulating many of these cellular events through the interaction with different molecular partners and/or post-translational modifications,2,12,13 which are features typical of many intrinsically disordered proteins (IDPs) such as the connexin CT domain.14–18 From a thermodynamic perspective, the entropic cost of coupled folding and binding a molecular partner results in high-specificity, low-affinity interactions, which are advantageous in signaling and regulatory functions for IDPs.19 Additionally, many IDPs have a conformational preference consistent with the bound state.20 Altering the conformational preference of an IDP can modulate the favorability of a molecular partner interaction. One cellular event regulating protein function that causes significant structural rearrangement and is disproportionately common for IDPs is phosphorylation.21,22 Connexins, with the exception of Cx26, are differentially phosphorylated at numerous CT residues at various times throughout their life cycle.23–27 Another cellular event that can influence the structure of IDPs is a change in pH.28 Generally, IDPs contain few hydrophobic residues and are enriched in residues carrying a net charge; this combination prevents a high degree of folding at physiological pH. Conversely, IDPs typically gain structure under highly acidic or basic conditions.28
Gap junction channels are not passive pores, but are highly regulated and respond to the cellular environment to open, close, or turnover as needed. Unlike many other ion channels for which there are selective agents to characterize the function of one ionic current in a complex environment, there are few highly specific chemical tools that modulate gap junction channels. Some reagents can modify the function of these channels; however, most lead to nonspecific and complex effects.29 Peptide-based approaches have been used to block formation of gap junction channels,29,30 but the result is to render the gap junctions closed, independent of any regulatory trigger. As a key player in the regulation of gap junctions, the CT presents itself as a target for manipulation intended to modify function. A critical step toward unlocking the therapeutic potential of the connexin CT domain is defining residues that can form and conditions that can manipulate the secondary structure. In addition, we do not yet fully understand the mechanisms that make each gap junction channel different. Investigation of the most divergent domain between connexins, the CT domain, would be a prime location to develop high-affinity binding pharmacophores that regulate specific connexins; missing is the structural information in normal and diseased states.
Toward accomplishing this goal, biophysical methods were used to structurally characterize Cx26, Cx32, Cx37, Cx40, Cx43, Cx45, and Cx50 CT domains alone and when attached to their fourth transmembrane (TM4) domain. Studies compared the influence of 2,2,2-trifluoroethanol, pH, phosphorylation, and mutations on the TM4-CxCT structure. Membrane tethering was necessary to elicit any change in CT structure. While pH modestly influences the CT structure, a major structural change was associated with phosphomimetic substitutions.
RESULTS
Sample Conditions
In this study, CD and NMR experiments were performed on soluble and membrane-tethered connexin CT domains (Table I). The connexins in this investigation were selected to represent the three major subfamilies (α: Cx37, Cx40, Cx43, and Cx50; β: Cx26 and Cx32; γ: Cx45). Most of them have well-characterized protein partners that interact with their CT domain, contain phosphorylation modification(s) in their CT domain, and are linked to human disease(s).3,31 Data were collected at either physiological (pH 7.5) or acidic (pH 5.8) conditions. Intracellular acidification, which is a major consequence of tissue ischemia, leads to closure, degradation, and altered expression of gap junction channels.32–34 Data were also collected at various concentrations of TFE. The cosolvent TFE is a helix-stabilizing compound that is readily used in protein folding and structural studies, as a tool for probing the helical propensity of proteins and peptides.35,36 At low levels (<50%), TFE will stabilize regions with intrinsic α-helical character and, at high levels (>50%), can induce an α-helical conformation.37 We previously used 10% TFE to stabilize innate α-helical content and improve NMR spectra for the TM4-Cx43CT.38–40 Importantly, 10% TFE did not induce α-helical structure.39 Finally, the estimated amount of α-helical content for any CT construct was reproducible within the same preparation and differed by no more than 1–2% when using the same CT construct from a different preparation.
Table I.
Sequence of the Connexin Constructs Used in This Study
| Isoform | Soluble CT | Membrane-tethered CT residues | Species |
|---|---|---|---|
| Cx26 | R216-V226 | D179-V226 | Homo sapiens |
| Cx32 | C217-C283 | D178-C283 | Rattus norvegicus |
| Cx37 | C233-V333 | D197-V333 | Mus musculus |
| Cx40 | S251-V356 | N194-V356 | Rattus norvegicus |
| Cx43 | V236-I382 | D197-I382 | Rattus norvegicus |
| Cx45 | K265-I396 | D219-I396 | Mus musculus |
| Cx50 | S232-I440 | D200-I440 | Mus musculus |
Secondary Structure of Soluble Connexin CT Constructs
CD spectroscopy in the far-UV spectral region (190–250 nm) was used to gain insight into the secondary structure of the soluble CT domain from different connexin isoforms at pH 7.5 and 5.8 (Figure 1 and Table I, Soluble CT). Of note, CD data for the soluble Cx37CT, Cx40CT, Cx43CT, and Cx45CT domains have been published and were placed in Supporting Information, Figure 1 for easier comparison with the soluble Cx26CT, Cx32CT, and Cx50CT domains.14,41,42 CD spectra of the soluble Cx26CT at both pH 7.5 and pH 5.8 have a minimum at 197 nm (negative MRE value) and a maximum around 222 nm (positive MRE value) indicating a random coil structure. The CD spectra of the soluble Cx32CT and Cx50CT domains at pH 5.8 and pH 7.5 also present a high proportion of random coil structure. However, in comparison to the Cx26CT domain, the right shift of the major absorption minima (between 199 and 202 nm) and the decrease in signal at 222 nm (negative MRE value) are due to the contribution of α-helix signal at 208 and 222 nm, respectively. This suggests the presence of a small amount of a very dynamic α-helix. These data also indicate that pH has little-to-no effect on the secondary structure of soluble connexin CT domains.
FIGURE 1.
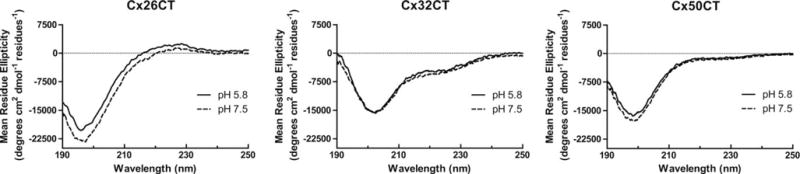
Secondary structural analysis of soluble connexin CT domains. CD spectra of the soluble Cx26CT, Cx32CT, and Cx50CT domains in 1× PBS at pH 5.8 and pH 7.5.
CD measurements were next carried out in various concentrations of TFE (Figure 2, pH 7.5 and Figure 3, pH 5.8; published data at pH 7.5 (Cx45CT) and pH 5.8 (Cx40CT, Cx43CT, and Cx45CT) are in Supporting Information, Figure 2). In general, increasing amounts of TFE at both pH 5.8 and 7.5, in varying degrees, caused a right shift in the ~200 nm peaks, a decrease in the 222 nm values, and an increase in the 195 nm values, which indicates additional α-helical content for each of the CT domains. TFE had the least influence on the Cx26CT (least α-helical) structure, and TFE had the greatest influence on the Cx32CT and Cx50CT structures. These results suggest that TFE can stabilize the innate α-helical structure and/or residues susceptible to α-helical formation for these connexin isoforms.
FIGURE 2.
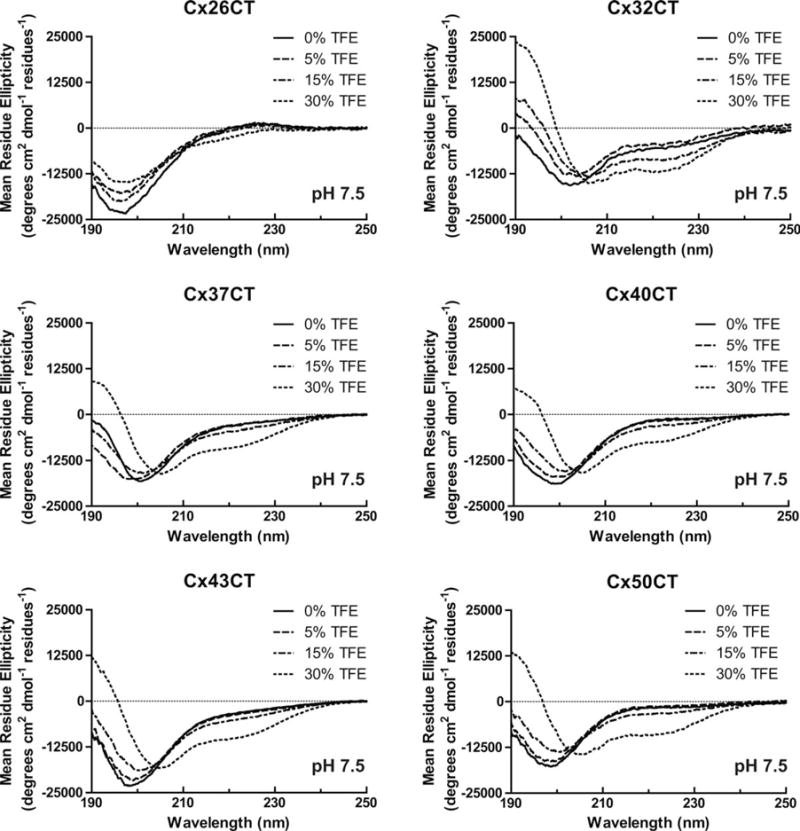
Secondary structural analysis of soluble connexin CT domains in 2,2,2-trifluoroethanol (TFE) at pH 7.5. CD spectra of the soluble Cx26CT, Cx32CT, Cx37CT, Cx40CT, Cx43CT, and Cx50CT domains in 0%, 5%, 15%, and 30% TFE, 1× PBS, and at pH 7.5.
FIGURE 3.
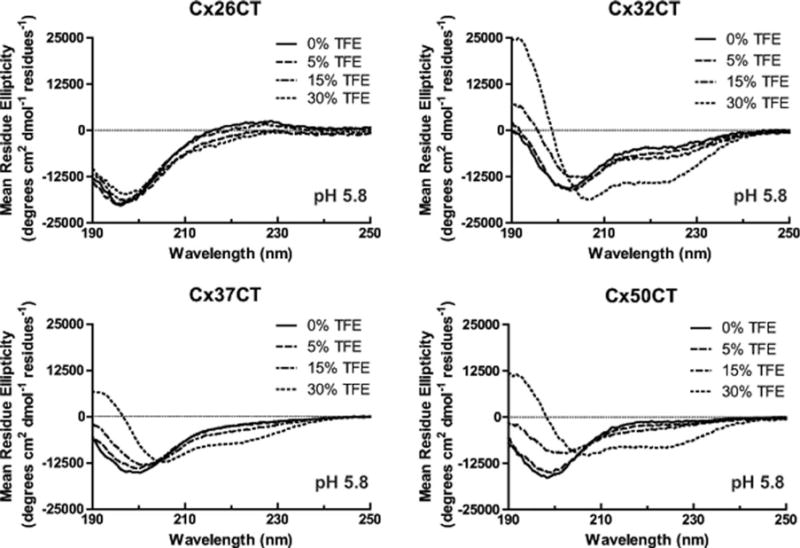
Secondary structural analysis of soluble connexin CT domains in 2,2,2-trifluoroethanol (TFE) at pH 5.8. CD spectra of the soluble Cx26CT, Cx32CT, Cx37CT, and Cx50CT domains in 0%, 5%, 15%, and 30% TFE, 1× PBS, and at pH 5.8.
We have previously shown that calmodulin induces α-helical structure within the Cx32CT domain.15 An explanation for the increase in α-helical content with the treatment of TFE as observed by CD is stabilization of the Cx32CT residues involved in binding calmodulin. To test this possibility, we identified the Cx32CT residues that were induced to form the α-helical structure in the presence of TFE by NMR. 15N-HSQC spectra of the Cx32CT were collected as a function of TFE concentration (Figure 4A). The 15N-HSQC is a two-dimensional NMR experiment where each amino acid (except proline) gives one signal (or chemical shift) that corresponds to the N–H amide group. These signals are sensitive to the chemical environment, and even small changes in structure and/or dynamics can change the chemical shift of an amino acid. The addition of TFE from 5% to 25% caused a majority of the Cx32CT amino acids to shift upfield (lower chemical shift value) and several amino acids to shift and then disappear or “broaden beyond detection.” These effects are consistent with the formation of α-helical structure.43–45 Based upon these criteria, three distinct regions (C217-N226, F235-K250, and K260-S266) were identified to form α-helical structure (Figure 4B). These three regions that change in TFE treatments correspond to the same residues that are affected when calmodulin binds the Cx32CT domain.15
FIGURE 4.
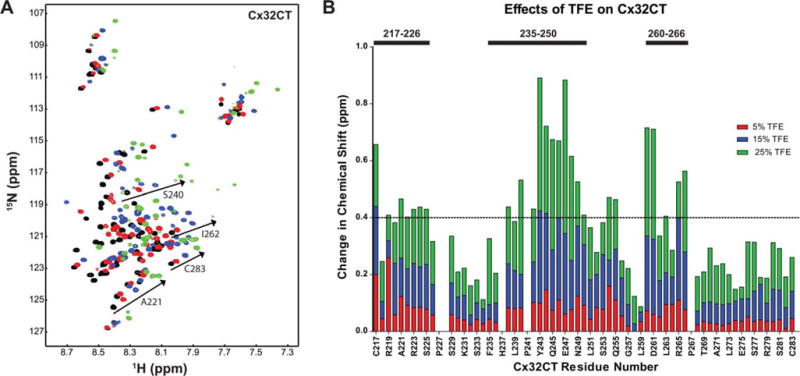
Identification of Cx32CT regions that have α-helical propensity. (A) NMR control spectrum, Cx32CT-only (black), has been overlapped with spectra obtained when the Cx32CT was in the presence of 5% (red), 15% (blue), and 25% (green) 2,2,2-trifluoroethanol (TFE). Spectra for the Cx32CT in the presence of 10%, 20%, and 30% TFE were also collected but not shown due to peak overlap. (B) From (A), each residue was plotted against their change in chemical shift (Δσ = √((ΔδHN)2 + (ΔδN/5))2) as a function of TFE concentration. The three regions that form α-helical structure in the presence of TFE are indicated with bars. The horizontal line indicates the chemical shift change cut-off at 0.4 ppm.
Secondary Structure of TM4-Tethered Connexin CT Constructs
CD spectroscopy was also used to determine the secondary structure of connexin CT domains when tethered to their TM4 domain (Figure 5 and Table I, membrane-tethered CT). CD data for the TM4-Cx43CT and TM4-Cx45CT domains at pH 7.5 and pH 5.8 have been published and placed in Supporting Information, Figure 3A for easier comparison with the other TM4 constructs.14,39 CD spectra for the TM4-Cx26CT, -Cx32CT, -Cx37CT, -Cx40CT, and -Cx50CT domains at pH 7.5 have two peak minima at ~208 and ~222 nm indicating the presence of α-helical structure. Under acidic conditions (pH 5.8, no TFE; Table II), the CT domain of each construct, except TM4-Cx40CT, had an overall small increase in α-helical content. A similar trend of more α-helical content was also observed for most of the constructs with treatment of TFE (5%→30%) at pH 7.5 (Figure 6 and Table III) and pH 5.8 (Figure 7 and Table II; published data for TM4-Cx43CT in Supporting Information, Figure 3B). The exceptions were TM4-Cx37CT at both pH values and TM4-Cx40CT at pH 7.5, which were unaffected by TFE. TFE had the greatest influence on the α-helical content of TM4-Cx45CT at both pH values and TM4-Cx50CT at pH 5.8. An important point, both pH and TFE have no effect on the linker and TM4 secondary structure (Figure 8).
FIGURE 5.
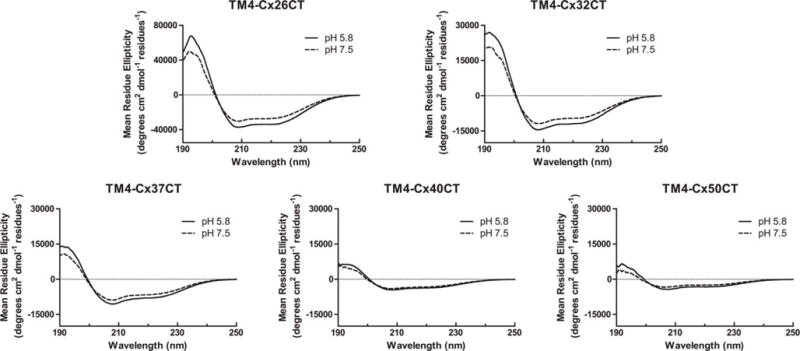
Secondary structural analysis of TM4-CxCT domains. CD spectra of TM4-Cx26CT, -Cx32CT, -Cx37CT, -Cx40CT, and -Cx50CT domains in MES buffer, 8% LPPG, and at pH 5.8 and pH 7.5.
Table II.
Mean Residue Ellipticity (MRE) Values at pH 5.8 for TM4-CxCT Domains
| Isoform | TFE (%) | First local minimum
|
Minimum at 222 nm
|
Totala/CT-only
|
|
|---|---|---|---|---|---|
| Wavelength (nm) | MRE | MRE | α-helical content (%/%) | ||
| TM4-Cx26CT | 0 | 208.8 | −37,208.4 | −33,569.4 | 64.9/18.6 (1.7)b (2.3)* |
| 5 | 208.7 | −39,574.6 | −35,057.7 | 67.1/20.8 | |
| 10 | 209.8 | −40,799.1 | −38,041.4 | 66.3/20.0 | |
| 15 | 208.7 | −43,163.2 | −37,000.5 | 68.9/22.6 | |
| 30 | 209.2 | −45,769.4 | −42,092.3 | 70.8/24.5 (2.2) | |
| TM4-Cx32CT | 0 | 208.1 | −14,522.3 | −11,797.9 | 36.9/11.7 (7.8) |
| 5 | 208.7 | −14,788.3 | −12,027.7 | 35.8/10.6 | |
| 10 | 208.5 | −14,269.4 | −11,587.9 | 35.3/10.1 | |
| 15 | 208.3 | −15,702.2 | −13,142.4 | 36.9/11.7 | |
| 30 | 208.7 | −15,476.2 | −13,553.2 | 37.8/12.6 (8.4) | |
| TM4-Cx37CT | 0 | 208.0 | −10,564.9 | −7909.7 | 22.6/3.0 (3.0) |
| 5 | 208.0 | −11,181.2 | −8517.3 | 22.0/2.4 | |
| 10 | 208.1 | −11,175.5 | −8465.8 | 22.8/3.2 | |
| 15 | 208.1 | −11,523.4 | −8827.0 | 23.3/3.7 | |
| 30 | 208.1 | −13,356.5 | −10,210.9 | 21.0/1.4 (1.4) | |
| TM4-Cx40CT | 0 | 208.5 | −4513.9 | −3639.9 | 14.8/0.0 (0.0) |
| 5 | 208.1 | −4650.1 | −3685.8 | 14.4/0.0 | |
| 10 | 208.7 | −5670.0 | −4598.4 | 16.5/0.0 | |
| 15 | 208.7 | −5386.1 | −4439.3 | 16.1/0.0 | |
| 30 | 207.8 | −5625.9 | −4617.1 | 18.2/1.9 (2.4) | |
| TM4-Cx43CT | 0 | 207.4 | −9620.9 | −6806.9 | 21.5/7.0 (10.4) |
| 5 | 207.6 | −11,481.8 | −8170.9 | 24.9/10.4 | |
| 10 | 207.7 | −11,117.2 | −7972.6 | 26.4/11.9 | |
| 15 | 207.6 | −11,416.0 | −8297.6 | 26.3/11.8 | |
| 30 | 207.6 | −12,999.3 | −9998.5 | 27.3/12.8 (19.0) | |
| TM4-Cx45CT | 0 | 208.3 | −11,067.3 | −9079.7 | 28.4/12.9 (18.1) |
| 5 | 208.0 | −10,850.7 | −8788.0 | 27.4/11.9 | |
| 10 | 208.3 | −11,478.2 | −9366.4 | 29.1/13.6 | |
| 15 | 208.1 | −12,276.9 | −9981.2 | 29.9/14.4 | |
| 30 | 208.4 | −14,936.6 | −12,477.7 | 36.9/21.4 (30.0) | |
| TM4-Cx50CT | 0 | 207.4 | −4316.78 | −3204.4 | 12.4/0.9 (1.8) |
| 5 | 206.9 | −4655.57 | −3426.1 | 14.1/2.6 | |
| 10 | 206.4 | −4773.65 | −3514.0 | 13.5/2.0 | |
| 15 | 207.2 | −4909.53 | −3732.5 | 13.9/2.4 | |
| 30 | 208.8 | −5771.22 | −4716.1 | 17.1/5.6 (11.4) | |
Determined using DichroWeb.
Estimate of the total number of CT residues structured as an α-helix. The contribution of the TM4 α-helix was subtracted from the total α-helix percentage, leaving the remaining α-helical content from the CT domain.
FIGURE 6.
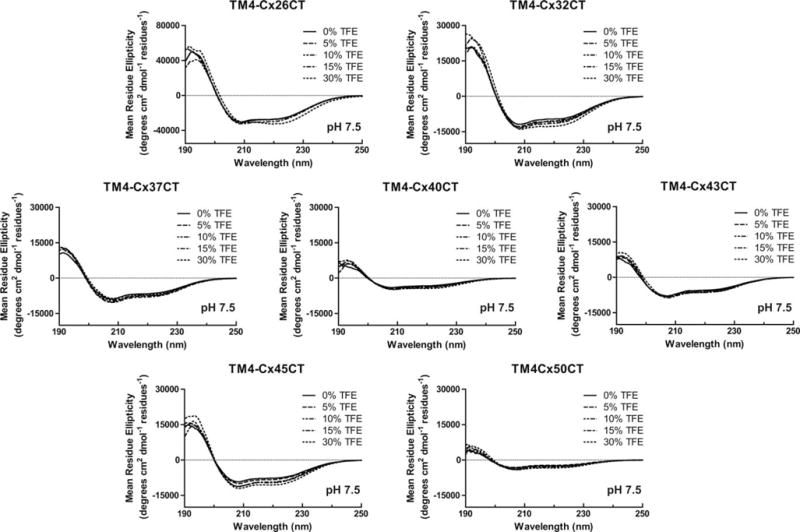
Secondary structural analysis of TM4-CxCT domains in 2,2,2-trifluoroethanol (TFE) at pH 7.5. CD spectra of TM4-Cx26CT, -Cx32CT, -Cx37CT, -Cx40CT, -Cx43CT, -Cx45CT, and -Cx50CT domains in 0%, 5%, 10%, 15%, and 30% TFE, MES buffer, 8% LPPG, and at pH 7.5.
Table III.
Mean Residue Ellipticity (MRE) Values at pH 7.5 for TM4-CxCT Domains
| Isoform | TFE (%) | First local minimum
|
Minimum at 222 nm
|
Totala/CT-only
|
|
|---|---|---|---|---|---|
| Wavelength (nm) | MRE | MRE | α-helical content (%/%) | ||
| TM4-Cx26CT | 0 | 208.8 | −30,335.5 | −26,678.9 | 60.0/13.7 (1.2)b |
| 5 | 208.9 | −31,188.1 | −26,874.9 | 60.9/14.6 | |
| 10 | 208.7 | −31,160.2 | −26,903.5 | 57.8/11.5 | |
| 15 | 209.4 | −32,470.1 | −29,004.2 | 56.7/10.4 | |
| 30 | 210.2 | −31,068 | −32,315.6 | 64.4/18.1 (1.6) | |
| TM4-Cx32CT | 0 | 207.8 | −11,847.7 | −9582.89 | 29.7/4.5 (3.0) |
| 5 | 207.8 | −12,835.4 | −10,243.6 | 31.1/5.9 | |
| 10 | 208.5 | −13,073 | −10,642.7 | 32.0/6.8 | |
| 15 | 208.2 | −13,565.2 | −11,151.4 | 32.0/6.8 | |
| 30 | 208.8 | −13,995.8 | −12,496.3 | 35.1/9.9 (6.6) | |
| TM4-Cx37CT | 0 | 208 | −8901.92 | −6454.69 | 18.4/0.0 (0.0) |
| 5 | 207.5 | −10,017.3 | −7408.12 | 18.1/0.0 | |
| 10 | 207.9 | −9416.02 | −7260.21 | 18.0/0.0 | |
| 15 | 207.8 | −10,269.5 | −7827.59 | 19.0/1.0 | |
| 30 | 208 | −8591.11 | −6939.2 | 18.2/0.0 (0.0) | |
| TM4-Cx40CT | 0 | 207.8 | −4007.67 | −3182.69 | 12.0/0.0 (0.0) |
| 5 | 208 | −4434.49 | −3549.43 | 14.1/0.0 | |
| 10 | 207.9 | −4540.67 | −3643.55 | 14.4/0.0 | |
| 15 | 208.5 | −4777.48 | −3958.42 | 14.5/0.0 | |
| 30 | 208.8 | −4830.4 | −4318.5 | 14.1/0.0 (0.0) | |
| TM4-Cx43CT | 0 | 207.2 | −7856.02 | −5348.97 | 17.4/2.9 (4.3) |
| 5 | 207.2 | −8010.42 | −5721.19 | 20.0/5.5 | |
| 10 | 207.3 | −8269.2 | −5900.64 | 22.0/7.5 | |
| 15 | 207.6 | −8644.65 | −6253.59 | 22.7/8.2 | |
| 30 | 207.5 | −8084.19 | −6271.77 | 21.1/6.6 (9.8) | |
| TM4-Cx45CT | 0 | 208.3 | −9245.19 | −7552.05 | 24.1/8.6 (12.0) |
| 5 | 208.2 | −9998.6 | −8072.77 | 25.6/10.1 | |
| 10 | 208.4 | −11,166.2 | −9102.24 | 28.0/12.5 | |
| 15 | 208.8 | −11,242.3 | −9443.91 | 29.4/13.9 | |
| 30 | 208.6 | −12,066.6 | −10,425.9 | 30.7/15.2 (21.3) | |
| TM4-Cx50CT | 0 | 206.3 | −3385.69 | −2481.02 | 10.1/0.0 (0.0) |
| 5 | 205.9 | −3178.23 | −2208.79 | 10.4/0.0 | |
| 10 | 207.6 | −3655.45 | −2583.12 | 11.7/0.2 | |
| 15 | 207.3 | −4103.86 | −2983.17 | 11.3/0.0 | |
| 30 | 207.9 | −4148.69 | −3287.84 | 12.8/1.3 (2.6) | |
Determined using DichroWeb.
Estimate of the total number of CT residues structured as an α-helix. The contribution of the TM4 α-helix was subtracted from the total α-helix percentage, leaving the remaining α-helical content from the CT domain.
FIGURE 7.
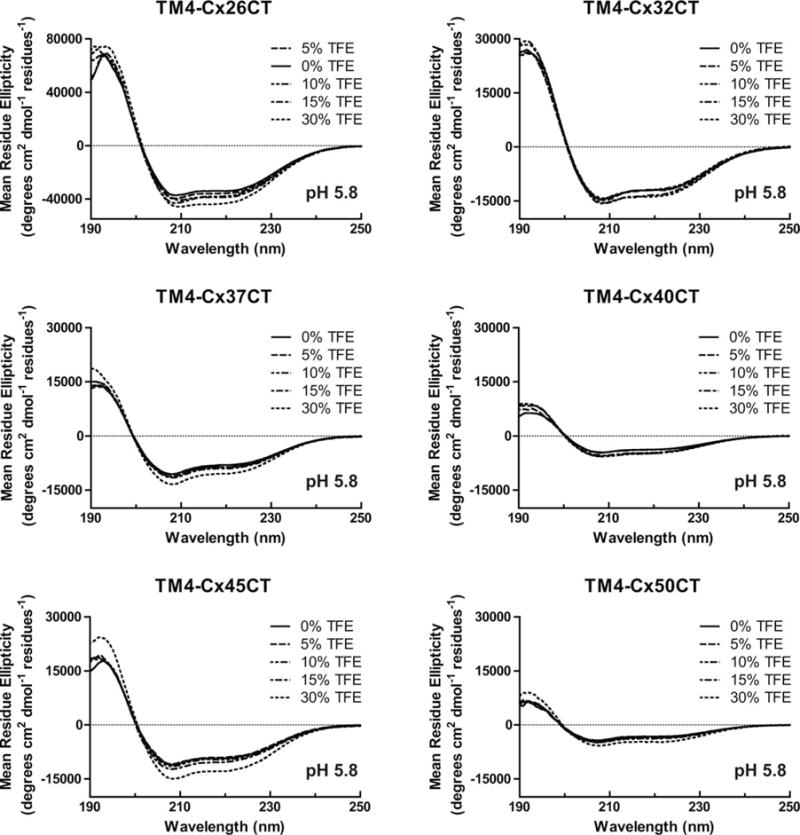
Secondary structural analysis of TM4-CxCT domains in 2,2,2-trifluoroethanol (TFE) at pH 5.8. CD spectra of TM4-Cx26CT, -Cx32CT, -Cx37CT, -Cx40CT, -Cx45CT, and -Cx50CT domains in 0%, 5%, 10%, 15%, and 30% TFE, MES buffer, 8% LPPG, and at pH 5.8.
FIGURE 8.
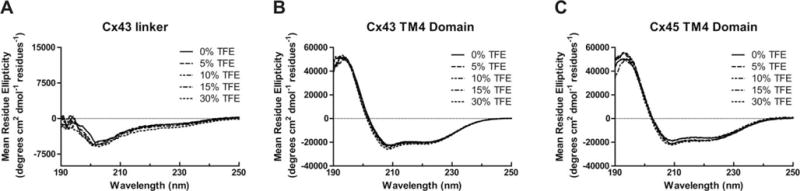
Effect of 2,2,2-trifluoroethanol (TFE) on the linker and TM4 domains. CD spectra of the (A) Cx43 linker, (B) Cx43 TM4 domain, and (C) Cx45 TM4 domain in 0%, 5%, 10%, 15%, and 30% TFE, MES buffer, 8% LPPG, and at pH 5.8.
Secondary Structure of TM4-Tethered Cx32 and Cx26 CT Mutants
Mutations in connexin genes are associated with a number of human diseases.3 In particular, Cx32 mutations cause X-linked Charcot-Marie-Tooth disease (CMTX) and Cx26 mutations account for ~50% of inherited childhood nonsyndromic recessive hearing loss (deafness is also associated with a number of congenital skin disorders). Cx32 and Cx26 mutations affect every portion of the proteins; here, we investigated a subset that either forms (Cx32: R219H, R230C, and F235C15,46–49) or does not form (Cx26: L214P and K224Q50,51) functional gap junction channels. The goal is to determine if one potential mechanism leading to the diseased state is a change in secondary structure. CD spectra of the TM4-Cx32CT mutants R219H and F235C at pH 7.5 contain a similar amount of α-helical content as wild-type with and without TFE (Figure 9 and Table IV, top). Conversely, the R230C mutant had a decrease in α-helical content alone (7.1%) or in the presence of TFE (e.g., 30% TFE, 6.1%) when compared to wild-type. At pH 5.8 (Figure 9 and Table IV, bottom), all three mutants had a decrease in α-helical content when compared to wild-type, with R219H completely losing its pH responsiveness. However, TFE enabled the final amount of α-helical content to be at comparable levels with wild-type.
FIGURE 9.
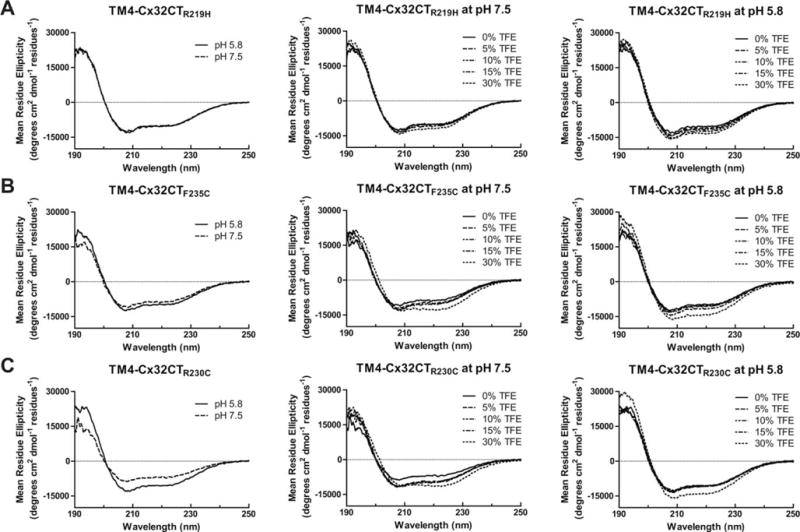
Secondary structural analysis of TM4-tethered Cx32CT CMTX mutants. CD spectra of TM4-Cx32CT mutants: (A) R219F, (B) F235C, and (C) R230C alone and in the presence of 0%, 5%, 10%, 15%, and 30% TFE, MES buffer, 8% LPPG, and at pH 7.5 and pH 5.8.
Table IV.
Mean Residue Ellipticity (MRE) Values for TM4-Cx32 Mutants
| Mutant | TFE (%) | First local minimum
|
Minimum at 222 nm
|
Mutant/wild-type
|
|
|---|---|---|---|---|---|
| Wavelength (nm) | MRE | MRE | α-helical contenta (%/%) | ||
| R219H (pH 7.5) | 0 | 207.8 | −12,530.0 | −9927.17 | 32.2/29.7 |
| 5 | 207.7 | −12,568.5 | −10,142.5 | 32.7/31.1 | |
| 10 | 208.1 | −13,948.0 | −10,517.5 | 34.6/32.0 | |
| 15 | 207.5 | −13,406.6 | −10,609.2 | 33.1/32.0 | |
| 30 | 208.1 | −14,313.3 | −11,706.5 | 34.8/35.1 | |
| F235C | 0 | 207.6 | −12,452.2 | −10,002.6 | 27.2/29.7 |
| 5 | 208.3 | −11,036.3 | −8606.9 | 27.9/31.1 | |
| 10 | 207.7 | −12,067.0 | −9631.32 | 30.0/32.0 | |
| 15 | 207.9 | −12,919.7 | −10,171.6 | 30.7/32.0 | |
| 30 | 208.3 | −13,264.3 | −12,797.9 | 35.2/35.1 | |
| R230C | 0 | 208.1 | −8742.33 | −6642.65 | 22.6/29.7 |
| 5 | 207.3 | −11,553.6 | −9285.26 | 29.1/31.1 | |
| 10 | 208.7 | −11,627.6 | −9443.29 | 28.1/32.0 | |
| 15 | 207.4 | −11,847.4 | −9827.88 | 29.1/32.0 | |
| 30 | 208.6 | −11,266.6 | −11,369.7 | 29.0/35.1 | |
| R219H (pH 5.8) | 0 | 207.8 | −13,036.5 | −10,106.4 | 33.1/36.9 |
| 5 | 206.7 | −14,228.3 | −10,799.7 | 35.0/35.8 | |
| 10 | 207.8 | −15,595.8 | −12,202.2 | 38.0/35.3 | |
| 15 | 208.4 | −14,373.3 | −11,403.0 | 34.1/36.9 | |
| 30 | 208.9 | −15,200.0 | −13,093.8 | 37.7/37.8 | |
| F235C | 0 | 207.6 | −12,410.2 | −9634.2 | 31.1/36.9 |
| 5 | 208.0 | −13,024.0 | −10,391.2 | 31.6/35.8 | |
| 10 | 207.5 | −13,102.5 | −10,448.6 | 31.6/35.3 | |
| 15 | 207.8 | −14,428.5 | −11,428.0 | 34.4/36.9 | |
| 30 | 208.5 | −16,281.5 | −13,789.1 | 37.8/37.8 | |
| R230C | 0 | 208.0 | −12,934.5 | −10,554.6 | 34.0/36.9 |
| 5 | 208.0 | −13,246.9 | −10,711.8 | 32.6/35.8 | |
| 10 | 208.0 | −13,315.1 | −10,649.4 | 33.2/35.3 | |
| 15 | 208.1 | −12,606.1 | −10,457.6 | 31.3/36.9 | |
| 30 | 208.0 | −15,756.6 | −13,806.9 | 38.6/37.8 | |
Determined using DichroWeb.
For the TM4-Cx26CT mutants L214P and K224Q, there was a large decrease in α-helical content as compared to wild-type at pH 7.5 and no TFE (Figures 10A and 10B and Table V, top). At pH 5.8 (no TFE), while there was an overall increase in the α-helical content, K224Q more closely resembled wild-type while L214P was significantly lower. TFE increased the α-helical content of L214P and K224Q to levels comparable with wild-type. Since the L214P mutation is located in the TM4 portion of Cx26, CD spectra of the TM domain only, with and without the mutation were also collected (Figure 10C). The TM domain only data strongly suggest that the pH-independent structural change observed in the TM4-Cx26CT L214P mutant is localized within the TM4 domain.
FIGURE 10.
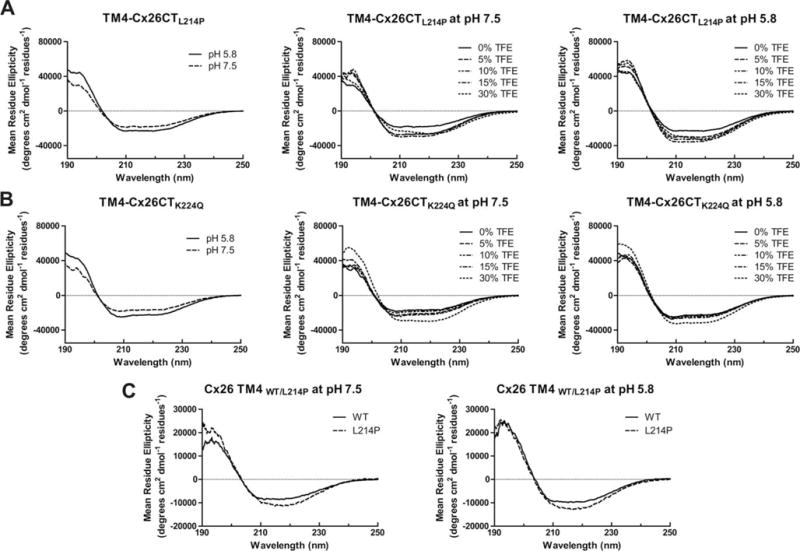
Secondary structural analysis of TM4-tethered Cx26CT nonsyndromic recessive hearing loss mutants. CD spectra of TM4-Cx26CT mutants (A) L214P and (B) K224Q alone and in the presence of 0%, 5%, 10%, 15%, and 30% TFE, MES buffer, 8% LPPG, and at pH 7.5 and pH 5.8. (C) CD Spectra of the Cx26 TM4 WT and L214P at pH 7.5 and 5.8.
Table V.
Mean Residue Ellipticity (MRE) Values for TM4-Cx26 Mutants
| Mutant | TFE (%) | First local minimum
|
Minimum at 222 nm
|
Mutant/wild-type
|
|
|---|---|---|---|---|---|
| Wavelength (nm) | MRE | MRE | α-helical contenta (%/%) | ||
| L214P (pH 7.5) | 0 | 210.3 | −18,844.7 | −17,872.6 | 44.2/60.0 |
| 5 | 210.5 | −27,498.3 | −26,284.4 | 62.7/60.9 | |
| 10 | 210.4 | −27,512.8 | −26,372.5 | 63.0/57.8 | |
| 15 | 210.4 | −30,011.3 | −28,779.1 | 64.9/56.7 | |
| 30 | 210.0 | −23,204.7 | −26,477.1 | 61.3/64.4 | |
| K224Q | 0 | 208.1 | −18,367.3 | −16,613.3 | 45.3/60.0 |
| 5 | 208.4 | −23,213.1 | −20,380.8 | 57.2/60.9 | |
| 10 | 208.6 | −20,160.5 | −17,594.9 | 59.1/57.8 | |
| 15 | 209.8 | −24,329.1 | −21,359.5 | 56.4/56.7 | |
| 30 | 209.9 | −29,079.6 | −29,287.3 | 67.7/64.4 | |
| L214P (pH 5.8) | 0 | 210.1 | −23,122.6 | −21,853.3 | 55.6/64.9 |
| 5 | 209.4 | −30,183.7 | −29,088.5 | 68.4/67.1 | |
| 10 | 210.4 | −32,918.3 | −30,952.7 | 69.1/66.3 | |
| 15 | 212.1 | −35,867.7 | −33,071.2 | 70.9/68.9 | |
| 30 | 212.9 | −30,496.6 | −32,621.8 | 62.2/70.8 | |
| K224Q | 0 | 208.0 | −24,502.0 | −22,203.1 | 61.4/64.9 |
| 5 | 208.7 | −25,288.7 | −22,080.1 | 59.2/67.1 | |
| 10 | 208.3 | −26,554.7 | −23,673.6 | 62.1/66.3 | |
| 15 | 208.6 | −27,384.7 | −24,630.1 | 63.8/68.9 | |
| 30 | 210.0 | −32,707.8 | −30,550.2 | 70.9/70.8 | |
Determined using DichroWeb.
Feasibility of Structural Analysis by NMR of the TM4-CxCT Constructs
Different connexin isoforms
2D NMR 15N-HSQC spectra were used to evaluate the sample properties of the TM4-CxCT constructs in LPPG micelles to determine the feasibility of solving their structures (Figure 11). Previous NMR studies identified that the His-tag, linker, and first 46 residues of the TM4-Cx43CT (D197-G242), which are the TM4 residues and residues immediately flanking the TM4, were not visible in the spectrum.40 Assignment of these residues was most likely complicated by the increased line widths associated with slower tumbling in the LPPG detergent micelles.40 The 15N-HSQC spectrum of the shortest construct, TM4-Cx26CT (D179-V226), only contains a few peaks. This observation is consistent with the area not observed (TM4 and adjacent residues) in the TM4-Cx43CT 15N-HSQC spectrum. The spectrum of TM4-Cx32CT, the next longest construct (D178-C283), also contained only a few number of peaks. A larger number of peaks, however, were observed in the TM4-Cx37CT, -Cx40CT, and -Cx50CT spectra. As was previously reported for the TM4-Cx43CT and TM4-Cx45CT domains (Supporting Information, Figure 4),14,38,39,52 the TM4-Cx37CT, Cx40CT, and Cx50CT domains are pure and contain a single conformation. This is indicated by the number of Gly (8, 11, and 11, respectively black circle) residues in the 15N-HSQC spectrum matching the number of Gly residues expected to be observed from their CT domain (excluding the linker, TM, and Gly residues within 5 residues of the TM4 domain). Another useful piece of information that can be obtained from an 15N-HSQC spectrum, based upon the spectral width, is an estimate of the amount of secondary structure present; the larger the peaks are spread out in the 1H ppm dimension equates to a larger amount of α-helical and/or β-sheet structure. The rank order of α-helical structure based upon CD spectra at pH 5.8 and 10% TFE is TM4-Cx45CT > TM4-Cx43CT > TM4-Cx37CT and TM4-Cx50CT > TM4-Cx40CT. This correlates with the rank order of spectral width from the 15N-HSQC spectra at pH 5.8 and 10% TFE, TM4-Cx45CT (1.1 ppm) > TM4-Cx43CT (1.0 ppm) > TM4-Cx37CT and TM4-Cx50CT (0.9 ppm) > TM4-Cx40CT (0.6 ppm). Altogether, the CD and NMR data portray the CT domain from different connexin isoforms as being predominately intrinsically disordered with small, but varying amounts of α-helical structure that can be influenced by changes in pH and TFE concentrations.
FIGURE 11.
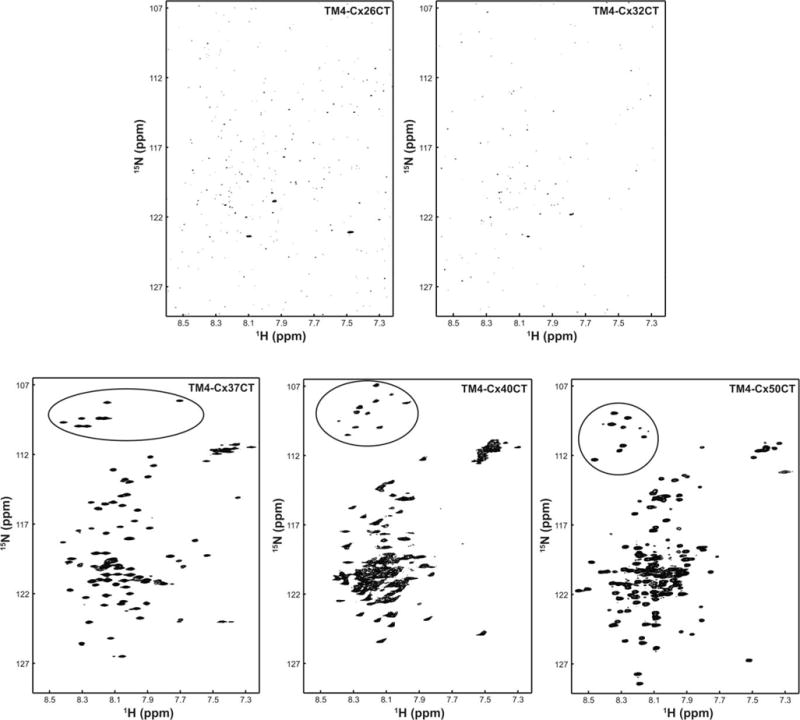
Structural analysis of TM4-CxCT domains by NMR. 15N-HSQC spectra were collected for the TM4-Cx26CT, -Cx32CT, -Cx37CT, -Cx40CT, and -Cx50CT domains in 10% TFE, MES buffer, 8% LPPG, and at pH 5.8. The black circles highlight the glycine residues.
Phosphorylated connexin isoforms
Using Asp substitution(s) to mimic phosphorylation, we determined that a single kinase (e.g., cdc2, PKA, and PKC) can alter the α-helical propensity of the Cx43CT when membrane-tethered (TM4-Cx43CT) at residues proximal and distal to the site(s) of modification.53 To model the next generation of molecules that could potentially regulate Cx43 function and determine if altered CT secondary structure by phosphorylation is a mechanism that modulates the binding affinity for protein partners involved in Cx43 regulation, here we characterized the CT structure with a phosphorylation pattern (i.e., multiple kinases) that more closely resembles connexins observed during gap junction channel assembly (CK1 and PKA; S325, S328, S330, S365D) and disassembly (Src, MAPK, and PKC; Y247, Y265, S255, S262, S279, S282, S368D). Importantly, studies have demonstrated simultaneous phosphorylation by CK1 and PKA (assembly) and Src, MAPK, and PKC (disassembly) residues with phospho-specific antibodies.23,54–57
CD was used to analyze the secondary structure of the TM4-Cx43CT assembly and disassembly constructs. Of note, we confirmed that Asp substitutions (like pH and TFE) only affect the CT structure (not TM453). The assembly and disassembly constructs have 25% and 40% greater α-helical content compared to wild-type, respectively (Figure 12A and Table VI). NMR was then used to map these structural changes caused by phosphorylation to specific CT residues using the resonance assignments of TM4-Cx43CT wild-type.53 Upon initial investigation, the number of peaks in the 15N-HSQC spectrum of the assembly and disassembly constructs indicates a single conformation (Figures 12B and 12C, top; 11 Gly in black circle). Examination on a residue-by-residue basis using the change in N–H chemical shift indicates that phosphorylation is having a global effect on the CT structure from both the assembly and disassembly constructs (Figures 12B and 12C, bottom). The greatest changes in chemical shift are localized to the residues around the phosphomimetic substitutions. Based on our previous study, we would predict these residues with changes in their N–H chemical shift have transitioned from random coil to fluctuating between random coil and α-helical structure.53
FIGURE 12.
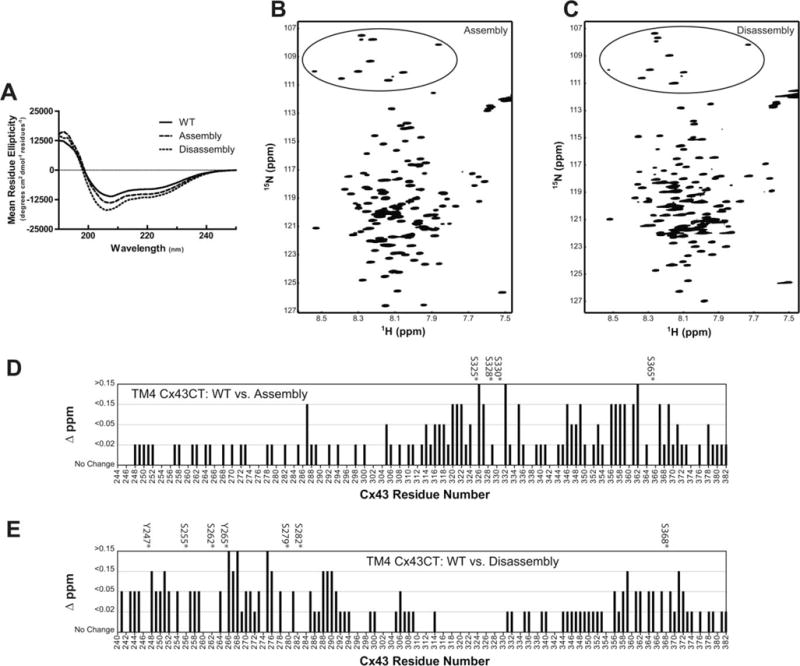
Structural analysis of multi-phosphorylated TM4-Cx43CT domains. A) CD and 15N-HSQC spectra of the (B) TM4-Cx43CT assembly(S325,328,330,365D) and (C) disassembly(Y247,265;S255,262,279,282,368D) phosphomimetic constructs in 10% TFE, MES buffer, 8% LPPG, and at pH 5.8. Black circles highlight the glycine residues. From the 15N-HSQC data, each (D) TM4-Cx43CT assembly(S325,328,330,365D) and (E) disassembly(Y247,265;S255,262,279,282,368D) residue was plotted against their change in chemical shift (Δσ =√((ΔδHN)2 + (ΔδN/5))2) as compared to TM4-Cx43CT wild-type. The asterisks denote the serine/tyrosine residues substituted for an aspartic acid.
Table VI.
Mean Residue Ellipticity (MRE) Values at pH 5.8 for TM4-Cx43CT Phosphomimetic Constructs
| Isoform
|
First local minimum
|
Minimum at 222 nm
|
Totala/CT-only
|
||
|---|---|---|---|---|---|
| TM4-Cx43CT | TFE (%) | Wavelength (nm) | MRE | MRE | α-helical contenta (%/%) |
| WT | 10 | 207.7 | −11,117.2 | −7972.6 | 26.4/11.9 (17.7)b |
| Assembly | 10 | 207.3 | −13,839.8 | −9988.5 | 35.4/20.9 (31.1) |
| Disassembly | 10 | 206.1 | −16,927.1 | −11,335.1 | 43.8/29.3 (43.6) |
Determined using DichroWeb.
Estimate of the total number of CT residues structured as an α-helix. The contribution of the TM4 α-helix was subtracted from the total α-helix percentage, leaving the remaining α-helical content from the CT domain.
Technical note
Several problems have contributed to the difficulty of elucidating the mechanisms by which kinases affect gap junction intercellular communication; the transient nature of a particular CT phosphorylation state, the ability of many kinases to phosphorylate more than one CT residue, the ability of various kinases to phosphorylate the CT at the same time, and the inability to precisely control which residue(s) are phosphorylated. Strategies employed to overcome these difficulties include the use of phospho-specific CT antibodies58 and short CT phosphopeptides.59,60 Also well appreciated is the use of Asp (or Glu) substitutions as a mimetic for phosphorylation. Asp substitutions allow for the precise control of the site(s) modified and the production of enough protein for biophysical studies. Moreover, we and others have demonstrated that an Asp can mimic a phosphorylated connexin residue in vivo (e.g., PKA, CK1, and MAPK23,61,62).
DISCUSSION
Soluble connexin CT domains have proven to be a useful model for defining regulatory features of gap junctions9,10,63–65; however, they may not be the best model system for investigating structure-based mechanisms. The clearest example is provided from the well-studied Cx43 isoform. The electron crystallography structure of the CT-truncated Cx43 mutant (terminates at T263) suggested that region S255-T263 adopts an α-helical structure.66 Our NMR data for the soluble CT indicated that this region was highly flexible and did not have α-helical structure.17 Additionally, not all the expected nuclear Overhauser effects (NOEs) were observed in the two α-helical regions of the soluble CT structure. The differences can be attributed to the constraints imposed when attached to the transmembrane domain. Evidence to support this hypothesis was initially provided from CD data indicating the TM4-Cx43CT has more α-helical content than can be attributed solely to the addition of the individual TM4 and soluble CT domains.40 Subsequent studies identified the need of the TM4 for a structural responsiveness of the Cx43CT domain to changes in pH and phosphorylation state.39,53 The alterations in the secondary structure were global, suggesting channel permeability could be modulated by a relay mechanism whereby the additional CT structure affects the orientation of the transmembrane α-helices thereby influencing pore size. Intercellular communication would also be affected if the binding affinity of molecular partners involved in regulation were modulated by a change in the conformational preference of the CT.
In order to provide confidence in the interpretation of the TM4-CxCT structural data provided herein, verification is needed that the pH and TFE changes in α-helical content are real. A compelling case for pH can be made based upon correlating the CD and NMR data performed on the soluble Cx43CT and TM4-Cx43CT. CD data identified that the soluble Cx43CT domain at pH 5.8 is ~7% α-helical (little-to-no α-helical at pH 7.5).42 The solution structure of the Cx43CT (pH 5.8) confirmed a small amount of α-helical structure and identified their location (~16% α-helical, A315-T326 and D340-A34817). CD data determined that the TM4-Cx43CT (pH 5.8, 10% TFE) was 26.4% α-helical. Using the prediction of transmembrane helices in proteins program TMHMM Server v. 2.0, Cx43 cryo-electron crystallography structure,66 and the Cx26 X-ray crystal structure67 to calculate the amount of α-helical residues in the TM4 of Cx43 (~15%), the remaining ~11.4% α-helical content would be attributed to the Cx43CT domain. The resonance assignments of the TM4-Cx43CT were used to predict the α-helical residues in the CT domain and confirmed this larger amount of α-helical structure than the soluble Cx43CT (~30%40).
Providing confidence that the findings herein with TFE have significance are studies involving the Cx32CT domain. We previously identified that the intrinsically disordered soluble Cx32CT domain becomes α-helical upon binding calmodulin.15 The Cx32CT residues affected by calmodulin correspond exactly to those affected in the presence of TFE; suggesting TFE is stabilizing CT residues that are induced to form α-helical structure upon binding a molecular partner. Additionally, our data would suggest that a decrease in pH would elicit a stronger binding affinity as less energy would be needed to induce the formation of α-helical structure by calmodulin. Previously, we characterized three Cx32CT CMTX mutants (R219H, R230C, and F235C) and identified that while they formed functional channels, they all showed reduced binding affinity for calmodulin.15 The data presented here suggest the observed decrease in binding affinity for these mutations is caused by a decreased ability to form α-helical structure. For example, the R230C mutant has less α-helical structure than wild-type at pH 7.5 and all three mutants have less α-helical structure than wild-type at pH 5.8.
Comparison of the CD and NMR data indicates that CD underestimates the total number of CT residues that are α-helical. With this in mind, and because CD is an average of the ensemble of protein conformations in solution, the absolute number of CT residues sampling α-helical structure for each TM4-CxCT construct is most likely larger than the numbers (in parenthesis, estimate) provided in Tables II and III. This number represents residues in a stable α-helical conformation (e.g., 2 = 2) and does not take into account if they are fluctuating between α-helical and random coil structures (e.g., 2 = 4 residues in 50% conformational exchange). Support for the later occurring is provided from the chemical shift index (CSI) and NOE data from the TM4-Cx43CT.40 CSI predicts up to 70 of the CT residues could be α-helical; however, a large number of the expected NOEs for these α-helical regions are not present. The lack of NOEs is due to the dynamic nature of the α-helical regions (a key attribute for the function of an IDP—high specificity/low affinity). Additionally, studies involving phosphorylation of the TM4-Cx43CT determined that increases in α-helical content were not due to stabilization of rigid secondary structure, rather phosphorylation shifted the conformational preference of random coil residues to form a dynamic α-helical structure.53 A key attribute for the function of an IDP is to lower the energetic cost in order to enable binding of a select molecular partner. We predict that solvent acidification (and TFE) is causing a similar effect as phosphorylation to increase α-helical structure. Together with the observation that pH and TFE do not affect the His-tag/linker/remaining extracellular loop 2 residues or TM4 domain, the data strongly suggest that the CD can be used to predict α-helical content in order to compare the CT domain from connexin isoforms.
The Cx43 cryoelectron and Cx26 X-ray crystallography structures66,67 suggest a small number of CT residues may extend the TM4 α-helix into the cytoplasm. Based on this information, we expect at physiological pH with no phosphorylation and no interaction with a molecular partner, the α-helical content of the Cx26CT, Cx32CT, Cx37CT, Cx40CT, and Cx50CT domains will be limited to the TM4-CT interface (Tables II and III, in parenthesis), unlike the Cx43CT and Cx45CT domains. TFE, which our data suggest is stabilizing inherent α-helical structure (that phosphorylation and molecular partners would accomplish), is increasing the amount of α-helical structure outside the region juxtaposed to the membrane, for the Cx32CT, Cx43CT, and Cx45CT domains. Interesting, TFE has no effect on the Cx26CT structure which has no known phosphorylation sites or binding partners. Under acidic pH conditions (no TFE), the Cx32, Cx43, and Cx45 isoforms gain CT α-helical structure outside the membrane interface. TFE increases the α-helical content for the Cx32CT, Cx43CT, and Cx45CT domains, as well as the Cx50CT domain.
Our studies have identified that tethering of a connexin CT to its TM4 is necessary for any structural analysis of the CT domain. While pH modestly influences the CT structure, phosphorylation has the most dramatic effect. Since the CT domain of most connexins are phosphorylated throughout their life cycle, future studies using phospho-TM4-CxCT isoforms will be critical toward understanding the role that structure plays in regulating gap junction function. With a better understanding of the phosphorylation pattern of all connexins, not just Cx43, easier will be the identification of regions (i.e., form α-helical structure) that can be exploited to aid in the design of chemical modifiers to regulate the function of gap junctions.
MATERIALS AND METHODS
Plasmid Construction
DNA encoding the different connexin TM4-CxCT domains (Table I) was cloned by PCR and ligated into the E. coli pET-14b expression vector (N-terminal 6× His-tag, ampicillin resistance; Novagen). The soluble Cx50CT domain was cloned, using PCR, into the bacterial expression vector pGEX-6P-2 (Amersham Biosciences). This vector contains the coding region of glutathione S-transferase fused to the cleavage site for the PreScission protease (GE Heathcare). All constructs were verified by the University of Nebraska Medical Center’s DNA Sequencing Core Facility.
TM-CxCT Protein Expression and Purification
Expression plasmids for the TM4-Cx26CT (and mutants K224Q and L214P), TM4-Cx32CT (and mutants R230C, R219H, and F235C), TM4-Cx37CT, TM4-Cx40CT, TM4-Cx43CT (and assembly (CK1 and PKA; S325, S328, S330, S365D) and disassembly (Src, MAPK, and PKC; Y247, Y265, S255, S262, S279, S282, S368D) constructs), and TM4-Cx50CT were transformed into the C41(DE3) E. coli strain while TM4-Cx45CT was transformed into the C41(DE3) E. coli strain containing the Rosetta2(DE3)pLysS plasmid (Table I). All cells were then inoculated in Luria–Bertani (LB) medium or enriched minimal media.68 Cultures were incubated at 37°C with continuous agitation. At an optical density of 0.6 at 600 nm, 0.5 mM isopropyl β-D-thiogalactopyranoside was added, and growth was allowed to proceed for 4 h (typical final optical density ~1.6). The cells were harvested by centrifuging (1,000g for 30 min), washed with 1× phosphate-buffered saline (PBS), and their pellet weight recorded before storing at −20°C.
Cells were resuspended in 1× PBS (5 mL/g cells), mixed with bacterial protease inhibitor cocktail (250 μL/10 g cells; Sigma-Aldrich), and disrupted with three passages through an EmulsiflexC3 (Avestin) at 15,000 psi. Cell debris were removed by centrifugation (1,000g for 30 min) and a pellet containing the inclusion bodies was collected by high-speed centrifugation of the supernatant (25,000g for 1 h). The pellet was resuspended in 6 M urea, 1× PBS (pH 8.0), 1% Triton X-100, 1 mM β-mercaptoethanol, and 20 mM imidazole and placed on a rocker at 4°C overnight. The suspension was centrifuged again (25,000g for 1 h) and the supernatant was loaded onto a HisTrap HP affinity chromatography column using an ÄKTA FPLC (GE Health-care). The TM4-CxCT polypeptides were eluted at 300 mM imidazole using a step gradient of 20, 40, 80, 100, 300, and 500 mM imidazole. Fractions containing TM4-CxCT were identified by SDS-PAGE, pooled, and dialyzed overnight at 4°C using a 10 kDa Slide-A-Lyzer dialysis cassette (Pierce) against 1 M urea, 1% Triton X-100, 1 mM DTT, and 1 mM EDTA. In some cases, the optimal precipitate occurred when the dialysis buffer above was gradually reduced from 1 M urea and 1% Triton X-100 to water only (20% intervals, 2 h). The precipitate was collected and centrifuged (300g for 5 min), washed three times with MES buffer (20 mM MES buffer, 1 mM DTT, 1 mM EDTA, and 50 mM NaCl, pH 5.8), and resuspended in 500 μL of 20 mM MES buffer (pH 5.8 or 7.5) and 8% 1-palmitoyl-2-hydroxy-sn-glycero-3-[phospho-RAC-(1-glycerol)] (LPPG) at 42°C (overnight). The TM4-Cx50CT construct never precipitated and MES buffer/LPPG was added to the solution after dialysis against water. The concentrations of TM4-Cx37CT, TM4-Cx40CT, TM4-Cx43CT, TM4-Cx45CT, and TM4-Cx50CT were determined using a Jasco J-815 Circular Dichroism Spectrophotometer (high tension voltage to optical density conversion at 280 nm in spectra analysis) and/or a NanoDrop 1000 UV–visible spectrophotometer (Thermo Scientific) at 280 nm. The concentrations of TM4-Cx32CT, TM4-Cx26CT, and their mutants were determined using a NanoDrop 2000/2000c spectrophotometer (Thermo Scientific) at 205 nm. All proteins were confirmed for purity and analyzed for degradation by SDS-PAGE.
Soluble CxCT Protein Expression and Purification
The soluble Cx32, Cx37, Cx40, Cx43, Cx45, and Cx50 CT domains (Table I) were expressed and purified as described previously.14,15,41,69 The soluble Cx26CT peptide was purchased from LifeTein. All polypeptides were equilibrated in 1× PBS at pH 5.8 or pH 7.5 and 1 mM DTT and confirmed for purity by SDS-PAGE.
Circular Dichroism (CD) Measurements
CD experiments were performed using a Jasco J-815 spectrophotometer fitted with a Peltier temperature control system. Spectra for the soluble CxCT domains were recorded at 7°C (for Cx45CT 25°C), in 1× PBS (no LPPG detergent present) at pH 5.8 or 7.5. Spectra for the TM4-CxCT domains (100–200 μM) were recorded at 42°C in the MES buffer (with 8% LPPG) at pH 5.8 or 7.5. Additional CD spectra were collected in the presence of 2,2,2-trifluoroethanol (TFE; 5%, 10%, 15%, and 30% (v/v)). Of note, we have identified that LPPG, which is needed to solubilize the membrane-tethered CT, does not affect the structure of the soluble CT domain.14 The membrane-tethered CT experiments were performed in MES buffer with 8% LPPG. MES buffer was used because it is optimal for NMR experiments using cryo-probes.70 We have identified that phosphate buffer (i.e., used for the soluble CT) has a similar CD profile as the MES buffer.39 For each sample, 5 scans (wavelength range: 190–250 nm; response time, 1 s; scan rate, 50 nm/min; bandwidth 1.0 nm) were collected using a 0.01 cm quartz cell and processed using Spectra Analysis (Jasco). The concentrations for samples used for CD were as follows: soluble CT – Cx26, 590 μM; Cx32, 85 μM; Cx37, 150 μM; Cx40, 120 μM; Cx43, 125 μM; Cx50, 125 μM; TM4-tethered CT-Cx26 wild-type and mutants, 100 μM; Cx32 wild-type and mutants, 100 μM; Cx37, 150 μM; Cx40, 245 μM; Cx45, 100 μM; Cx50, 200 μM.
CD Data Analysis
DicroWeb analysis using the CONTINLL program with the SP175 reference set was used to estimate the total amount of α-helical content for each TM4-tethered construct (Tables (II–V)).71–73 The percentage of regular and distorted α-helices and β-sheets, β-turns, and random structures, which totaled 100% for all peptides (and were accepted only if the root mean square deviation (rmsd) between the observed and calculated value was below 0.1) was combined and averaged across experiments; however, predicted β-sheet, β-turn, and random coil structure content are not presented here. α-helical structure has a higher correlation coefficient and less RMS deviation than for other secondary structures, given that β-sheets appear mostly unordered in aqueous solution74 (no CD spectra show peak maxima at about 198 nm and peak minima about 215 nm, characteristic of beta sheet content). We therefore emphasize only the extent of α-helical structure in each peptide.
Nuclear Magnetic Resonance
NMR data were acquired at 42°C using a 600 MHz Varian INOVA NMR spectrometer fitted with a cryoprobe at the University of Nebraska Medical Center’s NMR Shared Resource Facility. Gradient-enhanced two-dimensional 15N-HSQC experiments were used to observe backbone amide resonances of 15N-labeled TM4-CxCT domains in MES buffer and 8% LPPG. The concentrations for TM4-tethered CT samples used for NMR were each ~500 μM. Data were acquired with 1024 complex points in the direct dimension and 128 complex points in the indirect dimension. Sweep widths were 8000 Hz in the proton dimension and 1720 Hz in the nitrogen dimension. NMR spectra were processed using NMRPipe75 and analyzed using NMRView.76
Supplementary Material
Acknowledgments
Contract grant sponsor: United States Public Health Service
Contract grant number: GM072631
Contract grant sponsor: Nebraska Research Initiative funding for the Nebraska
Center for Structural Biology
Contract grant sponsor: Eppley Cancer Center Support
Contract grant number: P30CA036727
Footnotes
Additional Supporting Information may be found in the online version of this article.
Reviewing Editor: Stephen Blacklow
References
- 1.Herve JC, Derangeon M. Gap-junction-mediated cell-to-cell communication. Cell Tissue Res. 2013;352:21–31. doi: 10.1007/s00441-012-1485-6. [DOI] [PubMed] [Google Scholar]
- 2.Nielsen MS, Axelsen LN, Sorgen PL, Verma V, Delmar M, Holstein-Rathlou NH. Gap junctions. Comprehensive Physiology. 2012;2:1981–2035. doi: 10.1002/cphy.c110051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pfenniger A, Wohlwend A, Kwak BR. Mutations in connexin genes and disease. Eur J Clin Invest. 2011;41:103–116. doi: 10.1111/j.1365-2362.2010.02378.x. [DOI] [PubMed] [Google Scholar]
- 4.Laird DW. The gap junction proteome and its relationship to disease. Trends Cell Biol. 2010;20:92–101. doi: 10.1016/j.tcb.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 5.Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36:1171–1186. doi: 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herve J, Bourmeyster N, Sarrouilhe D, Duffy H. Gap junctional complexes: From partners to functions. Progress in Biophysics and Molecular Biology. 2007;94:29–65. doi: 10.1016/j.pbiomolbio.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 7.Thevenin AF, Kowal TJ, Fong JT, Kells RM, Fisher CG, Falk MM. Proteins and mechanisms regulating gap-junction assembly, internalization, and degradation. Physiology (Bethesda) 2013;28:93–116. doi: 10.1152/physiol.00038.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moreno AP, Chanson M, Elenes S, Anumonwo J, Scerri I, Gu H, Taffet SM, Delmar M. Role of the carboxyl terminal of connexin43 in transjunctional fast voltage gating. Circ Res. 2002;90:450–457. doi: 10.1161/hh0402.105667. [DOI] [PubMed] [Google Scholar]
- 9.Morley GE, Taffet SM, Delmar M. Intramolecular interactions mediate pH regulation of connexin43 channels. Biophys J. 1996;70:1294–1302. doi: 10.1016/S0006-3495(96)79686-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anumonwo JM, Taffet SM, Gu H, Chanson M, Moreno AP, Delmar M. The carboxyl terminal domain regulates the unitary conductance and voltage dependence of connexin40 gap junction channels. Circ Res. 2001;88:666–673. doi: 10.1161/hh0701.088833. [DOI] [PubMed] [Google Scholar]
- 11.Revilla A, Castro C, Barrio LC. Molecular dissection of transjunctional voltage dependence in the connexin-32 and connexin-43 junctions. Biophys J. 1999;77:1374–1383. doi: 10.1016/S0006-3495(99)76986-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palatinus JA, Rhett JM, Gourdie RG. The connexin43 carboxyl terminus and cardiac gap junction organization. Biochim Biophys Acta. 2012;1818:1831–1843. doi: 10.1016/j.bbamem.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butkevich E, Hulsmann S, Wenzel D, Shirao T, Duden R, Majoul I. Drebrin is a novel connexin-43 binding partner that links gap junctions to the submembrane cytoskeleton. Curr Biol. 2004;14:650–658. doi: 10.1016/j.cub.2004.03.063. [DOI] [PubMed] [Google Scholar]
- 14.Kopanic JL, Al-mugotir MH, Kieken F, Zach S, Trease AJ, Sorgen PL. Characterization of the connexin45 carboxyl-terminal domain structure and interactions with molecular partners. Biophys J. 2014;106:2184–2195. doi: 10.1016/j.bpj.2014.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stauch K, Kieken F, Sorgen P. Characterization of the structure and intermolecular interactions between the connexin 32 carboxyl-terminal domain and the protein partners synapse-associated protein 97 and calmodulin. J Biol Chem. 2012;287:27771–27788. doi: 10.1074/jbc.M112.382572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bouvier D, Kieken F, Kellezi A, Sorgen PL. Structural changes in the carboxyl terminus of the gap junction protein connexin 40 caused by the interaction with c-Src and zonula occludens-1. Cell Commun Adhes. 2008;15:107–118. doi: 10.1080/15419060802014347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sorgen PL, Duffy HS, Sahoo P, Coombs W, Delmar M, Spray DC. Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J Biol Chem. 2004;279:54695–54701. doi: 10.1074/jbc.M409552200. [DOI] [PubMed] [Google Scholar]
- 18.Locke D, Kieken F, Tao L, Sorgen PL, Harris AL. Mechanism for modulation of gating of connexin26-containing channels by taurine. J Gen Physiol. 2011;138:321–339. doi: 10.1085/jgp.201110634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dogan J, Gianni S, Jemth P. The binding mechanisms of intrinsically disordered proteins. Physical chemistry chemical physics : PCCP. 2014;16:6323–6331. doi: 10.1039/c3cp54226b. [DOI] [PubMed] [Google Scholar]
- 20.Hsu WL, Oldfield CJ, Xue B, Meng J, Huang F, Romero P, Uversky VN, Dunker AK. Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Sci. 2013;22:258–273. doi: 10.1002/pro.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 22.Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Solan JL, Marquez-Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J Cell Biol. 2007;179:1301–1309. doi: 10.1083/jcb.200707060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moreno AP, Lau AF. Gap junction channel gating modulated through protein phosphorylation. Prog Biophys Mol Biol. 2007;94:107–119. doi: 10.1016/j.pbiomolbio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delmar M, Coombs W, Sorgen P, Duffy HS, Taffet SM. Structural bases for the chemical regulation of Connexin43 channels. Cardiovasc Res. 2004;62:268–275. doi: 10.1016/j.cardiores.2003.12.030. [DOI] [PubMed] [Google Scholar]
- 26.Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta. 2005;1711:154–163. doi: 10.1016/j.bbamem.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 27.Laird DW. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim Biophys Acta. 2005;1711:172–182. doi: 10.1016/j.bbamem.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 28.Smith MD, Jelokhani-Niaraki M. pH-induced changes in intrinsically disordered proteins. Methods Mol Biol. 2012;896:223–231. doi: 10.1007/978-1-4614-3704-8_14. [DOI] [PubMed] [Google Scholar]
- 29.Evans WH, Boitano S. Connexin mimetic peptides: specific inhibitors of gap-junctional intercellular communication. Biochem Soc Trans. 2001;29:606–612. doi: 10.1042/bst0290606. [DOI] [PubMed] [Google Scholar]
- 30.Boitano S, Evans WH. Connexin mimetic peptides reversibly inhibit Ca(2+) signaling through gap junctions in airway cells. Am J Physiol Lung Cell Mol Physiol. 2000;279:L623–630. doi: 10.1152/ajplung.2000.279.4.L623. [DOI] [PubMed] [Google Scholar]
- 31.Betsuyaku T, Nnebe NS, Sundset R, Patibandla S, Krueger CM, Yamada KA. Overexpression of cardiac connexin45 increases susceptibility to ventricular tachyarrhythmias in vivo. Am J Physiol Heart Circ Physiol. 2006;290:H163–171. doi: 10.1152/ajpheart.01308.2004. [DOI] [PubMed] [Google Scholar]
- 32.Inserte J, Ruiz-Meana M, Rodriguez-Sinovas A, Barba I, Garcia-Dorado D. Contribution of delayed intracellular pH recovery to ischemic postconditioning protection. Antioxid Redox Signal. 2011;14:923–939. doi: 10.1089/ars.2010.3312. [DOI] [PubMed] [Google Scholar]
- 33.Morel S, Kwak BR. Roles of connexins in atherosclerosis and ischemia-reperfusion injury. Current pharmaceutical biotechnology. 2012;13:17–26. doi: 10.2174/138920112798868638. [DOI] [PubMed] [Google Scholar]
- 34.Smyth JW, Zhang SS, Sanchez JM, Lamouille S, Vogan JM, Hesketh GG, Hong T, Tomaselli GF, Shaw RM. A 14-3-3 mode-1 binding motif initiates gap junction internalization during acute cardiac ischemia. Traffic. 2014;15:684–699. doi: 10.1111/tra.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fort AG, Spray DC. Trifluoroethanol reveals helical propensity at analogous positions in cytoplasmic domains of three connexins. Biopolymers. 2009;92:173–182. doi: 10.1002/bip.21166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Libich DS, Harauz G. Solution NMR and CD spectroscopy of an intrinsically disordered, peripheral membrane protein: evaluation of aqueous and membrane-mimetic solvent conditions for studying the conformational adaptability of the 18.5 kDa isoform of myelin basic protein (MBP) Eur Biophys J. 2008;37:1015–1029. doi: 10.1007/s00249-008-0334-8. [DOI] [PubMed] [Google Scholar]
- 37.Reiersen H, Rees AR. Trifluoroethanol may form a solvent matrix for assisted hydrophobic interactions between peptide side chains. Protein Eng. 2000;13:739–743. doi: 10.1093/protein/13.11.739. [DOI] [PubMed] [Google Scholar]
- 38.Kellezi A, Grosely R, Kieken F, Borgstahl GE, Sorgen PL. Purification and reconstitution of the connexin43 carboxyl terminus attached to the 4th transmembrane domain in detergent micelles. Protein Expr Purif. 2008;59:215–222. doi: 10.1016/j.pep.2008.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grosely R, Kieken F, Sorgen PL. Optimizing the solution conditions to solve the structure of the Connexin43 carboxyl terminus attached to the 4(th) transmembrane domain in detergent micelles. Cell Commun Adhes. 2010;17:23–33. doi: 10.3109/15419061.2010.487956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grosely R, Kieken F, Sorgen PL. (1)H, (1)(3)C, and (1)(5)N backbone resonance assignments of the connexin43 carboxyl terminal domain attached to the 4th transmembrane domain in detergent micelles. Biomol NMR Assign. 2013;7:299–303. doi: 10.1007/s12104-012-9432-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nelson TK, Sorgen PL, Burt JM. Carboxy terminus and pore-forming domain properties specific to Cx37 are necessary for Cx37-mediated suppression of insulinoma cell proliferation. Am J Physiol Cell Physiol. 2013;305:C1246–1256. doi: 10.1152/ajpcell.00159.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bouvier D, Spagnol G, Chenavas S, Kieken F, Vitrac H, Brownell S, Kellezi A, Forge V, Sorgen PL. Characterization of the structure and intermolecular interactions between the connexin40 and connexin43 carboxyl-terminal and cytoplasmic loop domains. J Biol Chem. 2009;284:34257–34271. doi: 10.1074/jbc.M109.039594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wishart DS, Sykes BD, Richards FM. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 44.Bouvier D, Kieken F, Sorgen PL. (1)H, (13)C, and (15)N backbone resonance assignments of the carboxyl terminal domain of Connexin40. Biomol NMR Assign. 2007;1:155–157. doi: 10.1007/s12104-007-9044-x. [DOI] [PubMed] [Google Scholar]
- 45.Barbar E. NMR characterization of partially folded and unfolded conformational ensembles of proteins. Biopolymers. 1999;51:191–207. doi: 10.1002/(SICI)1097-0282(1999)51:3<191::AID-BIP3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 46.Yum SW, Kleopa KA, Shumas S, Scherer SS. Diverse trafficking abnormalities of connexin32 mutants causing CMTX. Neurobiol Dis. 2002;11:43–52. doi: 10.1006/nbdi.2002.0545. [DOI] [PubMed] [Google Scholar]
- 47.Castro C, Gomez-Hernandez JM, Silander K, Barrio LC. Altered formation of hemichannels and gap junction channels caused by C-terminal connexin-32 mutations. J Neurosci. 1999;19:3752–3760. doi: 10.1523/JNEUROSCI.19-10-03752.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang Y, Sirkowski EE, Stickney JT, Scherer SS. Prenylation-defective human connexin32 mutants are normally localized and function equivalently to wild-type connexin32 in myelinating Schwann cells. J Neurosci. 2005;25:7111–7120. doi: 10.1523/JNEUROSCI.1319-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang GS, de Miguel M, Gomez-Hernandez JM, Glass JD, Scherer SS, Mintz M, Barrio LC, Fischbeck KH. Severe neuropathy with leaky connexin32 hemichannels. Annals of neurology. 2005;57:749–754. doi: 10.1002/ana.20459. [DOI] [PubMed] [Google Scholar]
- 50.Mese G, Londin E, Mui R, Brink PR, White TW. Altered gating properties of functional Cx26 mutants associated with recessive non-syndromic hearing loss. Hum Genet. 2004;115:191–199. doi: 10.1007/s00439-004-1142-6. [DOI] [PubMed] [Google Scholar]
- 51.Samanich J, Lowes C, Burk R, Shanske S, Lu J, Shanske A, Morrow BE. Mutations in GJB2, GJB6, and mitochondrial DNA are rare in African American and Caribbean Hispanic individuals with hearing impairment. Am J Med Genet A. 2007;143A:830–838. doi: 10.1002/ajmg.a.31668. [DOI] [PubMed] [Google Scholar]
- 52.Kopanic JL, Al-Mugotir M, Zach S, Das S, Grosely R, Sorgen PL. An Escherichia coli strain for expression of the connexin45 carboxyl terminus attached to the 4th transmembrane domain. Frontiers in pharmacology. 2013;4:1–8. doi: 10.3389/fphar.2013.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grosely R, Kopanic JL, Nabors S, Kieken F, Spagnol G, Al-Mugotir M, Zach S, Sorgen PL. Effects of phosphorylation on the structure and backbone dynamics of the intrinsically disordered connexin43 C-terminal domain. J Biol Chem. 2013;288:24857–24870. doi: 10.1074/jbc.M113.454389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Solan JL, Lampe PD. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 2014;588:1423–1429. doi: 10.1016/j.febslet.2014.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261–272. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Srisakuldee W, Jeyaraman MM, Nickel BE, Tanguy S, Jiang ZS, Kardami E. Phosphorylation of connexin-43 at serine 262 promotes a cardiac injury-resistant state. Cardiovasc Res. 2009;83:672–681. doi: 10.1093/cvr/cvp142. [DOI] [PubMed] [Google Scholar]
- 57.Zhou L, Kasperek EM, Nicholson BJ. Dissection of the molecular basis of pp60(v-src) induced gating of connexin 43 gap junction channels. J Cell Biol. 1999;144:1033–1045. doi: 10.1083/jcb.144.5.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sosinsky GE, Solan JL, Gaietta GM, Ngan L, Lee GJ, Mackey MR, Lampe PD. The C-terminus of Connexin43 adopts different conformations in the golgi and gap junction as detected with structure specific antibodies. Biochem J. 2007;408:375–385. doi: 10.1042/BJ20070550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saidi Brikci-Nigassa A, Clement MJ, Ha-Duong T, Adjadj E, Ziani L, Pastre D, Curmi PA, Savarin P. Phosphorylation controls the interaction of the connexin43 C-terminal domain with tubulin and microtubules. Biochemistry. 2012;51:4331–4342. doi: 10.1021/bi201806j. [DOI] [PubMed] [Google Scholar]
- 60.Chen J, Pan L, Wei Z, Zhao Y, Zhang M. Domain-swapped dimerization of ZO-1 PDZ2 generates specific and regulatory connexin43-binding sites. EMBO J. 2008;27:2113–2123. doi: 10.1038/emboj.2008.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Remo BF, Qu J, Volpicelli FM, Giovannone S, Shin D, Lader J, Liu FY, Zhang J, Lent DS, Morley GE, Fishman GI. Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ Res. 2011;108:1459–1466. doi: 10.1161/CIRCRESAHA.111.244046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson KE, Mitra S, Katoch P, Kelsey LS, Johnson KR, Mehta PP. Phosphorylation on Ser-279 and Ser-282 of connexin43 regulates endocytosis and gap junction assembly in pancreatic cancer cells. Mol Biol Cell. 2013;24:715–733. doi: 10.1091/mbc.E12-07-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kieken F, Mutsaers N, Dolmatova E, Virgil K, Wit AL, Kellezi A, Hirst-Jensen BJ, Duffy HS, Sorgen PL. Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ Res. 2009;104:1103–1112. doi: 10.1161/CIRCRESAHA.108.190454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Quinn MP, Palatinus JA, Harris BS, Hewett KW, Gourdie RG. A peptide mimetic of the connexin43 carboxyl terminus reduces gap junction remodeling and induced arrhythmia following ventricular injury. Circ Res. 2011;108:704–715. doi: 10.1161/CIRCRESAHA.110.235747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stergiopoulos K, Alvarado JL, Mastroianni M, Ek-Vitorin JF, Taffet SM, Delmar M. Hetero-domain interactions as a mechanism for the regulation of connexin channels. Circ Res. 1999;84:1144–1155. doi: 10.1161/01.res.84.10.1144. [DOI] [PubMed] [Google Scholar]
- 66.Unger VM, Kumar NM, Gilula NB, Yeager M. Three-dimensional structure of a recombinant gap junction membrane channel. Science. 1999;283:1176–1180. doi: 10.1126/science.283.5405.1176. [DOI] [PubMed] [Google Scholar]
- 67.Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature. 2009;458:597–602. doi: 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- 68.Weber DJ, Gittis AG, Mullen GP, Abeygunawardana C, Lattman EE, Mildvan AS. NMR docking of a substrate into the X-ray structure of staphylococcal nuclease. Proteins. 1992;13:275–287. doi: 10.1002/prot.340130402. [DOI] [PubMed] [Google Scholar]
- 69.Duffy HS, Sorgen PL, Girvin ME, O’Donnell P, Coombs W, Taffet SM, Delmar M, Spray DC. pH-dependent intramolecular binding and structure involving Cx43 cytoplasmic domains. J Biol Chem. 2002;277:36706–36714. doi: 10.1074/jbc.M207016200. [DOI] [PubMed] [Google Scholar]
- 70.Kelly AE, Ou HD, Withers R, Dotsch V. Low-conductivity buffers for high-sensitivity NMR measurements. J Am Chem Soc. 2002;124:12013–12019. doi: 10.1021/ja026121b. [DOI] [PubMed] [Google Scholar]
- 71.Whitmore L, Wallace BA. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32:W668–673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Whitmore L, Wallace BA. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 73.van Stokkum IH, Spoelder HJ, Bloemendal M, van Grondelle R, Groen FC. Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal Biochem. 1990;191:110–118. doi: 10.1016/0003-2697(90)90396-q. [DOI] [PubMed] [Google Scholar]
- 74.Viguera AR, Jimenez MA, Rico M, Serrano L. Conformational analysis of peptides corresponding to beta-hairpins and a beta-sheet that represent the entire sequence of the alpha-spectrin SH3 domain. J Mol Biol. 1996;255:507–521. doi: 10.1006/jmbi.1996.0042. [DOI] [PubMed] [Google Scholar]
- 75.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 76.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.