

Abstract
Bone-specific transcription of the osteocalcin (OC) gene is regulated principally by the Runx2 transcription factor and is further stimulated in response to 1α,25-dihydroxyvitamin D3 via its specific receptor (VDR). The rat OC gene promoter contains three recognition sites for Runx2 (sites A, B, and C). Mutation of sites A and B, which flank the 1α,25-dihydroxyvitamin D3-responsive element (VDRE), abolishes 1α,25-dihydroxyvitamin D3-dependent enhancement of OC transcription, indicating a tight functional relationship between the VDR and Runx2 factors. In contrast to most of the members of the nuclear receptor family, VDR possesses a very short N-terminal A/B domain, which has led to the suggestion that its N-terminal region does not contribute to transcriptional enhancement. Here, we have combined transient-overexpression, coimmunoprecipitation, in situ colocalization, chromatin immunoprecipitation, and glutathione S-transferase pull-down analyses to demonstrate that in osteoblastic cells expressing OC, VDR interacts directly with Runx2 bound to site B, which is located immediately adjacent to the VDRE. This interaction contributes significantly to 1α,25-dihydroxyvitamin D3-dependent enhancement of the OC promoter and requires a region located C terminal to the runt homology DNA binding domain of Runx2 and the N-terminal region of VDR. Together, our results indicate that Runx2 plays a key role in the 1α,25-dihydroxyvitamin D3-dependent stimulation of the OC promoter in osteoblastic cells by further stabilizing the interaction of the VDR with the VDRE. These studies demonstrate a novel mechanism for combinatorial control of bone tissue-specific gene expression. This mechanism involves the intersection of two major pathways: Runx2, a “master” transcriptional regulator of osteoblast differentiation, and 1α,25-dihydroxyvitamin D3, a hormone that promotes expression of genes associated with these terminally differentiated bone cells.
The rat osteocalcin (OC) gene encodes a bone-specific protein that is induced in osteoblasts with the onset of mineralization of the extracellular matrix, at late stages of differentiation (17). Bone-specific expression of the OC gene is regulated principally by the Runx2 transcription factor (1, 4), which has been proven to be essential for bone formation. Elimination of the Runx2 gene causes developmental defects in osteogenesis (10), and hereditary mutations in this gene are linked to specific ossification defects, as observed in cleidocranial dysplacia (16). The rat OC gene promoter contains three recognition sites for Runx2 interactions, site A (−605 to −595), site B (−438 to −430), and site C (−138 to −130). Mutation of all three Runx2 sites results in significantly reduced OC expression in bone-derived cells (8). Another key regulatory element that controls OC gene expression is recognized by the 1α,25-dihydroxyvitamin D3 receptor (VDR) complex upon ligand activation. This 1α,25-dihydroxyvitamin D3-responsive element (VDRE) is located in the distal region of the rat OC promoter (−465 to −437) and functions as an enhancer to increase OC transcription (11). Binding of the ligand 1α,25-dihydroxyvitamin D3 induces conformational changes in the ligand binding domain (LBD) of the receptor that enable it to interact with several coactivators of the p160 family, such as NcoA-1/SRC-1, NCoA-2/GRIP/TIF2/SRC-2, and ACTR/SRC-3, that are critical for transcriptional activation (3, 19). In addition, the multisubunit DRIP (for vitamin D receptor-interacting protein) complex binds to the VDR in response to the ligand (21, 22). This interaction occurs through a single motif on the VDR in much the same manner as for the coactivators and also results in transcriptional enhancement (20). Recently, a novel ATP-dependent chromatin-remodeling complex that binds to the VDR, termed WINAC (for WSTF [Williams syndrome transcription factor]-including nucleosome assembly complex), has been reported (9). WINAC shares components with two other chromatin-remodeling complexes, SWI/SNF and ISWI, and has been proposed to mediate recruitment of unliganded VDR to target genes. Nevertheless, subsequent interaction of the targeted VDR with transcriptional coregulators requires the presence of 1α,25-dihydroxyvitamin D3.
It has been indicated that in contrast to most steroid receptors, VDR does not contain an AF-1 transactivation domain at the N-terminal end (19). VDR possesses a very short (18-amino-acid) A/B domain, which would not be large enough to bind coactivators and function as a transactivation motif. Nevertheless, the possibility that the N-terminal region of VDR could be contributing to transcriptional enhancement by interacting with cell-specific transcription factors has not been formally evaluated.
Several lines of evidence suggest a tight functional relationship between Runx2 and the VDR complex in the regulation of OC gene transcription. Thus, it has been reported that mutation of Runx2 sites A and B (which flank the VDRE) abolishes 1α,25-dihydroxyvitamin D3 enhancement of OC promoter activity (8). It was also shown, both in vitro and in intact osteoblastic cells, that VDR and Runx2 factors interact simultaneously with their cognate sequences located in close proximity within the distal region of the OC promoter (VDRE and Runx2 site B, respectively) (18, 24). In addition, we have reported that in intact osteoblastic cells, the transcriptional coactivator p300 is recruited to the OC promoter by Runx2, where it up-regulates both basal and 1α,25-dihydroxyvitamin D3-enhanced OC promoter activity (24). p300 has been shown to interact, either directly or through other molecules such as the members of the p160 family of coactivators, with nuclear receptors in a ligand-dependent manner (19). Therefore, it has been suggested that Runx2-mediated recruitment of p300 to the OC promoter may facilitate the subsequent interaction of p300 with the VDR upon ligand stimulation (24). Thus, there is a need to address whether specific protein-protein interactions facilitate formation of these multisubunit complexes at 1α,25-dihydroxyvitamin D3-responsive promoters.
Here, we show that in osteoblastic cells expressing OC, VDR interacts directly with Runx2 bound to Runx2 site B. This protein-protein interaction involves a domain located C terminal to the runt homology DNA binding region of Runx2 and the N-terminal end of VDR. Together, our results indicate that Runx2 plays a key role in the 1α,25-dihydroxyvitamin D3-dependent stimulation of the OC gene promoter in osteoblastic cells by directly stabilizing the binding of the VDR to the VDRE. This is the first demonstration that the N-terminal domain of VDR mediates protein-protein interactions that result in formation of transcriptionally active complexes.
MATERIALS AND METHODS
GST pull-down assays.
The fusion proteins glutathione S-transferase (GST)-Runx2, GST-Runx2Δ376, GST-Runx2Δ361, GST-Runx2Δ230, GST-Runx2(209-361), GST-VDR, GST-VDRΔ1-111, GST-VDRΔ1-21, GST-VDRΔ22, GST-VDRΔ165, GST-VDRΔ111, and GST-VDRS51G were obtained by expression in Escherichia coli BL21 as previously reported (5, 18, 24). GST-free VDR proteins were obtained by cleaving GST-VDR with 1.0 U of factor Xa (New England Biolabs, Beverly, Mass.) at 4°C overnight. The pGEX-VDRΔ1-111 plasmid, coding for a mutated version of VDR in which amino acids 1 to 111 have been deleted, was generated by PCR amplification with a plasmid containing the full-length VDR gene as a template and the primers 5′-CGCACGCGTCACACTACGGAGGAGGAGGCCTTGAAGGACAGTCTGCGGC-3′ (forward) and 5′-GCCACGATGCGGCCGCATCAGGAGATCTCATTCCCCAAACACTTCG-3′ (reverse). The PCR product was then cleaved with SalI and NotI and subsequently subcloned in the pGEX5X3 vector (Pharmacia Biotech, Uppsala, Sweden). The pGEX-VDRΔ111 plasmid, coding for the first 111 amino acids of the N terminus of VDR fused to GST, was generated by PCR with the primers 5′-TTCCCGGGGATATCACATGGAGGCAATGGC-3′ (forward) and 5′-AAGGCCTCGTCGACCTTCCGCTTCAGGAT-3′ (reverse). The PCR product was cleaved with EcoRV and SalI and subsequently cloned into the pGEX5X3 vector (Pharmacia Biotech), which was previously cleaved with SmaI and SalI. The pGEX-VDRΔ1-21 plasmid, coding for a mutated version of VDR in which residues 1 to 21 have been deleted, was generated by PCR amplification with a plasmid containing the full-length VDR gene as a template and the primers 5′ CTTTGACCGGAACGTCGACCGGATCTGTGGGGTG-3′ (forward) and 5′-GCCACGATGCGGCCGCATCAGGAGATCTCATTCCCCAAACACTTCG-3′ (ASVDRNotI, reverse). The PCR product was then cleaved with SalI and NotI and cloned into the pGEX5X3 vector. The pGEX-VDRΔ22 plasmid, coding for the first 22 amino acids of the N terminus of VDR fused to GST, was generated by PCR with the primers 5′-CGCACGCGTCGACACTACATGGAGGCAATGGCGGCCAGCACTTCCCTGCC-3′ (SVDRSalI, forward) and 5′-GGTCTCCAGCGGCCGCACAGATTCAGGGCACGTTCC-3′ (reverse). The PCR product was then cleaved with SalI and NotI and cloned into the pGEX5X1 vector. The pGEX-VDRS51G plasmid, coding for a mutated version of VDR in which serine 51 has been mutated to glycine, was generated by PCR amplification with a plasmid containing the full-length VDR gene mutated as a template and the primers SVDRSalI (forward) and ASVDRNotI (reverse). The PCR product was then cleaved with SalI and NotI and cloned into the pGEX5X1 vector. The pGEX-VDRΔ165 plasmid, coding for the first 165 amino acids of the N terminus of VDR fused to GST, was generated by PCR with the primers SVDRSalI (forward) and 5′-GTCTGGAGTGCGGCCGCGAAGGTCAGCTCCCTCCACC-3′ (reverse). The PCR product was then cleaved with SalI and NotI and cloned into the pGEX5X1 vector. The pGEX-Runx2(209-361) plasmid, coding for the Runx2 domain between residues 209 and 361 fused to GST, was generated by cleaving the pCDNA-Runx2(209-361) plasmid with BamHI and XhoI and subsequently cloning into the pGEX4T1 vector. GST pull-down assays were carried out as described before (24). Coprecipitated Runx2, VDR, RXRα, and SRC-1 proteins were revealed by Western blotting with specific antibodies (M-70 for Runx2, C-20 for VDR, and D-20 for RXRα [Santa Cruz Biotechnology] and clone 1135 for SRC-1 [Upstate Biotechnology]). GST-VDR and GST-VDRΔ111 proteins were revealed by Western blotting with anti-VDR polyclonal antibodies directed against the C terminus of the VDR protein (C-20; Santa Cruz Biotechnology). To detect GST-VDRΔ111 proteins, a specific rat monoclonal antibody directed against the N-terminal region of VDR (AB-1; Oncogene Research) was used. In some cases (see the figure legends), GST-fused proteins were detected by using anti-GST polyclonal antibodies (Pharmacia Biotech).
His pull-down assay.
Twenty microliters of Ni2+-nitrilotriacetic acid agarose beads (Qiagen Inc., Valencia, Calif.) was incubated with 1.5 μg of His-Runx2 in a 500-μl final volume of binding buffer (20 mM Tris-Cl [pH 8.0], 100 mM KCl, 1 mM dithiothreitol [DTT], 1 mM EDTA, 0.5% NP-40, 0.05 mM phenylmethylsulfonyl fluoride, and 1 μg of bovine serum albumin per μl) for 45 min at 4°C with gentle agitation. The beads were collected by centrifugation at 9,000 × g for 30 s at 4°C and then incubated with 1.5 μg of purified recombinant protein (e.g., GST-VDR or GST-RXRα) for 2 h at 4°C with gentle agitation. Following extensive washes with buffer (20 mM Tris-Cl [pH 8.0], 100 mM KCl, 1 mM DTT, 1 mM EDTA, 0.5% NP-40, and 0.05 mM phenylmethylsulfonyl fluoride), the beads were resuspended in 20 μl of loading buffer and incubated at 95°C for 5 min. The samples were then fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (10% acrylamide), and the presence of GST-VDR and Runx2 proteins was revealed by Western blotting with specific antibodies (Santa Cruz).
Mammalian expression constructs.
Luciferase reporter constructs driven by the wild-type rat OC promoter (pOC-LUC) or a promoter in which the distal Runx2 sites A and/or B or the proximal Runx2 site C has been mutated (pmAB-LUC, pmA-LUC, pmB-LUC, and pmC-LUC, respectively) were described previously (8, 24). To generate the eukaryotic expression vector pCDNA-VDRΔ1-21, the pGEX-VDRΔ1-21 plasmid was cleaved with BamHI and NotI and subsequently cloned into the pCDNA 3.1HisA vector. The expression vector pCDNA-VDRΔ165 was generated by cutting the pGEX-VDRΔ165 plasmid with BamHI and NotI and subsequently cloning the insert into the pCDNA 3.1HisA vector. The pCMVβ plasmid, coding for the enzyme β-galactosidase and driven by cytomegalovirus (CMV) promoter, was purchased from Clontech (Palo Alto, Calif.).
Cell cultures, transient transfections, and reporter assays.
ROS 17/2.8 and ROS 24/1 osteoblastic cells were cultured as described previously (12). ROS17/2.8 cells express OC and respond to 1α,25-dihydroxyvitamin D3 with a three- to fivefold increase in OC expression, while ROS 24/1 cells express neither OC nor VDR. ROS 17/2.8 cells were plated in six-well plates (35-mm-diameter wells) and transiently transfected with 1 μg of pOC-LUC, pmAB-LUC, pmA-LUC, pmB-LUC, or pmC-LUC and 0.5 μg of CMV empty vector. In the VDR and Runx2 forced-expression experiments, cells were cotransfected with the expression vector as described before (24) at the concentrations described in the figure legends. The total amount of exogenous DNA was maintained at 9 μg/well with salmon sperm DNA (Invitrogen, Carlsbad, Calif.). Cells were transfected by using Lipofectamine Plus reagent (Invitrogen) according to the manufacturer's instructions. The pCMVβ plasmid was included as internal control for transfection efficiency. Cells were harvested at 24 h after transfection and assayed for luciferase (Promega, Madison, Wis.) and β-galactosidase (Clontech) activities in a luminometer (TD20/20; Turner Designs Institute, Sunnyvale, Calif.).
Immunoprecipitations.
Nuclear extracts from ROS 17/2.8 or ROS 24/1 cells cultured in the presence or absence of 10−8 M 1α,25-dihydroxyvitamin D3 for 4 h were prepared by a modified version of the Dignam method (6). Coimmunoprecipitation experiments were performed as described before (24). VDR was immunoprecipitated with an anti-VDR monoclonal antibody (Oncogene Research, Boston, Mass.), and the coimmunoprecipitated Runx2 was detected by Western blotting with an anti-Runx2 polyclonal antibody (Santa Cruz Biotechnology).
In situ colocalization analyses.
ROS 17/2.8 cells were plated at a density of 105 cells per well on gelatin-coated coverslips in six-well plates. Cells were cultured for 40 to 48 h in F12 medium containing 1% charcoal-stripped serum and treated with 10−8 M 1α,25-dihydroxyvitamin D3 for 45 min or 24 h. Untreated cells were used as control. For immunofluorescence, cells were washed twice with ice-cold phosphate-buffered saline (PBS) and fixed with 3.7% formaldehyde for 10 min on ice. Cells were then permeabilized for 20 min with PBS containing 0.5% Triton X-100. To detect endogenous Runx2 and VDR, cells were incubated with a mouse monoclonal antibody against Runx2 (dilution of 1:1,000) (29) and a rabbit polyclonal antibody against VDR (dilution of 1:200) for 1 h. Fluorochrome-conjugated Alexa antibodies (Invitrogen) raised in goat against mouse or rabbit were used as secondary antibodies (dilution of 1:800). Cells were then stained with DAPI (4′,6′-diamidino-2-phenylindole) to detect nuclei. Coverslips were mounted on glass slides with VectaShield mounting medium (Vector Laboratories, Burlingame, Calif.) and stored at −20°C until analyzed. A Zeiss Axioplan microscope equipped with a charge-coupled device was used to acquire images. The Metamorph imaging software was used to analyze images.
ChIP and Re-ChIP assays.
Chromatin immunoprecipitation (ChIP) studies were performed as described earlier (24, 25), with modifications. All of the steps were performed at 4°C. ROS 17/2.8 cell cultures (100-mm-diameter plates) were incubated for 5 min with 1% formaldehyde and gentle agitation. The cross-linking was stopped by treatment with 0.125 M glycine for 5 min. The cells were then washed with 10 ml of PBS, scraped off in the same volume of PBS, and collected by centrifugation at 1,000 × g for 5 min. The cell pellet was resuspended in 3 ml of lysis buffer (50 mM HEPES [pH 7.8], 20 mM KCl, 3 mM MgCl2, 0.1% NP-40, and a cocktail of proteinase inhibitors) and incubated for 10 min on ice. The cell extract was then collected by centrifugation at 1,000 × g for 5 min, resuspended in 3.0 ml of sonication buffer (50 mM HEPES [pH 7.9], 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% deoxycholate acid, 0.1% SDS, and a cocktail of proteinase inhibitors) and incubated for 10 min on ice. To reduce the length of the chromatin fragments to approximately 500 bp (confirmed by electrophoretic analysis and PCR), the extract was sonicated with a Misonix sonicator (model 3000), using 10 15-s pulses at 30% power. After centrifugation at 16,000 × g, the supernatant was collected, frozen in liquid nitrogen, and kept at −80°C. An aliquot was kept for A260 measurements. Cross-linked extracts (6 U A260) were resuspended in sonication buffer to a final volume of 500 μl. The samples were plecleared by incubation with 30 μl of protein A/G-agarose beads preblocked with bovine serum albumin (Santa Cruz Biotechnology) for 15 min at 4°C with agitation. After centrifugation at 1,000 × g for 5 min, the supernatant was collected and immunoprecipitated with either an anti-Runx2 polyclonal antibody (Santa Cruz Biotechnology), an anti-VDR monoclonal antibody (Oncogene Research), an anti C/EBP β polyclonal antibody (Santa Cruz Biotechnology), or an anti X-press monoclonal antibody (Invitrogen). The immunocomplexes were recovered by the addition of 30 μl of protein A-agarose beads and subsequent incubation for 1 h at 4°C with agitation. The complexes were washed twice with sonication buffer, twice with sonication buffer plus 500 mM NaCl, twice with LiCl buffer (100 mM Tris-HCl [pH 8.0], 500 mM LiCl, 0.1% NP-40, and 0.1% deoxycholic acid), and twice with dialysis buffer (2 mM EDTA and 50 mM Tris-HCl [pH 8.0]), with the solution incubated at each washing for 5 min at 4°C. The protein-DNA complexes were then eluted by incubation with 100 μl of elution buffer (50 mM NaHCO3 and 1% SDS) for 15 min at 65°C. After centrifugation at 1,000 × g for 5 min, the supernatant was collected and incubated with 10 μg of RNase A per ml at 65°C to reverse the cross-linking. The proteins were then digested with 200 μg of proteinase K per ml for 2 h at 50°C. The DNA was recovered by phenol-chloroform extraction and ethanol precipitation with tRNA (5 μg/ml) as a precipitation carrier. The PCR conditions and the primers used were reported previously (23, 24). The Re-ChIP assays were performed as described earlier (13). The immunoprecipitated complexes obtained by ChIP were eluted by incubation for 30 min at 37°C in 25 μl of 10 mM DTT. After centrifugation, the supernatant was diluted 20 times with sonication buffer and subjected to the ChIP procedure.
RESULTS
The distal Runx site B is required for 1α,25-dihydroxyvitamin D3-enhanced OC promoter activity.
Previous reports have suggested cooperativity between the Runx2 and VDR factors in the 1α,25-dihydroxyvitamin D3-dependent transcriptional enhancement of OC gene expression in osteoblastic cells (8, 18, 24). To gain understanding of the molecular bases for a functional relationship between these two primary regulators, we assessed the contribution of the distal Runx2 sites in OC promoter responsiveness to the 1α,25-dihydroxyvitamin D3.
As shown in Fig. 1, single or combined mutations of the distal Runx2 binding sites (site A, −605 to −595; site B, −440 to −435) which flank the VDRE (−465 to −440) inhibit 1α,25-dihydroxyvitamin D3-enhancement of OC promoter activity. Hence, when we transiently transfected the osteoblastic cells ROS17/2.8 with OC promoter-luciferase reporter constructs carrying mutations in either Runx2 site A, site B, or both sites, which prevents Runx2 interaction, a significant inhibition in the 1α,25-dihydroxyvitamin D3-dependent stimulation of the OC promoter activity was detected (Fig. 1). Similarly, transient overexpression of Runx2 in ROS 17/2.8 cells resulted in both stimulation of OC promoter basal activity and up-regulation of the 1α,25-dihydroxyvitamin D3-dependent enhancement of this promoter (Fig. 2). This Runx2-dependent up-regulation requires the distal Runx2 elements and the VDRE (data not shown).
FIG. 1.

The Runx2 sites are required for 1α,25-dihydroxyvitamin D3-dependent enhancement of the OC promoter. (A) Schematic representation of various constructs containing wild-type or mutated versions of the rat OC gene promoter controlling the luciferase reporter gene. Each Runx2 site is indicated with an open box, and the mutated Runx2 sites are marked with an X. The VDRE is marked with a black box. pOC-LUC, wild-type rat OC gene promoter; pOC-LUCmAB, rat OC gene promoter with mutated Runx2 sites A and B; pOC-LUCmB, rat OC gene promoter with mutated Runx2 site B; pOC-LUCmA, rat OC gene promoter with mutated Runx2 site A; pOC-LUCmC, rat OC gene promoter with mutated Runx2 site C. (B) Each pOC-LUC construct (1 μg) was transiently transfected into ROS 17/2.8 cells, and luciferase activity was measured to evaluate responsiveness to 24 h of 1α,25-dihydroxyvitamin D3 [1α,25-(OH)2 vitamin D3] treatment. The effect of 1α,25-dihydroxyvitamin D3 is reported as fold stimulation of basal promoter activity levels. All transfections were normalized by cotransfection of the pCMV-β-galactosidase expression plasmid and represent several independent experiments, each assayed in triplicate. Each bar represents the mean ± standard error of the mean (n = 3; P < 0.03). WT, wild type.
FIG. 2.

Runx2 up-regulates basal and 1α,25-dihydroxyvitamin D3-enhanced OC gene promoter activity. (A) Schematic representation of the construct containing the rat OC gene promoter controlling the luciferase reporter gene (pOC-LUC) utilized in this analysis. See the legend to Fig. 1 for an explanation of the symbols. (B) ROS 17/2.8 cells were transiently cotransfected with a Runx2 expression plasmid (2 μg) and the pOC-LUC reporter construct (1 μg). The presence (+) or absence (−) of the Runx2 plasmid is shown below the bars. Luciferase activity was assayed after culturing the cells for 24 h in the presence (black bars) or absence (white bars) of 10−8 M 1α,25-dihydroxyvitamin D3 [1α,25-(OH)2D3]. The data were normalized to values for pCMV-β-galactosidase activity as an internal control. Each bar represents the mean ± standard error of the mean (n = 6; P < 0.03).
In agreement with previous reports (8) a mutation in Runx2 site C, which is located within the proximal promoter region (−138 to −130), did not prevent 1α,25-dihydroxyvitamin D3-dependent up-regulation of the OC promoter activity (Fig. 1). However, it appears that this site also contributes to 1α,25-dihydroxyvitamin D3 responsiveness, as the OC promoter construct containing a mutated Runx2 site C exhibited reduced 1α,25-dihydroxyvitamin D3-dependent stimulation compared to that shown by the wild-type OC promoter.
Together, these results indicate that although all three Runx2 sites contribute to full responsiveness of the OC promoter to 1α,25-dihydroxyvitamin D3, Runx2 site B is absolutely required. Interestingly, site B is located immediately adjacent to the VDRE, suggesting that the Runx2 and VDR factors could be components of a multisubunit complex bound to the OC promoter following ligand stimulation.
Runx2 and VDR are components of the same nuclear complex in osteoblastic cells treated with 1α,25-dihydroxyvitamin D3.
To assess whether Runx2 and the VDR are components of the same nuclear protein complexes in bone-derived cells cultured in the presence of 1α,25-dihydroxyvitamin D3, we combined coimmunoprecipitation and Western blot analyses. VDR was immunoprecipitated from nuclear extracts of ROS 17/2.8 osteoblastic cells treated with 10−8 M 1α,25-dihydroxyvitamin D3 for 4 h (Fig. 3A, lower panel, lane 2) but not from cells cultured in the absence of the hormone (Fig. 3A, lower panel, lane 3). This immunoprecipitation was specific, as VDR was not detected when a nonspecific mouse immunoglobulin G fraction was used in the reaction (Fig. 3A, lower panel, lane 4) or when nuclear extracts from ROS 24/1 osteoblastic cells, which express neither VDR nor OC (12, 14), were analyzed (Fig. 3A, lower panel, lane 5). More importantly, the bone-specific transcription factor Runx2 was present in the material immunoprecipitated from osteoblastic ROS 17/2.8 cells treated with 1α,25-dihydroxyvitamin D3 (Fig. 3A, upper panel, lane 2), strongly indicating that Runx2 and VDR are components of the same nuclear complex. Under our experimental conditions, Runx2 protein levels remained constant in both control and 1α,25-dihydroxyvitamin D3-treated ROS 17/2.8 and ROS 24/1 cells (Fig. 3B, upper panel, compare lane 1 with lane 2 and lane 3 with lane 4), and VDR was enriched only in the nuclear fraction of ROS 17/2.8 cells treated with 1α,25-dihydroxyvitamin D3 (Fig. 3B, middle panel, compare lanes 1 and 2).
FIG. 3.

Runx2 and VDR are components of the same nuclear protein complexes in osteoblastic cells expressing OC. (A) Nuclear extracts (N.E.) (200 μg) from ROS 17/2.8 or ROS 24/1 cells cultured in the presence of 10−8 M 1α,25-dihydroxyvitamin D3 [1α,25-(OH)2D3] for 4 h were immunoprecipitated (IP) with an anti-VDR (αVDR) monoclonal antibody (Oncogene) or with nonspecific mouse immunoglobulin G fraction (Santa Cruz Biotechnology). Immunoprecipitated complexes were fractionated by SDS-PAGE (8% acrylamide) and analyzed by Western blotting (WB) with anti-Runx2 (upper panel) or anti-VDR (lower panel) antibodies (Santa Cruz Biotechnology). (B) Aliquots (15 μg) of N.E. samples from the ROS 17/2.8 or ROS 24.1 cells shown in panel A were analyzed by Western blotting with anti-Runx2 (upper panel) or anti-VDR (middle panel) antibodies. To control for equal loading, the membrane was reblotted against TFIIB (lower panel). The different combinations of antibodies and N.E. included in each lane are indicated at the top of the blots. The positions of molecular markers are shown to the left of the blots.
To further demonstrate that Runx2 and VDR interact within the nuclei of osteoblastic cells, we determined whether both factors colocalize in ROS 17/2.8 cells cultured in the presence or absence of 1α,25-dihydroxyvitamin D3. As shown in Fig. 4, treatment with 1α,25-dihydroxyvitamin D3 resulted in nuclear enrichment of VDR and, most importantly, in colocalization with Runx2. Thus, the punctuated pattern normally shown by Runx2 in ROS 17/2.8 cells (28) overlapped significantly (40%) with the nuclear signal associated with VDR upon ligand stimulation (Fig. 4).
FIG. 4.

Runx2 and VDR colocalize in the nuclei of osteoblastic cells. Untreated ROS 17/2.8 cells (control) or cells treated with 1α,25-dihydroxyvitamin D3 were processed for in situ immunofluorescence as described in Materials and Methods. A weak nuclear-cytoplasmic signal for VDR is detected in the control cells, while Runx2 exhibits a characteristic punctate nuclear staining. The overlap between the two signals is undetectable as assessed by the measure colocalization function of Metamorph software (top panels). The treatment of cells with 1α,25-dihydroxyvitamin D3 for 45 min (middle panels) or 24 h (bottom panels) significantly increases nuclear staining for the VDR and the subsequent overlap with the Runx2 signal (10 and 40% overlap, respectively). The DAPI panel shows the stained nuclei in the field.
Runx2-VDR interaction is mediated by specific protein domains.
The presence of Runx2 and VDR within the same nuclear complexes raised the possibility that both factors were associated through a direct protein-protein interaction. To evaluate this possibility, we carried out GST and His pull-down analyses. Recombinant full-length GST-Runx2, GST-RXRα, His-Runx2, and GST-VDR proteins were produced in E. coli and tested for their ability to interact in vitro. GST-VDR bound His-Runx2 in the absence of 1α,25-dihydroxyvitamin D3 and RXRα, the specific heterodimer partner of VDR (Fig. 5B, lane 3) (26, 27). Addition of 1α,25-dihydroxyvitamin D3 (Fig. 5B, lane 4) or bacterially produced recombinant RXRα (Fig. 5B, lane 5) did not affect significantly the Runx2-VDR interaction. Similarly, His-Runx2 bound to an Ni2+ resin specifically retained GST-VDR (Fig. 5C, lanes 4 and 6). These interactions are specific, as neither His-Runx2 nor GST-VDR was precipitated by the resins (Fig. 5B, lane 2, and C, lane 3) or GST alone (data not shown). In addition, Runx2 did not interact with GST-RXRα (Fig. 5B, lane 6), further demonstrating the specificity of the VDR-Runx2 interaction.
FIG. 5.

Runx2 and VDR interact in vitro. Full-length Runx2 and VDR were expressed as His- and GST-tagged fusion proteins, respectively, in bacteria, and their ability to interact in vitro was evaluated by pull-down assays. (A) Schematic representation of the His-Runx2 and GST-VDR recombinant proteins. The runt homology domain of Runx2, localized between amino acid residues 92 and 220, is indicated with a black box. The zinc finger motif of VDR (DBD), localized between amino acid residues 24 and 91, and the LBD of VDR, localized between residues 227 and 422, are also indicated. (B) GST-VDR (1.5 μg) bound to 20 μl of glutathione-Sepharose resin was incubated with His-Runx2 (1.5 μg) and/or GST-RXRα (1.5 μg) for 2 h at 4°C. Precipitated proteins were then analyzed by Western blotting with anti-Runx2 (upper panel) and anti-VDR (lower panel) antibodies. (C) His-Runx2 (1.5 μg) bound to 20 μl of Ni2+-nitrilotriacetic acid (NTA) resin was incubated with GST-VDR (1.5 μg) and/or GST-RXRα (1.5 μg) for 2 h at 4°C. Precipitated proteins were analyzed by Western blotting with specific antibodies against VDR (upper panel) and Runx2 (lower panel). The different combinations of recombinant proteins and the presence of 10−8 M 1α,25-dihydroxyvitamin D3 [1α,25-(OH)2D3] are indicated at the top of each panel. The positions of molecular mass markers are shown at the left of the blots.
Using the same approach, we next defined the domains required for the Runx2-VDR protein-protein interaction. As shown in Fig. 6, it was found that the N terminus of VDR is necessary for its interaction with Runx2. Thus, GST-Runx2 was unable to precipitate a VDRΔ1-111 mutant protein (Fig. 6A), in which amino acid residues 1 to 111 had been deleted (Fig. 6B, compare lanes 7 and 8). In addition, we determined that these 111 amino acid residues from the N terminus of VDR (Fig. 7A) are sufficient for interaction with Runx2, as the GST-VDRΔ111 protein, which contains this domain, precipitated Runx2 with high affinity (Fig. 7B, upper panel, lane 4).
FIG. 6.

VDR requires a specific domain to interact with Runx2. (A) Schematic representation of the GST-Runx2 and GST-free VDR recombinant proteins utilized in these GST pull-down experiments. See the legend to Fig. 5 for an explanation of the symbols. The VDRΔ1-111 protein corresponds to a mutated VDR form in which amino acid residues 1 to 111, which include the zinc finger domain, have been deleted. (B) GST-Runx2 (1.5 μg) bound to 20 μl of glutathione-Sepharose beads was incubated with 1.5 μg of either GST-free VDR or GST-free VDRΔ1-111 for 2 h at 4°C. Precipitated VDR (upper panel) and GST-Runx2 (lower panel) proteins were then determined by Western blotting. The combinations of proteins used in each binding reaction are indicated at the top of the blots. The positions of molecular mass markers are shown to the left of the blots.
FIG. 7.

The N terminus of VDR is sufficient for interaction with Runx2. (A) Schematic representation of the His-Runx2 and GST-VDR recombinant proteins utilized in these GST pull-down experiments. See the legend to Fig. 5 for an explanation of the symbols. The GST-VDRΔ111 protein corresponds to a mutated form of VDR that lacks amino acid residues 112 to 427. The GST-VDRΔ1-21 protein corresponds to a mutated form of VDR that lacks amino acid residues 1 to 21. The GST-VDRΔ22 protein corresponds to a mutated form of VDR that lacks amino acid residues 23 to 427. (B) GST-VDR (1.5 μg) or GST-VDRΔ111 (1.5 μg) bound to glutathione-Sepharose beads were incubated with 1.5 μg of His-Runx2 for 2 h at 4°C. Precipitated Runx2 (upper panel) and GST-VDR (lower panel) proteins were visualized by Western blotting with anti-Runx2 and anti-GST antibodies, respectively. (C) GST-VDRΔ111 (1.5 μg) and GST-VDRΔ22 (1.5μg) bound to glutathione-Sepharose beads were incubated with 1.5 μg of His-Runx2 for 2 h at 4°C. Precipitated Runx2 (upper panel) and GST-VDR (lower panel) proteins were visualized by Western blotting with anti-Runx2 and anti-GST antibodies, respectively. (D) GST-VDRS51G corresponds to a mutated VDR protein in which serine residue 51 has been changed to glycine. GST-VDRS51G (1.5 μg), GST-VDR (1.5 μg), and GST-VDRΔ1-21 (1.5 μg) were incubated with His-Runx2 protein and analyzed as described above. The combinations of proteins used in each binding reaction are indicated at the top of the blots. The positions of molecular mass markers are shown to the left of the blots.
It has been indicated that VDR does not possess a proper N-terminal domain, as it includes a very short (18-residue) A/B region, which would be not large enough to function as a transactivation domain (19). We then assessed whether this short A/B segment was sufficient to bind Runx2. A GST-VDRΔ22 protein, which contains only the first 21 residues from the N terminus of VDR, did not precipitate Runx2 (Fig. 7C, lane 4). In contrast, a VDRΔ1-21 mutant protein, which lacks these first 21 residues, bound to Runx2 with high affinity, equivalent to that exhibited by the wild-type receptor molecule (Fig. 7C, compare lanes 3 and 4). These results indicate that the short N-terminal A/B domain of VDR is not required for its interaction with Runx2. Our findings also indicate that VDR binds to Runx2 through a domain located between residues 22 and 111, which corresponds to the DNA binding region of the receptor (Fig. 7A).
To determine whether maintenance of the structural motifs at the DNA binding domain (DBD) of VDR is required to interact with Runx2, we mutated the serine 51 residue to glycine (S51G). This S51 is located within an α-helix between the two zinc finger motifs that form the DBD of VDR. Hsieh et al. have shown that the mutation S51G prevents binding of VDR to DNA and significantly reduces VDR-mediated transactivation (7). This effect would be the result of an increase in flexibility at this region and therefore a decrease in the structural properties that are necessary for the correct function of the receptor. The VDRS51G mutant protein was unable to interact with Runx2, thus indicating that changes in the structural features of the DBD of VDR prevent interaction with Runx2 (Fig. 7D, lane 5). The interaction of Runx2 with the zinc finger-containing DBD of VDR is specific, as other members of the steroid receptor family, such as RXR, do not bind Runx2 (Fig. 3, lane 6).
The VDR-Runx2 interaction was further examined by use of GST pull-down experiments with recombinant GST-VDR and nuclear extracts from ROS 17/2.8 cells, which are enriched in Runx2 (Fig. 3B). Both VDR proteins, GST-VDRΔ111 and GST-VDR, were able to precipitate Runx2 from the nuclear extracts with similar efficiency (Fig. 8B, second panel, compare lanes 3 and 6, respectively). This protein-protein interaction was independent of 1α,25-dihydroxyvitamin D3, as GST-VDR brought down equivalent amounts of Runx2 in either the absence or presence of 10−8 M 1α,25-dihydroxyvitamin D3 (Fig. 8B, second panel, compare lanes 6 and 7). As expected, the presence of the ligand was absolutely necessary for the interaction between GST-VDR and the transcriptional coactivator SRC-1 present in the nuclear extracts from ROS 17/2.8 cells (Fig. 8C, upper panel, compare lanes 6 and 7). This ligand-dependent interaction requires the C-terminal LBD of VDR (19), as the GST-VDRΔ1-111 mutant receptor protein, which lacks the N-terminal region of VDR, was capable of recruiting SRC-1 only in the presence of 1α,25-dihydroxyvitamin D3 (Fig. 8C, upper panel, compare lanes 4 and 5). Accordingly, the GST-VDRΔ111 mutant receptor, which lacks the LBD, was unable to precipitate SRC-1 in either the absence or presence of 1α,25-dihydroxyvitamin D3 (data not shown). Because SRC-1 appears to bind with higher efficiency to the truncated form VDRΔ1-111 (Fig. 8C, compare lanes 5 and 7), we determined whether binding of Runx2 to VDR reduces the interaction between the receptor and SRC-1. Nuclear extracts from ROS 17/2.8 cells were enriched in Runx2 by addition of purified His-Runx2 protein (2 μg) and then incubated with GST-VDR. As expected, a higher Runx2 concentration in the nuclear extracts was reflected by a significantly larger amount of the Runx2 precipitated by GST-VDR (Fig. 8D, compare lanes 3 and 4). Nevertheless, this increased Runx2 concentration did not inhibit the ligand-dependent VDR-SRC-1 interaction (Fig. 8D, compare lanes 3 and 4).
FIG. 8.

Runx2 and VDR are components of multisubunit nuclear complexes in osteoblastic cells. (A) Schematic representation of the GST-VDR proteins used in the GST pull-down assays. See the legend to Fig. 5 for an explanation of the symbols. (B) One hundred fifty micrograms of nuclear extract (N.E.) from ROS 17/2.8 cells was incubated with either GST-VDR, GST-VDRΔ111, or GST-VDRΔ1-111 (1 μg of each) bound to glutathione-Sepharose beads. Binding reactions were carried out in the presence (+) or absence (−) of 10−8 M 1α,25-dihydroxyvitamin D3 [1α,25-(OH)2D3]. The precipitated proteins were resolved by SDS-PAGE (10% acrylamide) and revealed byWestern blotting with specific antibodies against RXRα (upper panel), Runx2 (second panel), VDR (third and fourth panels), or GST (lower panel). The different VDR protein forms were detected as follows: GST-VDR and GST-VDRΔ1-111 (third panel) with a specific antibody against the C terminus of VDR; GST-VDRΔ111 (fourth panel) with an antibody against the N terminus of VDR. The asterisk indicates a cleavage product of the GST-VDR protein. (C) Aliquots of the precipitated protein samples shown in panel B were resolved by SDS-PAGE (7% acrylamide) and analyzed by Western blotting detecting the presence of SRC-1 (upper panel) as well as GST-VDR and GST-VDRΔ1-111 (lower panel). (D) One hundred fifty micrograms of nuclear extract from ROS 17/2.8 cells was enriched in Runx2 by adding 2 μg of purified His-Runx2 (lanes 3 and 4). The samples were then incubated with GST-VDR and analyzed by Western blotting as described above. The combinations of proteins used in each binding reaction are indicated at the top of the blots. The positions of molecular mass markers are shown to the left of the blots.
In addition, we confirmed that heterodimerization of GST-VDR with the RXRα present in the ROS17/2.8 nuclear extracts was a 1α,25-dihydroxyvitamin D3-dependent process, as no RXRα protein is detected in the precipitate when the incubation was carried out in the absence of the ligand (Fig. 8B, second panel, compare lanes 4 and 5 and lanes 6 and 7). Together, our results indicate that VDR is able to bind Runx2 through its N-terminal domain within the context of the nuclear protein fraction.
We next analyzed whether expression of the N-terminal domain of VDR affects the activity of the OC promoter within osteoblastic cells. Constructs coding for wild-type VDR and two VDR mutant forms (Fig. 9A) were expressed in ROS 17/2.8 cells and analyzed for their ability to either enhance or inhibit OC promoter activity. As the first 111 residues of VDR do not include a nuclear localization signal, we expanded the VDR N-terminal domain to residue 165, hence generating a truncated VDR protein that includes its natural nuclear localization signal. This VDRΔ165 protein is expressed and binds Runx2 with an efficiency similar to that of VDRΔ111 (data not shown). As shown in Fig. 9B, we reproducibly found that the OC promoter activity was slightly increased when wild-type VDR or VDRΔ1-22 was expressed. In contrast, expression of VDRΔ165 significantly reduced the OC promoter activity in a dose-dependent manner. Together these results indicate that expression of the Runx2 binding domain of VDR, which lacks the LBD and hence is unable to recruit transcriptional coactivators, interferes with the functional coupling between VDR and Runx2 at the OC promoter.
FIG. 9.

Expression of the VDRΔ165 domain inhibits OC promoter activity in osteoblastic cells. (A) Schematic representation of the construct carrying the full-length OC promoter in front of the luciferase reporter gene (pOC-LUC) and the expression plasmids encoding wild-type VDR (pcDNA-VDR) and the N-terminal domain of VDR (pcDNAΔ165). (B) pOC-LUC and VDR expression plasmids were cotransfected in ROS 17/2.8 cells cultured in the presence (black bars) or absence (white bars) of 1α,25-dihydroxyvitamin D3 [1α,25-(OH)2D3] for 24 h. The presence or absence of each plasmid is shown below the bars. pOC-LUC was added at a concentration of 1 μg/well (35 mm), whereas pcDNAΔ165 and pcDNA-VDR were added at 0.25 and 0.5 μg/well. Luciferase activity was evaluated as described in the legend to Fig. 1. Each bar represents the mean ± standard error of the mean (n = 6; P < 0.03).
We also mapped the region of Runx2 responsible for the interaction with VDR. For that purpose, a series of Runx2 mutant proteins in which C-terminal sequences had been progressively deleted (Fig. 10A) were expressed in E. coli and tested for their ability to bind full-length VDR. GST-Runx2 full-length protein, GST-Runx2Δ376, and GST-Runx2Δ361 were all capable of precipitating VDR (Fig. 10B, upper panel, lanes 3, 4, and 5, respectively). However, GST-Runx2Δ376 and GST-Runx2Δ361 appeared to bind purified VDR with less affinity than full-length Runx2, as the signal corresponding to bound VDR in the Western blot was significantly decreased (Fig. 10B upper panel, compare lanes 3 and 5). This reduced VDR signal is not due to the loading of uneven concentrations of the different GST-Runx2 recombinant proteins, as equivalent levels of the isoforms were detected by Western blotting in each reaction mix (Fig. 10B, middle panel, compare lanes 3, 4, and 5). Also, all of these protein-protein interactions were specific, as neither the Sepharose resin nor the purified GST protein retained detectable levels of VDR (Fig. 10B, lower panel, lanes 2 and 6, respectively). GST-Runx2Δ361 contained the minimal protein domain required to interact with VDR in vitro, as the GST-Runx2Δ230 deletion mutant (Fig. 10C) was unable to precipitate the receptor protein (Fig. 10C, upper panel, compare lanes 3 and 4).
FIG. 10.

Runx2 requires a specific domain to interact with VDR. (A) Schematic representation of the GST-Runx2 and VDR proteins used in the GST pull-down assays. GST-Runx2Δ376, GST-Runx2Δ361, and GST-Runx2Δ230 correspond to mutated Runx2 proteins that lack amino acid residues 377 to 513, 362 to 513, and 231 to 513, respectively. (B) A 1.5-μg amount (each) of GST-Runx2, GST-Runx2Δ376, and GST-Runx2Δ361 proteins bound to glutathione-Sepharose beads was incubated with 1.5 μg of GST-free VDR. The precipitated VDR (upper panel) and Runx2 (middle panel) proteins were determined by Western blotting with specific antibodies. The asterisks mark cleavage products of GST-Runx2Δ376. (C) GST pull-down assays were repeated as for panel B but with GST-Runx2 and GST-Runx2Δ230, which lacks most of the C-terminal region of Runx2. The precipitated proteins were detected by Western blotting as indicated for panel B. The combinations of proteins used in each binding reaction are indicated at the top of the blots. The positions of molecular mass markers are shown to the left of the blots.
These results were also confirmed by repeating the GST pull-down analyses but with nuclear extracts from ROS 17/2.8 osteoblastic cells instead of purified VDR. Both full-length GST-Runx2 and GST-Runx2Δ361 had the ability to precipitate the nuclear VDR present in ROS 17/2.8 cells treated with 1α,25-dihydroxyvitamin D3 (Fig. 11B, upper panel, lanes 9 and 10, respectively). In contrast, no VDR protein was detected in the precipitate when nuclear extracts from untreated cells were analyzed (Fig. 11B, upper panel, lanes 7 and 8) or when the extracts were incubated with GST-Sepharose (Fig. 11B, upper panel, lanes 3 and 4). As we found with purified recombinant VDR, GST-Runx2Δ230 was unable to precipitate the endogenous VDR present in nuclear extracts (Fig. 11C, upper panel, lanes 6 and 7), further indicating that a sequence located between residues 230 and 361 of Runx2 is absolutely required for interacting with VDR. Nevertheless, this Runx2 domain alone (residues 209 to 361) is not sufficient for an interaction with VDR, as the GST-Runx2(209-361) fusion protein, which lacks the Runx2 DBD, was unable to precipitate VDR, either purified or from nuclear extracts, in GST pull-down assays (data not shown). Interestingly, and in contrast to what we found with purified VDR, GST-Runx2 and GST-Runx2Δ361 bound the VDR present in nuclear extracts from 1α,25-dihydroxyvitamin D3-treated ROS17/2.8 cells with equivalent affinity (Fig. 11B, upper panel, lanes 9 and 10). This result suggests that there may be another component present in the nuclear extracts but absent in our assay with purified proteins that facilitates or further stabilizes the GST-Runx2Δ361-VDR interaction.
FIG. 11.

GST-Runx2 interacts with nuclear VDR. (A) GST-Runx2 proteins. (B and C) The ability of GST-Runx2 proteins to interact with the VDR present in nuclear extracts (N.E.) from ROS 17/2.8 cells treated with 10−8 M 1α,25-dihydroxyvitamin D3 [1α,25-(OH)2D3] for 4 h or with vehicle was evaluated by GST pull-down assays. (B) GST-Runx2 and GST-Runx2Δ361 (1.5 μg each) bound to Sepharose beads (lower panel) were incubated with 150 μg of nuclear extracts from ROS 17/2.8 cells for 2 h at 4°C. The precipitated VDR was then detected by Western blotting (upper panel). (C) Similarly, GST-Runx2 and GST-Runx2Δ230 (1.5 μg each) (lower panel) were incubated with 150 μg of nuclear extracts from ROS 17/2.8 cells, and the precipitated VDR was detected by Western blotting (upper panel). The combinations of proteins used in each binding reaction are marked at the top of the blots. The positions of molecular mass markers are shown to the left of the blots.
Together, these results confirm that Runx2 functionally interacts with VDR to up-regulate OC promoter activity following 1α,25-dihydroxyvitamin D3 stimulation.
Enhanced interaction of the Runx2 and VDR factors with the distal OC promoter in intact osteoblastic cells incubated with 1α,25-dihydroxyvitamin D3.
We have shown that in 1α,25-dihydroxyvitamin D3-treated osteoblastic cells, VDR binds to a recognition site in the distal region of the OC promoter (24). Therefore, by use of ChIP analyses with specific antibodies against Runx2 and VDR, we assessed whether upon ligand stimulation VDR-Runx2 interaction could stabilize the association of both factors with the OC promoter sequence. These antibodies were used in whole-cell lysates to precipitate sonicated chromatin that was obtained after formaldehyde cross-linking of DNA to transcription factors in intact ROS 17/2.8 cells cultured in the absence or presence of 1α,25-dihydroxyvitamin D3 for 4 h. DNA fragments that coprecipitated with the transcription factors were purified and used as templates for PCR with specific primers directed against the distal or proximal OC promoter regions (Fig. 12A, segments a and b, respectively). We adjusted the number of PCR cycles to remain within the linear range of PCR amplification.
FIG. 12.
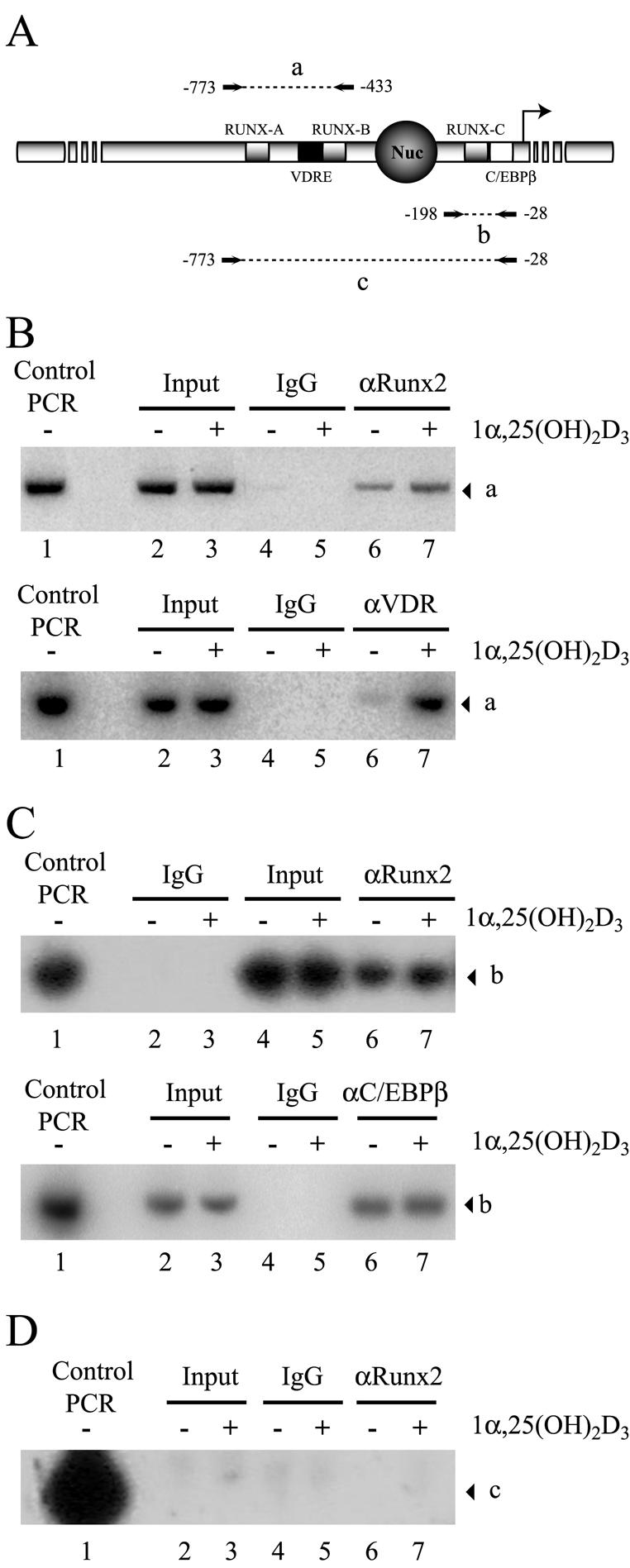
Enhanced binding of Runx2 and VDR to the distal OC promoter in 1α,25-dihydroxyvitamin D3-treated osteoblastic cells. (A) Schematic representation of the rat OC gene promoter, indicating the relative positions of binding sites for VDR, Runx2, and C/EBPβΒ transcription factors. The combination of primers used in the PCR amplification of immunoprecipitated DNA fragments is indicated, and the amplified fragments are marked by letters a (−773 to −433), b (−198 to −28), and c (−773 to −28). Nuc, positioned nucleosome. (B to D) ChIP of formaldehyde-cross-linked chromatin from ROS17/2.8 cells cultured in the presence (+) or absence (−) of 1α,25-dihydroxyvitamin D3 [1α,25-(OH)2D3], using anti-Runx2 antibody (B [upper panel], C [upper panel], and D), anti-VDR (B [lower panel]), and anti-C/EBPβ (C [lower panel]). Control PCR indicates the signal obtained when the pOC-3.4 plasmid, containing 1.1 kb of rat OC promoter, was used as a template. IgG, immunoglobulin G.
To control for an efficient sonication that generates chromatin fragments with an average size of less than 500 bp, we analyzed the samples with a set of primers that can amplify the OC promoter only when the DNA segments are larger than 780 bp (Fig. 12A, segment c). As shown in Fig. 12D, the absence of amplification products with these primers confirms successful sonication.
Runx2 was associated with the distal and proximal OC promoter in ROS 17/2.8 cells (Fig. 12B and C). Interestingly, we detected a small but significant (twofold) increase in the PCR signal of chromatin samples from 1α,25-dihydroxyvitamin D3-treated cells that were precipitated with the Runx2 antibody and amplified with primers directed against the distal OC promoter region (Fig. 12B, upper panel, compare lanes 6 and 7). This increased association of Runx2 correlates with the ligand-dependent interaction of VDR (Fig. 12B, lower panel, lanes 6 and 7), indicating that binding of Runx2 to the distal OC promoter is further stabilized by 1α,25-dihydroxyvitamin D3 treatment. In contrast, Runx2 association with the proximal OC promoter region remained unchanged in osteoblastic cells incubated with 1α,25-dihydroxyvitamin D3 for 4 h (Fig. 12C, upper panel). Similarly, binding of C/EBPβ, another key regulator of OC expression in osteoblastic cells (6), was not affected by this hormone treatment (Fig. 12B, lower panel).
It was previously shown that forced expression of the N-terminal domain of VDR (VDRΔ165) inhibits the OC promoter activity (Fig. 9). To further demonstrate that this inhibitory effect is due to a direct interaction of VDRΔ165 with the distal OC promoter, it was necessary to show that this truncated VDR version binds to the OC VDRE. For that purpose, we overexpressed the VDRΔ165 protein (X-press tagged) in ROS 17/2.8 cells and then detected its association with the distal OC promoter by ChIP, using either anti-X-press or anti-VDR antibodies directed against the N-terminal end of the receptor (Fig. 13A). Under our experimental conditions, we were able to overexpress high levels of the VDRΔ165 protein in ROS 17/2.8 cells (Fig. 13B, lanes 3 and 4). Interestingly, this high expression resulted in interaction of VDRΔ165 with the distal OC promoter in both the presence and absence of 1α,25-dihydroxyvitamin D3 (Fig. 13C, lanes 8 and 9). The interaction of VDRΔ165 reduced the ligand-dependent enhanced association of the endogenous VDR with the VDRE in cells treated with 1α,25-dihydroxyvitamin D3 (Fig. 13C, compare lanes 10 and 11), indicating that both forms of the receptor bind to the same sequence and compete for their interaction. Binding of VDRΔ165 did not reduce the association of Runx2 with the distal OC promoter (Fig. 13C, lanes 12 and 13); however, it blocked the increase in Runx2 interaction that occurs following 1α,25-dihydroxyvitamin D3 treatment (Fig. 12B). Together, these results indicate that VDRΔ165 inhibits the OC promoter activity by binding to the VDRE and thereby blocking formation of transcriptionally active complexes. This effect is especially relevant following 1α,25-dihydroxyvitamin D3 treatment, as the VDRΔ165 protein lacks the LBD that interacts with transcriptional coactivators.
FIG. 13.

The VDRΔ165 domain binds to the OC promoter and competes with VDR for interaction with the OC VDRE. (A) Schematic representation of the ChIP analysis at the OC gene promoter in cells that overexpress the VDRΔ165 domain (VDR DBD). The transcription factors bound to the promoter are indicated, and the white star indicates the ligand 1α,25-dihydroxyvitamin D3 [1α,25-(OH)2D3]. The anti-X-press (αX-press) antibody used in the ChIP and the X-press tag present in the VDR DBD are also indicated. (B) Aliquots (15 μg) of whole cellular extracts (W.C.E.) from ROS 17/2.8 cells transiently transfected with the pcDNA-VDRΔ165 or pcDNA3.1His plasmid were analyzed by Western blotting with anti-Runx2 (upper panel) or anti-VDR (lower panel) antibodies. To control for equal loading, the membrane was reblotted against GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (middle panel). The different combinations of antibodies and W.C.E. included in each lane are indicated at the top of the blots. The positions of molecular size markers are shown to the left of the blots. (C) ChIP of formaldehyde-cross-linked chromatin from ROS17/2.8 cells that overexpress the VDRΔ165 domain (lanes 4, 5, and 8 to 13) cultured in the presence (+) or absence (−) of 1α,25-dihydroxyvitamin D3, using anti X-press antibody (lanes 6 to 9), anti-VDR antibody (lanes 10 and 11), and anti-Runx2 antibody (lanes 12 and 13). ROS17/2.8 cells transiently transfected with the pcDNA3.1His empty plasmid were used as a negative control for ChIP with anti-X-press antibody (lanes 6 and 7). Control PCR indicates the signal obtained when the pOC-3.4 plasmid, containing 1.1 kb of rat OC promoter, was used as a template. The DNA fragment corresponding to the distal OC gene promoter amplified by PCR is marked by the letter a (see Fig. 12A for an illustration). IgG, immunoglobulin G.
Following a similar approach, we next evaluated whether Runx2 and VDR are bound simultaneously to the distal OC promoter in osteoblastic cells expressing this gene. We performed two sequential immunoprecipitations of the cross-linked ROS 17/2.8 cell extracts with both anti-Runx2 and anti-VDR antibodies and then determined by PCR whether OC promoter sequences were present in the final precipitate (Re-ChIP). When the cell extracts were first immunoprecipitated with anti-VDR antibodies and the collected samples were subsequently reprecipitated with anti-Runx2 antibodies (Fig. 14A), the distal OC promoter was found only in the samples from ROS 17/2.8 cells treated with 1α,25-dihydroxyvitamin D3 (Fig. 14C, lane 7), further indicating that in the absence of 1α,25-dihydroxyvitamin D3, the receptor is not bound to the promoter. Accordingly, when the samples were first precipitated with anti-Runx2 antibodies and then reprecipitated with anti-VDR antibodies (Fig. 14B), the OC promoter was detected mainly in the samples from 1α,25-dihydroxyvitamin D3-treated cells (Fig. 14D, lane 7), strongly indicating that following incubation with 1α,25-dihydroxyvitamin D3, both Runx2 and VDR proteins are bound simultaneously to the distal OC promoter.
FIG. 14.

Runx2 and VDR bind simultaneously to the distal OC promoter in osteoblastic cells treated with 1α,25-dihydroxyvitamin D3. (A and B) Schematic representation of the Re-ChIP analysis at the rat OC gene promoter. The different transcription factors bound to the promoter are indicated, and the white star indicates the ligand 1α,25-dihydroxyvitamin D3 [1α,25-(OH)2D3]. The antibodies used in each ChIP step are indicated at the left of each panel. (C and D) Re-ChIP of formaldehyde-cross-linked chromatin from ROS17/2.8 cells cultured in the presence (+) or absence (−) of 1α,25-dihydroxyvitamin D3, using anti-VDR (αVDR) antibody followed by anti-Runx2 antibody (C) or anti-Runx2 antibody followed by anti-VDR antibody (D). Control PCR indicates the signal obtained when the pOC-3.4 plasmid, containing 1.1 kb of rat OC promoter, was used as a template. The DNA fragment corresponding to the distal OC gene promoter amplified by PCR is marked by the letter a. IgG, immunoglobulin G.
DISCUSSION
Here we have shown that Runx2 overexpression not only up-regulates OC promoter basal activity but also further enhances responsiveness of this promoter to 1α,25-dihydroxyvitamin D3. Importantly, we determined biochemically that Runx2 and VDR are components of the same nuclear complexes in osteoblastic cells expressing OC and cultured in the presence of 1α,25-dihydroxyvitamin D3. In addition, Runx2 and VDR were found by immunocytochemistry to colocalize in the nuclei of ROS 17/2.8 cells upon treatment with 1α,25-dihydroxyvitamin D3. A direct Runx2-VDR protein-protein interaction was confirmed by GST-pull down analyses with purified recombinant VDR and Runx2 proteins or endogenous factors in nuclear extracts from ROS 17/2.8 osteoblastic cells. Using this same approach, we defined the domain of each protein involved in the Runx2-VDR interaction. Thus, it was found that the N-terminal region of VDR (residues 22 to 111) was necessary and sufficient for the interaction with Runx2 and that a region C terminal to the runt homology DBD of Runx2 was necessary, although not sufficient, for interaction with the VDR. We also determined that 1α,25-dihydroxyvitamin D3 stimulation results in both VDR binding and increased Runx2 association to the distal OC promoter region in intact ROS 17/2.8 cells.
This Runx2-VDR interaction may be highly relevant for the stabilization of the VDR complex as it binds to the OC promoter VDRE upon ligand stimulation (2, 18). Several lines of evidence had previously indicated a direct correlation between binding of Runx2 to the distal OC promoter region and the ability of this promoter to respond to 1α,25-dihydroxyvitamin D3 (8, 18, 24). It has been established that the OC VDRE functions as an enhancer element to increase OC basal transcription in bone-derived cells (11). Moreover, the VDR/RXR complex requires chromatin remodeling to interact with the distal OC promoter. Thus, in osteoblastic cells expressing OC, the VDRE is located within a distal DNase I-hypersensitive site which is present at the OC promoter in the absence of 1α,25-dihydroxyvitamin D3. Packaging the OC VDRE into nucleosomal particles (18) or loss of the distal DNase I-hypersensitive site due to chromatin hyperacetylation (15) abolishes both binding of the VDR/RXR heterodimer and 1α,25-dihydroxyvitamin D3-dependent OC promoter up-regulation. Interestingly, binding of Runx2 to the OC promoter is required for the changes in chromatin structure that accompany transcriptional activity in osteoblasts. Thus, mutation of Runx2 sites (A, B, and C) inhibits formation of the DNase I-hypersensitive sites, significantly reduces OC promoter activity, and blocks 1α,25-dihydroxyvitamin D3 enhancement (8). Thus, the VDR-Runx2 interaction demonstrates a tight relationship between tissue-specific and steroid-responsive transcriptional regulators in osteoblastic cells.
By combining several experimental approaches, including ChIP, coimmunoprecipitation, and GST pull-down analyses, we have recently demonstrated that in osteoblastic cells expressing OC, the coactivator p300 can be recruited to the OC promoter by Runx2, where it up-regulates both basal and 1α,25-dihydroxyvitamin D3-enhanced OC expression (24). Moreover, unpublished results from our group indicate that p300-dependent up-regulation of 1α,25-dihydroxyvitamin D3-enhanced OC promoter activity requires a functional Runx2 site B, which is located immediately adjacent to the VDRE. p300 can bind simultaneously to several transcription factors and form large multimolecular protein complexes that regulate transcription. Among these factors are the nuclear steroid receptors, which upon ligand stimulation bind p300 directly or through other coactivator molecules such as SRC-1 (19). Here, we have shown that upon 1α,25-dihydroxyvitamin D3 stimulation, VDR interacts with the SRC-1 molecules present in nuclear extracts from ROS 17/2.8 cells. This interaction requires the presence of 1α,25-dihydroxyvitamin D3 and involves the LBD of VDR located at the C-terminal end of the protein. In addition, unpublished results from our group suggest that SRC-1 is a component of a multisubunit complex that is formed on the distal region of the OC promoter and that contributes to 1α,25-dihydroxyvitamin D3-dependent up-regulation of OC expression in osteoblastic cells. Together, these results provide a basis for postulating that following 1α,25-dihydroxyvitamin D3 treatment of bone-derived cells expressing OC, the p300 molecules recruited to the OC promoter by Runx2 can functionally interact with the VDR complex bound to the VDRE and together enhance basal OC transcription (Fig. 15).
FIG. 15.

Schematic view of OC gene promoter transcribing at basal levels (A) or enhanced by 1α,25-dihydroxyvitamin D3 (B). The OC gene promoter and its various regulatory elements are represented by different shadings. The circle indicates a positioned nucleosome (Nuc) flanked by a distal and proximal DNase I-hypersensitive sites (dDHS and pDHS). The different transcription factors bound to the promoter are indicated, and the white star indicates the ligand 1α,25-dihydroxyvitamin D3. The stimulatory effect of the transcription factors on the general transcription machinery is represented by arrows.
It has been generally believed that in contrast to most steroid receptors, the VDR does not possess a ligand-independent AF-1 transactivation motif at the N-terminal domain (19). These results indicate that the N terminus of VDR still plays an important regulatory role, as it allows interaction with Runx2, a key regulator of bone-specific transcription. Thus, our studies using the OC gene model define a new mechanism for tissue-specific control of transcription in bone cells. This mechanism involves the intersection of two major pathways: Runx2, a “master” transcriptional regulator of osteoblast differentiation, and 1α,25-dihydroxyvitamin D3, a hormone that promotes expression of genes associated with these terminally differentiated bone cells.
Acknowledgments
We thank Jose Gutierrez and Soraya Gutierrez for helpful discussions and critical reading of the manuscript.
This work was supported by grants from FONDECYT (1030749 to M.M. and 2010103 to R.P.), NIH-FIRCA grant TW00990 (to G.S. and M.M.), DIUC grant 201.031.090-1.4 (to M.M.), and NIH grants AR48818 and AR39588 (to G.S.) and DE12528 (to J.L.). F.C. has been supported by a fellowship from MECESUP UCH9903, and R.P. has been supported by a scholarship from Fundacion Andes.
The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Banerjee, C., L. R. McCabe, J. Y. Choi, S. W. Hiebert, J. L. Stein, G. S. Stein, and J. B. Lian. 1997. Runt homology domain proteins in osteoblast differentiation: AML3/CBFA1 is a major component of a bone-specific complex. J. Cell Biochem. 66:1-8. [DOI] [PubMed] [Google Scholar]
- 2.Breen, E. C., A. J. van Wijnen, J. B. Lian, G. S. Stein, and J. L. Stein. 1994. In vivo occupancy of the vitamin D responsive element in the osteocalcin gene supports vitamin D-dependent transcriptional upregulation in intact cells. Proc. Natl. Acad. Sci. USA 91:12902-12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Christakos, S., P. Dhawan, Y. Liu, X. Peng, and A. Porta. 2003. New insights into the mechanisms of vitamin D action. J. Cell Biochem. 88:695-705. [DOI] [PubMed] [Google Scholar]
- 4.Ducy, P., R. Zhang, V. Geoffroy, A. L. Ridall, and G. Karsenty. 1997. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89:747-754. [DOI] [PubMed] [Google Scholar]
- 5.Gutierrez, J., J. Sierra, R. Medina, M. Puchi, M. Imschenetzky, A. van Wijnen, J. Lian, G. Stein, J. Stein, and M. Montecino. 2000. Interaction of CBF alpha/AML/PEBP2 alpha transcription factors with nucleosomes containing promoter sequences requires flexibility in the translational positioning of the histone octamer and exposure of the CBF alpha site. Biochemistry 39:13565-13574. [DOI] [PubMed] [Google Scholar]
- 6.Gutierrez, S., A. Javed, D. K. Tennant, M. van Rees, M. Montecino, G. S. Stein, J. L. Stein, and J. B. Lian. 2002. CCAAT/enhancer-binding proteins (C/EBP) beta and delta activate osteocalcin gene transcription and synergize with Runx2 at the C/EBP element to regulate bone-specific expression. J. Biol. Chem. 277:1316-1323. [DOI] [PubMed] [Google Scholar]
- 7.Hsieh, J. C., P. W. Jurutka, S. Nakajima, M. A. Galligan, C. A. Haussler, Y. Shimizu, N. Shimizu, G. K. Whitfield, and M. R. Haussler. 1993. Phosphorylation of the human vitamin D receptor by protein kinase C. Biochemical and functional evaluation of the serine 51 recognition site. J. Biol. Chem. 268:15118-15126. [PubMed] [Google Scholar]
- 8.Javed, A., S. Gutierrez, M. Montecino, A. J. van Wijnen, J. L. Stein, G. S. Stein, and J. B. Lian. 1999. Multiple Cbfa/AML sites in the rat osteocalcin promoter are required for basal and vitamin D-responsive transcription and contribute to chromatin organization. Mol. Cell. Biol. 19:7491-7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kitagawa, H., R. Fujiki, K. Yoshimura, Y. Mezaki, Y. Uematsu, D. Matsui, S. Ogawa, K. Unno, M. Okubo, A. Tokita, T. Nakagawa, T. Ito, Y. Ishimi, H. Nagasawa, T. Matsumoto, J. Yanagisawa, and S. Kato. 2003. The chromatin-remodeling complex WINAC targets a nuclear receptor to promoters and is impaired in Williams syndrome. Cell 113:905-917. [DOI] [PubMed] [Google Scholar]
- 10.Komori, T., H. Yagi, S. Nomura, A. Yamaguchi, K. Sasaki, K. Deguchi, Y. Shimizu, R. T. Bronson, Y. H. Gao, M. Inada, M. Sato, R. Okamoto, Y. Kitamura, S. Yoshiki, and T. Kishimoto. 1997. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89:755-764. [DOI] [PubMed] [Google Scholar]
- 11.Lian, J. B., J. L. Stein, G. S. Stein, M. Montecino, A. J. van Wijnen, A. Javed, and S. Gutierrez. 2001. Contributions of nuclear architecture and chromatin to vitamin D-dependent transcriptional control of the rat osteocalcin gene. Steroids 66:159-170. [DOI] [PubMed] [Google Scholar]
- 12.Majeska, R. J., S. B. Rodan, and G. A. Rodan. 1980. Parathyroid hormone-responsive clonal cell lines from rat osteosarcoma. Endocrinology. 107:1494-1503. [DOI] [PubMed] [Google Scholar]
- 13.Metivier, R., G. Penot, M. Hubner, G. Reid, H. Brand, M. Kos, and F. Gannon. 2003. Estrogen receptor-α direct ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115:751-763. [DOI] [PubMed] [Google Scholar]
- 14.Montecino, M., J. Lian, G. Stein, and J. Stein. 1996. Changes in chromatin structure support constitutive and developmentally regulated transcription of the bone-specific osteocalcin gene in osteoblastic cells. Biochemistry 35:5093-5102. [DOI] [PubMed] [Google Scholar]
- 15.Montecino, M., B. Frenkel, A. J. van Wijnen, J. B. Lian, G. S. Stein, and J. L. Stein. 1999. Chromatin hyperacetylation abrogates vitamin D-mediated transcriptional upregulation of the tissue-specific osteocalcin gene in vivo. Biochemistry. 26:1338-1345. [DOI] [PubMed] [Google Scholar]
- 16.Otto, F., A. P. Thornell, T. Crompton, A. Denzel, K. C. Gilmour, I. R. Rosewell, G. W. H. Stamp, R. S. P. Beddington, S. Mundlos, B. R. Olsen, P. B. Selby, and M. J. Owen. 1997. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89:765-771. [DOI] [PubMed] [Google Scholar]
- 17.Owen, T. A., M. Aronow, V. Shalhoub, L. M. Barone, L. Wilming, M. S. Tassinari, M. B. Kennedy, S. Pockwinse, J. B. Lian, and G. S. Stein. 1990. Progressive development of the rat osteoblast phenotype in vitro: reciprocal relationships in expression of genes associated with osteoblast proliferation and differentiation during formation of the bone extracellular matrix. J. Cell Physiol. 143:420-430. [DOI] [PubMed] [Google Scholar]
- 18.Paredes, R., J. Gutierrez, S. Gutierrez, L. Allison, M. Puchi, M. Imschenetzky, A. van Wijnen, J. Lian, G. Stein, J. Stein, and M. Montecino. 2002. Interaction of the 1α,25-dihydroxyvitamin D3 receptor at the distal promoter region of the bone-specific osteocalcin gene requires nucleosomal remodelling. Biochem. J. 363:667-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rachez, C., and L. P. Freedman. 2000. Mechanisms of gene regulation by vitamin D(3) receptor: a network of coactivator interactions. Gene 246:9-21. [DOI] [PubMed] [Google Scholar]
- 20.Rachez, C., M. Gamble, C. P. B. Chang, G. B. Atkins, M. A. Lazar, and L. P. Freedman. 2000. The DRIP complex and SRC-1/p160 coactivators share similar nuclear receptor binding determinants but constitute functionally distinct complexes. Mol. Cell. Biol. 20:2718-2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rachez, C., B. D. Lemon, Z. Suldan, V. Bromleigh, M. Gamble, A. M. Naar, H. Erdjument-Bromage, P. Tempst, and L. P. Freedman. 1999. Ligand-dependent transcription activation by nuclear receptors requires the DRIP complex. Nature 398:824-828. [DOI] [PubMed] [Google Scholar]
- 22.Rachez, C., Z. Suldan, J. Ward, C. P. Chang, D. Burakov, H. Erdjument-Bromage, P. Tempst, and L. P. Freedman. 1998. A novel protein complex that interacts with the vitamin D3 receptor in a ligand-dependent manner and enhances VDR transactivation in a cell-free system. Genes Dev. 12:1787-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen, J., M. Montecino, L. B. Lian, G. S. Stein, A. J. Van Wijnen, and J. L. Stein. 2002. Histone acetylation in vivo at the osteocalcin locus is functionally linked to vitamin D-dependent, bone tissue-specific transcription. J. Biol. Chem. 277:20284-20292. [DOI] [PubMed] [Google Scholar]
- 24.Sierra, J., A. Villagra, R. Paredes, F. Cruzat, S. Gutierrez, A. Javed, G. Arriagada, J. Olate, M. Imschenetzky, A. J. van Wijnen, J. B. Lian, G. S. Stein, J. L. Stein, and M. Montecino. 2003. Regulation of the bone-specific osteocalcin gene by p300 requires Runx2/Cbfa1 and the vitamin D3 receptor but not p300 intrinsic histone acetyltransferase activity. Mol. Cell. Biol. 23:3339-3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soutoglou, E., and I. Talianidis. 2002. Coordination of PIC assembly and chromatin remodeling during differentiation-induced gene activation. Science 295:1901-1904. [DOI] [PubMed] [Google Scholar]
- 26.Staal, A., A. J. vanWijnen, J. C. Birkenhager, H. A. P. Pols, J. Prahl, H. DeLuca, M. P. Gaub, J. B. Lian, G. S. Stein, J. P. T. M. vanLeeuwen, and J. L. Stein. 1996. Distinct conformations of vitamin D receptor retinoid X receptor-alpha heterodimers are specified by dinucleotide differences in the vitamin D-responsive elements of the osteocalcin and osteopontin genes. Mol. Endocrinol. 10:1444-1456. [DOI] [PubMed] [Google Scholar]
- 27.Thompson, P. D., P. W. Jurutka, C. A. Haussler, G. K. Whitfield, and M. R. Haussler. 1998. Heterodimeric DNA binding by the vitamin D receptor and retinoid X receptors is enhanced by 1,25-dihydroxyvitamin D3 and inhibited by 9-cis-retinoic acid. Evidence for allosteric receptor interactions. J. Biol. Chem. 273:8483-8491. [DOI] [PubMed] [Google Scholar]
- 28.Zeng, C., A. J. van Wijnen, J. L. Stein, S. Meyers, W. Sun, L. Shopland, J. B. Lawrence, S. Penman, J. B. Lian, G. S. Stein, and S. W. Hiebert. 1997. Identification of a nuclear matrix targeting signal in the leukemia and bone-related AML/CBF-alpha transcription factors. Proc. Natl. Acad. Sci. USA 94:6746-6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang, Y. W., N. Yasui, K. Ito, G. Huang, M. Fujii, J. Hanai, H. Nogami, T. Ochi, K. Miyazono, and Y. Ito. 2000. A RUNX2/PEBP2alpha A/CBFA1 mutation displaying impaired transactivation and Smad interaction in cleidocranial dysplasia. Proc. Natl. Acad. Sci. USA 97:10549-10554. [DOI] [PMC free article] [PubMed] [Google Scholar]