

Abstract
Clinical and histologic similarities between various eczematous disorders point to a common efferent pathway. We demonstrate here that activated T cells infiltrating the skin in atopic dermatitis (AD) and allergic contact dermatitis (ACD) induce keratinocyte (KC) apoptosis. KCs normally express low levels of Fas receptor (FasR) that can be substantially enhanced by the presence of IFN-γ. KCs are rendered susceptible to apoptosis by IFN-γ when FasR numbers reach a threshold of approximately 40,000 per KC. Subsequently, KCs undergo apoptosis induced by anti-FasR mAb’s, soluble Fas ligand, supernatants from activated T cells, or direct contact between T cells and KCs. Apoptotic KCs show typical DNA fragmentation and membrane phosphatidylserine expression. KC apoptosis was demonstrated in situ in lesional skin affected by AD, ACD, and patch tests. Using numerous cytokines and anti-cytokine neutralizing mAb’s, we found no evidence that cytokines other than IFN-γ participate in this process. In addition, apoptosis-inducing pathways other than FasR triggering were ruled out by blocking T cell–induced KC apoptosis by caspase inhibitors and soluble Fas-Fc protein. Responses of normal human skin and cultured skin equivalents to activated T cells demonstrated that KC apoptosis caused by skin-infiltrating T cells is a key event in the pathogenesis of eczematous dermatitis.
Introduction
Eczematous disorders account for a large proportion of all skin disease, and constitute a major health problem worldwide. Eczematous dermatitis is characterized histologically by pathologic changes in the epidermis and a predominantly mononuclear cell infiltrate. The clinical features include itching, redness, papules, vesicles, and scaling. Because T cells constitute a large population of cellular infiltrate in eczematous dermatitis, a dysregulated, cytokine-mediated response of the immune system appears to be an important pathogenetic factor. Keratinocytes (KCs) within eczematous lesions exhibit an unusual expression of MHC class II antigens (e.g., HLA-DR), ICAM-1 (CD54), and IFN-γ inducible protein-10, all of which are strongly induced by IFN-γ (1, 2). ICAM-1 mediates strong adhesion between T cells and KCs (3). Thus it seems probable that cytokines and cell-surface molecules of the accumulated skin-infiltrating T cells affect KCs in situ.
Atopic dermatitis (AD) is a chronic skin disease manifested by eczematous skin lesions that affects more than 10% of children (4, 5). Lesional atopic skin clinically and histologically resembles allergic contact dermatitis (ACD), which may be mediated by predominant expression of the type 1 cytokine IFN-γ (6, 7). In AD, circulating allergen-specific memory/effector T cells expressing cutaneous lymphocyte-associated antigen homing receptor have been demonstrated to be activated in vivo, to regulate IgE by an IL-13–dominated cytokine pattern, and to delay eosinophil apoptosis by IL-5 (8–10). Allergen-specific T cells from skin and peripheral blood have shown a profound dysregulation of IL-4 and IFN-γ secretion, with an overproduction of IL-4 and reduced IFN-γ secretion in AD (11, 12). However, studies focused on the intralesional cytokine production revealed higher IFN-γ production than occurs in T cells from peripheral blood (13, 14). A sequential activation of type 2 and type 1 T cells in the pathogenesis of AD was therefore proposed (15–17). Recent results from a murine model of eczematous dermatitis suggests that both the type 2 cytokines IL-4 and IL-5, and the type 1 cytokine IFN-γ play important roles in the inflammation of the skin (18). Furthermore, injection of IFN-γ into the skin of human volunteers demonstrated that this cytokine can induce transient skin inflammation (19). Moreover, transgenic mice expressing IFN-γ in the epidermis have developed eczema spontaneously, and showed an increased contact hypersensitivity reaction (20).
In this study, we demonstrated that KC apoptosis is a major mechanism in the pathogenesis of eczematous disorders. T cells infiltrating the skin upregulate Fas receptor (FasR) on KCs, render them susceptible to apoptosis by IFN-γ, and induce apoptosis by Fas ligand (FasL) expressed on the T-cell surface.
Methods
Subjects.
Patients with acute AD, chronic AD, and acute ACD were included in the study. They were not receiving any systemic therapy for at least 2 weeks before biopsy and patch testing with house dust mite and nickel. AD patients had definite AD, diagnosed according to standard criteria (21). ACD for nickel was originally diagnosed by epicutaneous patch testing. The patch test reactions were 2+ according to the International Contact Dermatitis Research Group, showing erythema, induration, papules, and vesicles. Incisional skin biopsies were taken from lesional AD skin (acute AD, four biopsies; chronic AD, five biopsies), atopy patch tests (two biopsies), and ACD patch tests (four biopsies). Control skin was obtained from three healthy, nonatopic individuals. Informed consent was obtained from all subjects, and the study was approved by the Ethical Committee of Davos, Switzerland.
Reagents and Abs.
Recombinant human IL-12 and IL-5 were purchased from PharMingen (San Diego, California, USA). Recombinant human IL-13 was purchased from PeproTech Inc. (Rocky Hill, New Jersey, USA). Recombinant human IFN-γ, IL-2, IL-4, and TNF-α came from Novartis Pharmaceuticals (Basel, Switzerland). Ethidium bromide and PMA were purchased from Sigma Chemical Co. (St. Louis, Missouri, USA). Ionomycin was purchased from Calbiochem-Novabiochem Corp. (San Diego, California, USA). The mAb’s for flow cytometry were purchased from Beckman Coulter Intl. SA (Nyon, Switzerland), Immunotech (Marseilles, France), or PharMingen. Anti-CD14, anti-CD16, anti-CD19, and anti-CD45RA magnetic microbeads for magnetic activated cell sorting (MACS) were from Miltenyi Biotec (Bergisch Gladbach, Germany). Anti-CD2 (4B2 and 6G4) and anti-CD28 (15E4) mAb’s were from the Red Cross Blood Transfusion Service (Amsterdam, the Netherlands). Anti-CD3 came from clone CRL8001, obtained from American Type Culture Collection (Manassas, Virginia, USA). The activating, cross-linking anti-FasR mAb CH-11 and the anti-FasR mAb ZB4 (which inhibits apoptosis induced by CH-11) were purchased from Immunotech. The blocking anti–IFN-γ receptor-α chain (CD119) mAb was purchased from Genzyme Pharmaceuticals (Cambridge, Massachusetts, USA). Neutralizing anti–IL-4 mAb’s (8F12 and 3H4) and IFN-γ mAb 45-15 were provided by Novartis Pharmaceuticals. Anti–IL-13 mAb’s (JES8-5A2 and JES10-2F9) and the mutant IL-4 antagonist (Y124D) with IL-13– and IL-4–inhibitory activity were provided by DNAX Research Institute (Palo Alto, California, USA). The recombinant human soluble FasL and Fas-Fc protein (inhibiting the activity of FasL) were purchased from Alexis Corp. (San Diego, California, USA). The enzyme inhibitor (Z)-Val-Ala-DL-Asp-fluoromethylketone (Z-VAD-FMK) was from Bachem AG (Bubendorf, Switzerland).
KC culture.
Primary human KCs were obtained from punch biopsies from nonlesional, nonmedicated gluteal skin. The skin was split overnight in a PBS solution of sucrose and trypsin (0.1% sucrose, 0.25% trypsin, 1 mM EDTA; all reagents were from Sigma Chemical Co.) at 4°C. Epidermal sheets were removed from the dermis, and KC cell suspensions were cultured in a fully supplemented (5 μg/mL bovine insulin, 0.5 μg/mL hydrocortisone, 0.1 ng/mL human recombinant EGF, 30 μg/mL bovine pituitary extract, 100 μg/mL gentamicin, and 100 ng/mL amphotericin B), low calcium (0.15 mM Ca2+), serum-free KC growth medium (Clonetics Corp., San Diego, California, USA). Hydrocortisone and antibiotics were removed from the culture medium for the duration of experiments. All cell cultures were incubated at 37°C in a humidified atmosphere with 5% CO2. KCs that had undergone at least three passages were grown in either 6-well Transwell plates, 6-well plates, or 96-well flat-bottom plates (all from Corning-Costar Corp., Cambridge, United Kingdom) for different experiments. Single-cell suspensions of KCs were prepared by the addition of 0.025% trypsin and 0.01% EDTA for 5–15 minutes at 37°C, neutralized by adding 10% FCS (Sera-Lab Ltd., Sussex, United Kingdom).
Isolation of CD45RA+ and CD45RO+ T cells from peripheral blood and differentiation and characterization of CD4+ and CD8+ type 1 and type 2 T cells.
After 2–3 weeks of KC culture, PBMCs from the same patient were isolated by Ficoll (Biochrom KG, Berlin, Germany) density gradient centrifugation of peripheral venous blood. Cells were washed and resuspended in DMEM supplemented with 10 mM HEPES (both from Life Technologies AG, Basel, Switzerland), 5 mM EDTA, 2% FCS, 100 U/mL glutamine, and 100 μg/mL streptomycin (both from Life Technologies AG). CD45RA+ and CD45RO+ T cells were isolated with the MACS system according to the instructions of the manufacturer (Miltenyi Biotec). In brief, anti-CD14– and anti-CD19–depleted cells were incubated with MACS microbeads coated with anti-CD45RA and anti-CD16 mAb, and the T-cell fractions were recovered by sequential elution from the MACS column. CD4+ and CD8+ type 1 and type 2 T cells were generated from naive CD45RA+ T cells isolated from cord blood. The cells were incubated in the presence of either IL-12 (10 ng/mL) and neutralizing anti–IL-4 mAb (10 μg/mL) (for the generation of type 1 T cells) or IL-4 (25 ng/mL) and neutralizing anti–IL-12 mAb (10 μg/mL) (for the generation of type 2 T cells) in the presence of IL-2 (20 ng/mL) for 12 days. The T cells were stimulated with a combination of mAb’s (0.5 μg/mL anti-CD2, 1.0 μg/mL anti-CD3, and 0.5 μg/mL anti-CD28). Supernatants were harvested after 3 days for cytokine measurements. Phenotypes of type 1 and type 2 T-cell subsets were verified for cytokine production by ELISA of culture supernatants and intracellular cytokine flow cytometry analysis.
KC–T cell cocultures.
CD45RO+ T cells and differentiated type 1 and type 2 T cells were stimulated with a combination of anti-CD2, anti-CD3, and anti-CD28 mAb’s, plate-bound anti-CD3 (10 μg/mL), or PMA (10 ng/mL) and ionomycin (0.5 μM) for 24 hours, and then washed twice before coculture with KCs. In some experiments, type 2 T cells were stimulated in the presence of IL-12 (10 ng/mL). For studying the effects of T cells on KCs, T-cell supernatants were collected, diluted 50%, and added to KCs in 96-well plates. For coculture experiments, KCs were cultured in 96-well plates in the presence or absence of purified T cells at different ratios (generally 104 T cells and 3 × 104 KCs, ratio 1:3). For further coculture experiments, KCs and T cells were cultured in 6-well Transwell plates (Corning-Costar Corp.). The Transwells consist of a lower and an upper compartment, which are separated by a 10-μm-thick polycarbonate membrane with 0.4-μm pores. KCs (3 × 105) were transferred into the lower culture well and T cells (105) were added to the upper compartment.
Flow cytometry analysis.
IFN-γ receptor and IFN-γ–induced FasR (CD95) expression was determined on primary human KCs with anti–IFN-γ receptor and FasR mAb (ZB4), followed by phycoerythrin-conjugated (PE-conjugated) anti-mouse IgG. For phenotyping of effector T cell subsets, 5 × 105 cells were incubated at 4°C for 30 minutes, and then sequentially stained with anti-CD4–, anti-CD8– or CD45RO-PE. Stained cells were fixed in 2% paraformaldehyde. PE-conjugated mouse IgG1 was used as control. After washing, cells were analyzed on an EPICS XL-MCL flow cytometer (Beckman Coulter Intl. SA). The purity of the T-cell populations ranged from 90% to 96%. FasR counting on KC surfaces was done by indirect immune fluorescence staining with 50 μg/mL anti-FasR mAb (ZB4) followed by FITC-conjugated anti-mouse IgG (DAKO A/S, Glostrup, Denmark). Beads coated with known amounts of mouse mAb’s were used as standards (DAKO A/S) (22). The receptor molecules per cell were probably counted in lower numbers with this method than the classical Scatchard blot receptor determination would yield.
Viability and apoptosis detection.
The KC viability after exposure to anti-FasR mAb, soluble FasL, or after coculture with T cells was routinely evaluated by means of ethidium bromide (1 μM) uptake and flow cytometry. Membrane phosphatidylserine redistribution from the inner to the outer membrane leaflet takes place in apoptotic cells. Annexin V is a phosphatidylserine binding protein used to detect apoptotic cells. The technique was performed according to Vermes et al. (23). Briefly, cells were incubated with 1.0 μg/mL annexin V–FITC (R&D Systems Inc., Abingdon, United Kingdom) and 2.5 μg/mL propidium iodide (Sigma Chemical Co.) in calcium-containing binding buffer (HEPES with 0.25 mM CaCl2). DNA flow cytometry was performed according to Nicoletti et al. (24). In brief, cells were resuspended in hypotonic fluorochrome solution containing 50 μg/mL propidium iodide (Sigma Chemical Co.), 0.1% sodium citrate (Fluka Chemie AG, Buchs, Switzerland), and 0.1% (vol/vol) Triton-X 100 (Fluka Chemie AG), and then incubated at 4°C for 6 hours.
RNase protection assay.
Third-passage primary human KCs were treated with FasR mAb, recombinant human IFN-γ, and supernatant from stimulated CD45RO+ T cells diluted 50%. The KCs were lysed on the plate, and the RNA was isolated according to the RNeasy protocol (QIAGEN AG, Basel, Switzerland), which includes DNase digestion. The RNase protection assay was performed according to the protocol of the manufacturer (PharMingen). Briefly, all RNA obtained from 3 × 106 cells (3–4 μg) was hybridized overnight to the 32P-labeled RNA probe (hApo3c; PharMingen). Single-stranded RNA and free probe were digested by RNase A and RNase T1. Protected RNA was phenolized and precipitated, and then analyzed on a 6% denaturing polyacrylamide gel. Bands were visualized with phosphorous imaging (raytest; Schweiz AG, Urdorf, Switzerland).
Quantification of cytokines.
IL-4, IL-5, IL-13, and IFN-γ were determined by sandwich ELISA as described (9, 10). The sensitivity of IL-4 ELISA was at or below 20 pg/mL (mAb and IL-4 standard were provided by C.H. Heusser, Novartis Pharmaceuticals). The detection limit of the IL-5 ELISA was 50 pg/mL (mAb and IL-5 standard were from PharMingen). The detection limit of IL-13 ELISA was 100 pg/mL (mAb and IL-13 standard were from PharMingen). The sensitivity of the IFN-γ ELISA was at or below 10 pg/mL (mAb and IFN-γ standard were gifts from S.S. Alkan, Novartis Pharmaceuticals). Soluble FasL was detected by a commercial ELISA kit (MBL Co., Nagoya, Japan) with a sensitivity of 0.1 ng/mL.
Immunohistology.
The tissue samples were placed in Tissue-Tek OCT compound (Sakura Finetek Europe BV, Zoeterwoude, The Netherlands) and stored at –80°C. Four-micrometer cryostat sections were prepared on gelatin-coated slides (Merck AG, Dietikon, Switzerland). After air drying, sections were fixed in acetone for 10 minutes at 4°C. A three-step streptavidin-biotin complex peroxidase method (streptABComplex-peroxidase; DAKO A/S) was used. Briefly, sections were incubated with the primary mAb (CD4, CD8, and CD45R0 from DAKO A/S; FasL from Alexis Corp.; IFN-γ receptor from Genzyme Pharmaceuticals; and FasR, anti-perforin, and anti–granzyme B from Ancell Corp., Bayport, Minnesota, USA) at 4°C overnight. This was followed by incubation with biotin-conjugated rabbit anti-mouse IgG and preformed streptABComplex-peroxidase (both from DAKO A/S) at room temperature for 1 hour. Incubation with the peroxidase-specific substrate 3-amino-9-ethylcarbazole (Sigma Chemical Co.) was used for visualization, with hematoxylin counterstaining. For control purposes, the primary mAb was replaced by an irrelevant isotype-matched mAb that consistently yielded negative results.
Nick-end labeling of DNA from apoptotic KCs.
Apoptotic cells may be identified in situ by histochemical techniques with staining of double-stranded DNA breaks. Terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling (TUNEL) was performed as described (25). This technique is used widely, and has been found to be sensitive and to preferentially detect DNA breaks encountered in apoptosis. Briefly, TUNEL reaction mixture (Boehringer Mannheim GmbH, Mannheim, Germany) was added to the samples, which were incubated for 60 minutes at 37°C. Incorporated fluorescein was detected by anti-fluorescein Ab Fab fragments from sheep, conjugated with alkaline phosphatase.
Identification of apoptosis by HOECHST staining.
HOECHST staining was done according to Norris et al. (26) in KCs cultured in Transwell plates (Corning-Costar Corp.) and in lesional skin sections. After fixation with 4% paraformaldehyde and 200 mM dihydrogen phosphate (pH 7.0) overnight at 4°C, staining was performed with HOECHST 33342 dye (1 μg/mL; Sigma Chemical Co.) for 5 minutes. Stained sections were evaluated with an ultraviolet microscope (Axiovert 405M; Carl Zeiss AG, Feldbach, Switzerland).
Cocultures of normal human skin and skin equivalents with T cells.
Normal healthy human skin obtained during plastic surgery was cut into small pieces (approximately 6 mm × 3 mm). This skin was then placed in the upper compartment of Transwell plates, not allowing cell-cell contact, and was exposed for 3 days to heterologous unstimulated and stimulated CD45RO+ T cells that were placed in the lower compartment (106/well). From normal, healthy human skin obtained during plastic surgery, a three-dimensional whole-skin model was built up from dermal fibroblasts embedded in a biomatrix and multilayered epidermis with stratum corneum, according to the method of the reconstructed skin equivalent AST-2000 (CellSystems Biotechnologie Vertrieb GmbH, St. Katharinen, Germany) (27). The cultured skin equivalents, grown for 2 weeks at the air-liquid interface, were then exposed together with their inserts (not allowing cell-cell contact) for 3 days to heterologous unstimulated and stimulated CD45RO+ T cells. The skin pieces were then fixed in Bouin’s solution (Sigma Chemical Co.) and were further processed for staining with hematoxylin/eosin and TUNEL.
Statistical analysis.
Results are shown as mean ± SD. The paired Student’s t test was used for comparison of paired conditions.
Results
KC injury induced by activated T cells.
The histologic hallmark of eczematous disorders consists of a marked KC pathology. Acantholysis and spongiosis in the epidermis is characterized by impairment or loss of cohesion between KCs and an influx of fluid from the dermis, progressing to vesicle formation (Figure 1a and ref. 28). In preliminary experiments, we observed several morphologic changes in cultured primary human KCs (Figure 1b) in the coculture with autologous T cells. Resting, unstimulated T cells did not affect KC morphology (Figure 1c), whereas KCs cocultured with stimulated T cells were shrunken and rounded; they finally detached from the plates and died (Figure 1d). The type of KC death was investigated by demonstration of apoptotic features such as chromatin condensation and fragmented nuclei made visible by HOECHST staining (26). To eliminate the possibility that the apoptotic bodies were derived from T cells, KCs and T cells were separated in Transwell plates. As shown in Figure 1f, KC apoptosis was induced by soluble factors from stimulated T cells, whereas resting T cells did not show any effect (Figure 1e). These results demonstrated that KCs are target cells of activated T cells that invade the epidermis in the efferent phase of eczematous disorders, suggesting that KC injury and death may have a decisive contribution to the KC pathology seen in eczema.
Figure 1.
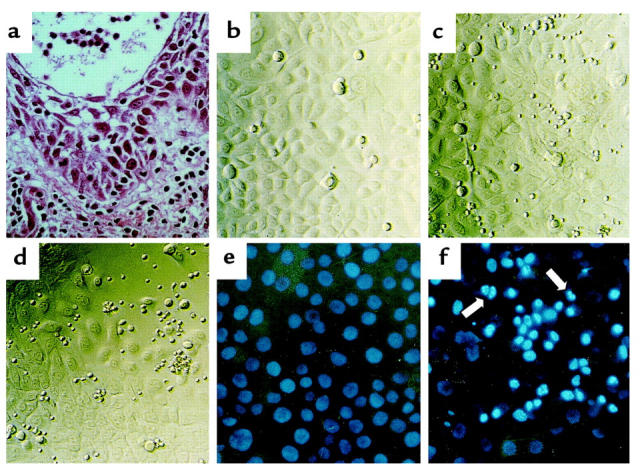
(a) Representative histologic findings of acute eczematous dermatitis. A dense subepidermal inflammatory infiltrate and marked epidermal acantholytic and spongiotic changes progress to vesicle formation. Hematoxylin/eosin staining. ×400. (b–d) Signs of KC injury after coculture with autologous T cells. Photomicrographs from 96-well plates with an inverted microscope equipped with phase contrast. ×200. (b) Intact monolayer of cultured third-passage primary human KCs. The KCs are relatively uniform in size and morphology. (c) Intact monolayer of KCs after 3 days in coculture with unstimulated autologous CD45RO+ T cells. (d) Partly destroyed monolayer of KCs after 3 days in coculture with autologous CD45RO+ T cells stimulated with anti-CD2, anti-CD3, and anti-CD28 mAb’s. (e and f) Induction of KC apoptosis in vitro. Identification of apoptotic nuclei with HOECHST staining. ×200. (e) Intact monolayer of KCs after 3 days of coculture in Transwell plates with unstimulated CD45RO+ T cells. (f) Induction of KC apoptosis after coculture in Transwell plates with stimulated CD45RO+ T cells. Note bright, condensed nuclei and nuclear fragmentation (arrows), signs of KC apoptosis.
Role of IFN-γ and FasR in KC apoptosis.
We then focused on the mechanism of KC death induced by activated T cells. FasR (CD95) is a 47-kDa membrane protein that transduces an apoptotic signal into the cell when triggered by its ligand (FasL) or by agonistic Abs (29, 30). KCs expressed detectable levels of FasR mRNA, which significantly increased after incubation with IFN-γ or with supernatants from stimulated CD45RO+ (memory/effector) T cells (Figure 2a). In contrast, FasL mRNA was not detected in the KCs. As shown in Figure 2b, unstimulated KCs showing low FasR expression were resistant to Fas-mediated killing. IFN-γ, which is known to induce growth arrest and the expression of squamous differentiation genes (31), showed no significant killing activity. However, KCs pretreated with IFN-γ (but not TNF-α, IL-4, IL-5, IL-12, or IL-13; data not shown) were efficiently killed by cross-linking of FasR with anti-FasR mAb or soluble FasL (Figure 2b; P < 0.05). In KCs, the sensitivity to Fas-induced death correlated very well with levels of cell-surface FasR (Figure 2c). Increasing doses of IFN-γ induced FasR expression on KCs. When the number of FasR molecules on KCs reached a threshold of approximately 40,000 per KC, the cells became susceptible to apoptosis.
Figure 2.

(a) Expression of FasR mRNA by primary human KCs. Lane 1: unprotected template. Lane 2: after 8 hours of KC culture. Lane 3: after 24 hours of KC culture. Lane 4: after 16 hours of KC culture followed by 8 hours with 1.0 μg/mL anti-FasR mAb. Lane 5: after 16 hours of KC culture followed by 8 hours with diluted (50%) supernatant (sup.) from stimulated CD45RO+ T cells. Lane 6: after 16 hours of KC culture followed by 8 hours with 1.0 ng/mL IFN-γ. A representative result of three experiments is shown. (b) IFN-γ and Fas-induced KC apoptosis. KC viability was monitored by ethidium bromide exclusion and flow cytometry. Treatments shown are control (KCs alone), 1 μg/mL activating anti-FasR mAb, 10 ng/mL IFN-γ, and 1 μg/mL anti-FasR mAb added 1 day after starting incubation with 10 ng/mL IFN-γ. AP < 0.05. (c) IFN-γ–induced FasR counts exhibit a threshold for KC apoptosis. KCs were pretreated with the indicated doses of IFN-γ; 1 μg/mL anti-FasR mAb was added 1 day after starting incubation with IFN-γ. KC viability was assessed by ethidium bromide exclusion and flow cytometry at day 3. FasR count 1 day after starting incubation with indicated doses of IFN-γ. (d) CD45RO+ T cell–induced KC death is inhibited by blocking IFN-γ. KC viability was monitored by ethidium bromide exclusion and flow cytometry. In the flow cytometry setting, KCs and T cells are gated according to forward and side scatter. Both cell populations were therefore monitored separately. Coculture of primary human KCs and autologous unstimulated or stimulated (with anti-CD2, anti-CD3, and anti-CD28 mAb) CD45RO+ T cells. AP < 0.05. Inhibition of CD45RO+ T cell–induced KC death by 1 μg/mL blocking anti–IFN-γ receptor mAb and 20 μg/mL neutralizing anti–IFN-γ mAb. Results in b–d represent mean ± SD of triplicate cultures from three different experiments. Control, KCs alone.
Induction and regulation of KC apoptosis by T cells.
Skin-infiltrating T cells belong primarily to the CD45RO+ subset (32, 33). In KC–T cell cocultures, we observed killing of KCs by stimulated autologous CD45RO+ T cells (Figure 2d; P < 0.05). Ligation of the T-cell receptor–CD3 complex by antigen peptides or mAb’s to T-cell receptor–CD3 triggers a series of activation events, eventually leading to cell proliferation and cytokine production (34, 35). It is known that anti-CD3 mAb cross-linking induces the expression of cell-surface and soluble FasL by T cells (36). Accordingly, KC killing was significant only after activation of T cells, either with phorbol ester and calcium ionophore, cross-linking of the T-cell receptor–CD3 complex by plate-bound anti-CD3 (data not shown), or a combination of anti-CD2, anti-CD3, and anti-CD28 mAb’s (Figure 2d; P < 0.05). Cytokine pretreatment of KCs was not necessary for death; however, it is apparent that sufficient IFN-γ was produced by the stimulated T cells themselves to upregulate KC FasR expression. As seen in Figure 2d, KC killing could be inhibited by pretreatment with either neutralizing anti–IFN-γ mAb or by the IFN-γ receptor–blocking mAb. Neutralizing mAb’s against IL-4, IL-5, and IL-13, and the mutant IL-4 antagonist showed no inhibitory activity (data not shown). We further examined whether FasR engagement on IFN-γ–treated KCs triggered apoptotic cell death. Stimulated T cells induced KC apoptosis as demonstrated by HOECHST staining (Figure 1f), by annexin V staining of membrane phosphatidylserine inversion in KC–T cell cocultures (Figure 3a), and by DNA fragmentation in Transwell cocultures with no cell-cell contact allowed (Figure 3b). This demonstrates that soluble FasL released from T cells induces apoptosis in KCs in the absence of cell-cell contact. KC apoptosis was again inhibited by neutralizing IFN-γ, blocking the IFN-γ receptor, and by pretreatment of KCs with the caspase inhibitor Z-VAD-FMK (37). To further test that FasR expression on KCs is triggered by FasL from stimulated T cells, we used the human Fas-Fc protein as a competitive inhibitor of FasL-FasR interactions (38). We found that the Fas-Fc protein effectively inhibited apoptosis of KCs induced by stimulated T cells (Figure 3, a and b).
Figure 3.

Induction and regulation of KC apoptosis in KC–T cell cocultures. (a) Coculture of primary human KCs and stimulated autologous CD45RO+ T cells. After 3 days of coculture, cells were stained with annexin V and subjected to flow cytometry. With the indicated stimuli, the number of apoptotic KCs increases and the cells show an increase in annexin V binding (open histograms). Filled histograms show live KCs. The percentage of apoptotic KCs is indicated at upper right. Panel 1: KC apoptosis induced by coculture with CD45RO+ T cells stimulated with anti-CD2, anti-CD3, and anti-CD28 mAb’s. Panel 2: Isotype control mAb. Panel 3: Blocking anti–IFN-γ receptor mAb (1 μg/mL). Panel 4: Fas-Fc protein (1 μg/mL). Panel 5: The caspase inhibitor Z-VAD-FMK (50 μM). Panel 6: Z-VAD-FMK (100 μM). (b) Coculture of primary human KCs and stimulated autologous CD45RO+ T cells in Transwell plates. After 3 days in coculture, permeabilized KCs were stained with propidium iodide and subjected to flow cytometry. Filled histograms demonstrate control KCs containing diploid DNA. The percentage of apoptotic KCs with hypodiploid DNA (open histograms) is shown in the upper right corner. Panel 1: Primary human KCs alone were cultured in the lower well. Panel 2: KCs were pretreated with 10 ng/mL IFN-γ for 24 hours, and KC apoptosis was determined 3 days after 1 μg/mL activating anti-FasR mAb was added. Panel 3: KC apoptosis induced by Transwell coculture with stimulated CD45RO+ T cells. Panel 4: Inhibition of KC apoptosis induced by IFN-γ and anti-FasR mAb, with 1 μg/mL ZB4 blocking mAb. Panels 5 and 6: Inhibition of KC apoptosis induced by Transwell coculture with stimulated CD45RO+ T cells, with 1 μg/mL Fas-Fc protein (panel 5), and with 10 μg/mL neutralizing anti–IFN-γ mAb (panel 6). Results in a and b are representative of three experiments.
Induction of KC apoptosis by type 1 and type 2 T cells.
To establish a more disease-related model for KC apoptosis in eczematous disorders, we investigated the effects of different phenotypes of stimulated T cells on KCs. Type 1 CD4+ and CD8+ T cells contain and secrete primarily the cytokine IFN-γ, and only low amounts of IL-4 and IL-5. In contrast, type 2 CD4+ and CD8+ T cells contain and secrete IL-4, IL-5, and IL-13, but only low amounts of IFN-γ (39). The type 1 CD4+ and CD8+ T cells were able to induce KC death (Figure 4a; P < 0.05). In contrast, type 2 T cells had no effect on KCs after differentiation. Additionally, we combined type 1 and type 2 T cells and measured KC apoptosis. This mixture induced apoptosis less than type 1 cells alone did, but apoptosis was still highly significant (data not shown). IL-12 is produced mainly by macrophages and dendritic cells, and promotes type 1 T-cell responses by selective upregulation of IFN-γ production by T cells (40). Stimulation of type 2 CD4+ or CD8+ T cells with IL-12 increased IFN-γ production and ablated IL-4 production (Figure 4b; P < 0.05). In this case, IL-12 stimulation of type 2 T cells enabled the induction of KC apoptosis (Figure 4c; P < 0.05). Supernatants from stimulated type 1 and type 2 T cells released soluble FasL; addition of IL-12 did not influence the secretion of soluble FasL (Figure 4d).
Figure 4.

Induction of KC apoptosis by type 1 and type 2 T cells. (a) Type 1 but not type 2 T cells mediated KC death. Coculture of primary human KCs and heterologous differentiated type 1 and type 2 T cells. AP < 0.05. (b) Cytokine content of differentiated and stimulated type 2 T cells as determined by ELISA. CD4+ T helper 2 (Th2) and CD8+ T cytotoxic 2 (Tc2) cells were stimulated with anti-CD2, anti-CD3, and anti-CD28 mAb’s or with a combination of anti-CD2, anti-CD3, and anti-CD28 mAb’s and IL-12. AP < 0.05. (c) Coculture of primary human KCs and heterologous differentiated type 2 T cells. KC viability was measured by ethidium bromide exclusion and flow cytometry at day 3. Control, KCs alone. CD4+ Th2 cells and CD8+ Tc2 cells were stimulated with anti-CD2, anti-CD3, and anti-CD28 mAb’s or with a combination of anti-CD2, anti-CD3, and anti-CD28 mAb’s and IL-12. AP < 0.05. (d) Levels of soluble FasL in T-cell supernatants as determined by ELISA. CD4+ Th1/Th2 and CD8+ Tc1/Tc2 cells were stimulated with anti-CD2, anti-CD3, and anti-CD28 mAb’s or with a combination of anti-CD2, anti-CD3, and anti-CD28 mAb’s and IL-12. Results shown represent three (a, c) to five (b, d) experiments and are shown as mean ± SD from triplicate cultures.
Features of inflammation in eczematous dermatitis.
The histologic appearance of acute lesional AD and ACD skin was defined by various degrees of dermal perivascular infiltration with mononuclear cells, mostly consisting of CD4+ T cells as well as CD8+ T cells (Figure 5, a–c). The expression of CD45RO on the T cells suggests a previous encounter with antigens; many of the T cells showed signs of intralesional activation as defined by membrane expression of HLA-DR (2). It should be noted here that T cells constitute the major cells in dermal infiltrates, and that some of the CD4+ and CD8+ T cells invade the epidermis (Figure 5, b and c). Therefore, membrane-bound FasL on T cells can trigger FasR on KCs by cell-cell contact. Figure 5d demonstrates FasL immunoreactivity on lesional T cells of acute AD. The basal and suprabasal compartment of the epidermis was strongly reactive for FasR, and the KCs showed high FasR surface expression in acute AD (Figure 5e). Similarly, ACD skin showed FasL expression on T cells and FasR expression on KCs (data not shown). Chronic AD lesions displayed weaker FasR immunoreactivity in basal KCs. Healthy control skin displayed no inflammatory infiltrate and no KC staining with anti-FasR mAb. KCs from healthy individuals and from AD and ACD patients expressed IFN-γ receptors on their surface in vitro and in vivo (Figure 5f and ref. 41).
Figure 5.
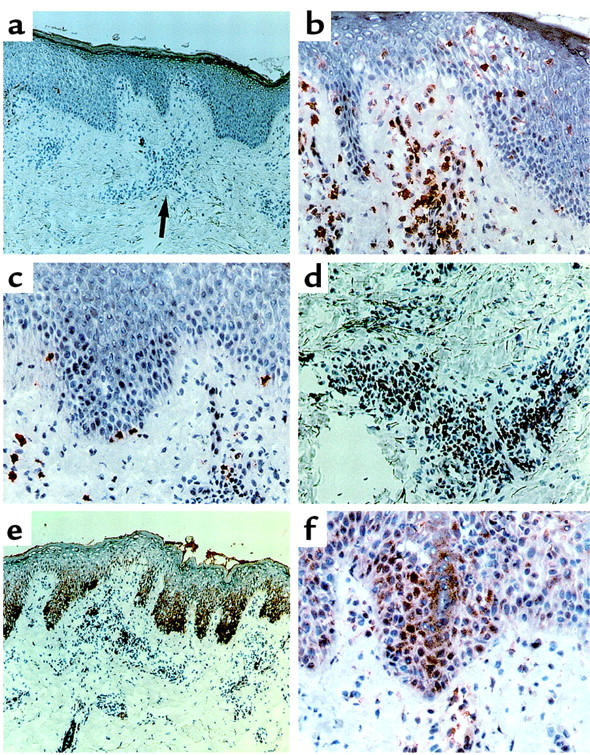
Features of inflammation in AD. Immunohistologic staining with 3-amino-9-ethylcarbazole substrate and counterstaining with hematoxylin. (a) Subepidermal inflammatory infiltrate, consisting mainly of lymphocytes. ×100. (b) CD4+ cells infiltrating the spongiotic epidermis. ×200. (c) CD8+ cells. ×200. (d) FasL+ cells in the dermal cellular infiltrate. ×200. (e) Strong immunoreactivity of FasR in the basal epidermis; weaker staining in the T-cell infiltrate. ×100. (f) Detection of IFN-γ receptor on KCs and infiltrating T cells. ×200.
Demonstration of KC apoptosis in AD, atopy patch tests, ACD, and cultures of healthy human skin and skin equivalents.
Skin biopsies from acute AD, chronic AD, atopy patch tests, and acute ACD were analyzed using the TUNEL technique (Figure 6, a and b) and HOECHST staining (Figure 6, c and d). Patch-test reactions to aeroallergens constitute a suitable model for the study of allergic inflammation in AD. They are specific for sensitized AD patients, and do not occur in healthy volunteers or in patients suffering from asthma or rhinitis (42). As a negative control, we used normal skin from healthy individuals. No TUNEL-stained cells were observed in the stratum basale or stratum spinosum of normal skin. TUNEL-stained nuclei were rarely seen in the lumen of spongiotic vesicles of acute AD and ACD. As shown in Figure 6, a and b, TUNEL-stained KCs are visible primarily in the cohesive epidermis (stratum spinosum) of acute lesions in which acantholysis and spongiosis had not progressed to vesicle formation. In chronic AD, TUNEL-stained KCs are visible at a lower frequency than in acute AD and ACD. HOECHST staining was used as a second method, complementing TUNEL staining (26, 43). In Figure 6, c and d, condensed and fragmented nuclei of apoptotic cells are demonstrated in acute AD and ACD lesions. Besides the demonstration of KC apoptosis in acute AD, chronic AD, atopy patch tests, and acute ACD, we also investigated apoptosis in cocultures of normal human skin of healthy individuals with stimulated T cells, and in cocultures of skin equivalents with stimulated T cells. We were able to demonstrate the induction of KC apoptosis by in vitro exposure of normal human skin (Figure 6e) and cultured skin equivalents (Figure 6, f and g) to stimulated CD45RO+ T cells in Transwell cocultures. In contrast, unstimulated CD45RO+ T cells had no effect.
Figure 6.

Demonstration of apoptotic KCs in AD, atopy patch tests, ACD, and in vitro induced eczematous dermatitis. Skin sections were subjected to TUNEL (a, b, e–g) or HOECHST staining (c, d) without counterstaining. (a) Healthy normal skin as a negative control (left); lesional skin of acute AD (right). Red condensed and partly fragmented nuclei indicate positive staining of apoptotic KCs (some of these are indicated by arrows). ×400. (b) Left: Acute ACD; 2+ patch test 48 hours after exposure to 5% nickel sulfate. Right: 2+ atopy patch test 72 hours after exposure to 10,000 protein nitrogen units of house dust mite. ×400. (c) Normal, healthy skin as a negative control. ×200. (d) Left: Lesional skin of acute AD. Right: Acute ACD. 2+ patch test 48 hours after exposure to 5% nickel sulfate. Apoptotic cells show condensed or fragmented nuclei (some of these are indicated by arrows). ×400. (e) Normal human skin after 3 days of in vitro culture with unstimulated CD45RO+ T cells. Left: No TUNEL-stained KCs are detectable. Right: Normal human skin after 3 days of in vitro culture exposed to stimulated CD45RO+ T cells. Features of spongiosis and numerous apoptotic KCs are detectable (arrows). ×200. (f) Cultured skin equivalent after 3 weeks of in vitro culture. Left: KC layers and the stratum corneum are visible (hematoxylin/eosin staining). Right: TUNEL staining. No TUNEL-stained nuclei are detectable. ×400. (g) Cultured skin equivalent after 3 days exposed to stimulated CD45RO+ T cells. There are signs of KC damage (left). With the TUNEL technique, several apoptotic nuclei are stained in the basal and suprabasal epidermis (arrows, right). Results shown in e–g are representative of two experiments with healthy skin and three experiments with cultured skin equivalents.
Discussion
This study demonstrates that Fas-induced KC apoptosis caused by skin-infiltrating T cells is a major mechanism in the pathogenesis of eczematous dermatitis. In recent years, much interest has focused on apoptosis as a mechanism to control cell numbers (44). The interactions between FasL and FasR are important in maintaining lymphoid cell numbers. However, it is likely that FasL-FasR interaction also plays a role in other important biologic processes. Various cells in the body constitutively express FasR, or can be induced to do so (45, 46). KCs express FasR (47, 48), and the sensitivity to Fas-induced KC death correlated very well with numbers of cell surface FasR in our study. An explanation for this may be that quantitative increases in FasR expression above a certain threshold level result in a signaling intensity that promotes apoptosis. On the other hand, it is possible that the signal transduction pathway for Fas-induced apoptosis is functionally inactive in unstimulated KCs. The threshold for IFN-γ in the induction of FasR on KCs appears to be in the range of 0.1–1.0 ng/mL.
FasL expression is usually limited to activated T cells, natural killer cells, and cells of certain immunologically privileged sites (49). T cells appear to be important players regulating inflammatory processes in eczematous dermatitis (6–18). This is supported by the observation that immunosuppressive drugs such as FK-506 and glucocorticoids, which block T-cell activation, are effective in treatment of eczematous disorders (4, 5). The coupling of Fas signaling to the death pathway involves receptor trimerization followed by the binding of adapter proteins and then the caspase enzyme cascade, ultimately resulting in apoptosis (29, 30). Our studies using the caspase inhibitor Z-VAD-FMK (37) show that caspase activation is also critical for Fas-mediated apoptosis of KCs. In addition, recombinant Fas-Fc protein (38) prevents T cell–mediated KC apoptosis. This protective effect indicates that the FasL-FasR interaction is responsible for T cell–mediated KC apoptosis. The lethal hit is delivered to KCs by FasL expressed on the surface of T cells that invade the epidermis, and probably by soluble FasL released from T cells. IFN-γ, which increases FasR expression on KCs, is a further prerequisite in T cell–mediated KC apoptosis. This is demonstrated by the fact that blocking of IFN-γ totally abrogates KC apoptosis induced by activated T cells. Partial contributions by other cytokines and apoptotic mechanisms were principally eliminated in experiments demonstrating neutralization of the effects by blocking IFN-γ and by using Fas-pathway antagonists. It has been proposed that the basal epidermis is resistant to apoptosis due to highly developed antiapoptotic defenses, which are reversible and decrease during differentiation (26). Adding IFN-γ causes cultured KCs to differentiate and lose resistance to apoptosis (31). Thus the increased susceptibility to killing by Fas ligation in vitro may be due to both increased FasR expression and reduced anti-apoptotic defenses.
Eczematous skin lesions with distinct etiology are associated with T-cell infiltration in the epidermis, contact between T cells and KCs, and a marked KC pathology (28, 50, 51). KCs in inflamed skin express HLA-DR and ICAM-1, which are not expressed on KCs of normal skin (1, 2). The only mediator known to date that induces HLA-DR on KCs is IFN-γ. Together, these data suggest a model of T cell–induced KC apoptosis in the pathogenesis of eczematous dermatitis. In an early step, T cells infiltrating the epidermis attach to KCs. Subsequently, the apoptosis signal is delivered to KCs by FasL expressed on the surface of T cells. In our experiments, FasL was detected only on the T-cell surface — not on KCs in AD, ACD, and patch-test lesions. However, in a study of toxic epidermal necrolysis, FasL expression on highly activated KCs was shown (52). In addition to FasL-FasR interaction, it is possible that activated T cells in eczematous dermatitis use granule-mediated killing by perforin and granzymes (53–55). However, granule-mediated cytotoxicity is not blocked by soluble Fas-Fc protein. Additionally, using immunohistology, we could not detect perforin or granzyme B in lesional skin of ACD and AD patients.
To establish a more disease-related model for KC apoptosis in eczematous dermatitis, we investigated the effects of different subsets of activated T cells on KCs. We demonstrated Fas-mediated apoptosis by both CD4+ and CD8+ type 1 T cells. There is accumulating evidence that not only CD4+, but also CD8+ T cells possess the potential to both stimulate IgE production and promote allergic inflammation (10, 56). ACD is regarded as a type 1 T cell–mediated phenomenon in vivo, based on a number of observations (6, 7). Several results suggest that type 1 and type 2 cytokines secreted from activated T cells are important for the pathogenesis of AD (17). Clinical and histologic features of AD are almost indistinguishable from those present in ACD. Recent studies found IFN-γ mRNA and protein highly expressed in eczematous skin of the vast majority of AD patients (15). IL-12 stimulation of type 2 T cells abolished IL-4 and induced IFN-γ production without major changes in IL-5 and IL-13 production. This explains the biphasic nature of the cytokine response observed in acute versus chronic AD lesions. Acute lesions and early response after epidermal allergen application were characterized with a predominant type 2 cytokine pattern (4, 16, 57). Increased levels of IL-12 mRNA was demonstrated in chronic AD lesions (58), and chronic lesions were characterized by increased IFN-γ and decreased IL-4 levels, with more or less constant IL-5 and IL-13 levels (13, 15, 16, 57, 58). We observed killing of KCs by type 2 T cells after stimulation in the presence of IL-12. A cause-and-effect relationship between IL-12 and IFN-γ expression may play a role in the conversion of type 2 T cells into IFN-γ–producing T cells that induce KC apoptosis in AD (15–17, 58). In addition, virus infections and bacterial superantigens of AD skin may lead to IFN-γ secretion of skin-infiltrating T cells (14, 58, 59). Furthermore, KCs exhibited a relatively low threshold for FasR expression in response to IFN-γ, in the range of 0.1–1.0 ng/mL.
Our results suggest that KC apoptosis is the initiating event in the development of the epidermal pathology seen in eczematous dermatitis. TUNEL and HOECHST staining showed that apoptotic KCs were present in the epidermis of patients with eczematous disorders. Most notably, KC apoptosis occurs in suprabasal cells, where spongiosis and acantholysis takes place. The basal layers of lesional epidermis had high levels of FasR, but it is likely that anti-apoptotic mechanisms prevent apoptosis of basal KCs (26). Apoptosis of individual KCs is the first event leading to disruption of epidermal continuity and vesicle formation. Damage to KCs leads to the loss of intercellular cohesion (acantholysis) and subsequent cleft formation. Fluid influx from the dermis and intercellular edema contributes to spongiosis, which is defined as widening of the intercellular space and a spongelike appearance of the epidermis (28). Based on our findings, the reason that not all the suprabasal KCs that express FasR undergo apoptosis uniformly in situ has to do with the activation of KCs induced by IFN-γ and the ligation of FasR on the KCs by FasL expressed on T cells infiltrating the epidermis. Apoptosis of epidermal KCs is known to be an important feature in lichenoid tissue reaction, fixed drug eruption, graft-versus-host reaction, and the effects of ultraviolet radiation (26, 60, 61).
Skin equivalents formed by KCs obtained from healthy skin and individuals with no skin disorders that are cocultured with fibroblasts embedded in collagen lattices represent promising tools for studies of skin pathology (27, 62). In cultured skin equivalents, KC differentiation and composition of the basement membrane are not exactly the same as in living epidermis. Therefore it is likely that the pro- and anti-apoptotic mechanisms operating in KCs are somewhat different in vivo, and basal KCs are probably more sensitive to apoptosis in skin equivalents. We also used the short-term culture of normal human skin in a medium maintaining KC viability to investigate the effects of activated T cells on epidermal KCs. The induction of KC apoptosis in normal human skin and cultured skin equivalents after exposure to activated T cells establishes the in vivo relevance of T cell–mediated, Fas-induced KC apoptosis. The knowledge of this molecular basis is pivotal in understanding the development of pathology in eczematous disorders, and opens a future for more focused therapeutic applications.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (TR460/1-1), the Swiss National Foundation (31.50590.97/1), and the Baumgarten Foundation (Zürich). We thank A. Speiser (Landspital Davos), J. Kammerer (Department of Pathology, Rätisches Kantonsspital Chur), R. Gillitzer, W. Dummer, A. Toksoy, H. Krenig, and C. Siedel (Department of Dermatology, University of Würzburg) for their work.
References
- 1.Singer KH, Tuck DT, Sampson HA, Hall RP. Epidermal keratinocytes express the adhesion molecule intracellular adhesion molecule-1 in inflammatory dermatoses. J Invest Dermatol. 1989;92:746–750. doi: 10.1111/1523-1747.ep12722441. [DOI] [PubMed] [Google Scholar]
- 2.Bieber T, Dannenberg B, Ring J, Braun-Falco O. Keratinocytes in lesional skin of atopic eczema bear HLA-DR, CD1a and IgE molecules. Clin Exp Dermatol. 1989;14:35–39. doi: 10.1111/j.1365-2230.1989.tb00880.x. [DOI] [PubMed] [Google Scholar]
- 3.Dustin ML, Singer KH, Tuck DT, Springer TH. Adhesion of T lymphoblasts to epidermal keratinocytes is regulated by interferon-γ and is mediated by intercellular adhesion molecule-1 (ICAM-1) J Exp Med. 1988;167:1323–1340. doi: 10.1084/jem.167.4.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leung DY. Pathogenesis of atopic dermatitis. J Allergy Clin Immunol. 1999;104(Suppl.):S99–S108. doi: 10.1016/s0091-6749(99)70051-5. [DOI] [PubMed] [Google Scholar]
- 5.Rudikoff D, Lebwohl M. Atopic dermatitis. Lancet. 1998;351:1715–1721. doi: 10.1016/S0140-6736(97)12082-7. [DOI] [PubMed] [Google Scholar]
- 6.Grabbe S, Schwarz T. Immunoregulatory mechanisms involved in elicitation of allergic contact hypersensitivity. Immunol Today. 1998;19:37–44. doi: 10.1016/s0167-5699(97)01186-9. [DOI] [PubMed] [Google Scholar]
- 7.Fong TA, Mosman TR. The role of IFN-γ in delayed-type hypersensitivity mediated by Th1 clones. J Immunol. 1989;143:2887–2893. [PubMed] [Google Scholar]
- 8.Santamaria Babi LF, et al. Circulating allergen-reactive T cells from patients with atopic dermatitis and allergic contact dermatitis express the skin-selective homing receptor, the cutaneous lymphocyte-associated antigen. J Exp Med. 1995;181:1935–1940. doi: 10.1084/jem.181.5.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akdis M, Akdis CA, Weigl L, Disch R, Blaser K. Skin-homing CLA+ memory T cells are activated in atopic dermatitis and regulate IgE by an IL-13 dominated cytokine pattern: IgG4 counter-regulation by CLA− memory T cells. J Immunol. 1997;159:4611–4619. [PubMed] [Google Scholar]
- 10.Akdis M, et al. Skin-homing (cutaneous-lymphocyte-associated-antigen positive) CD8+ T cells respond to superantigen and contribute to eosinophilia and IgE production in atopic dermatitis. J Immunol. 1999;163:466–475. [PubMed] [Google Scholar]
- 11.Leung DY. Role of IgE in atopic dermatitis. Curr Opin Immunol. 1993;5:956–962. doi: 10.1016/0952-7915(93)90112-6. [DOI] [PubMed] [Google Scholar]
- 12.Jujo K, Renz H, Abe J, Gelfand EW, Leung DY. Decreased IFN-γ and increased IL-4 production in atopic dermatitis promotes IgE synthesis. J Allergy Clin Immunol. 1992;90:323–331. doi: 10.1016/s0091-6749(05)80010-7. [DOI] [PubMed] [Google Scholar]
- 13.Akdis CA, et al. T cells and T cell-derived cytokines as pathogenetic factors in the nonallergic form of atopic dermatitis. J Invest Dermatol. 1999;113:628–634. doi: 10.1046/j.1523-1747.1999.00720.x. [DOI] [PubMed] [Google Scholar]
- 14.Herz U, Bunikowski R, Renz H. Role of T cells in atopic dermatitis. Int Arch Allergy Immunol. 1998;115:179–190. doi: 10.1159/000023899. [DOI] [PubMed] [Google Scholar]
- 15.Grewe M, Gyufko K, Schöpf E, Krutman J. Lesional expression of interferon-γ in atopic eczema. Lancet. 1994;343:25–26. doi: 10.1016/s0140-6736(94)90879-6. [DOI] [PubMed] [Google Scholar]
- 16.Thepen T, et al. Biphasic response against aeroallergen in atopic dermatitis showing a switch from an initial Th2 response to a Th1 response in situ: an immunocytochemical study. J Allergy Clin Immunol. 1996;97:828–837. doi: 10.1016/s0091-6749(96)80161-8. [DOI] [PubMed] [Google Scholar]
- 17.Grewe M, et al. A role for Th1 and Th2 cells in the immunopathogenesis of atopic dermatitis. Immunol Today. 1998;19:359–361. doi: 10.1016/s0167-5699(98)01285-7. [DOI] [PubMed] [Google Scholar]
- 18.Spergel JM, Mizoguchi E, Oettgen H, Bhan AK, Geha RS. Roles of Th1 and Th2 cytokines in a murine model of allergic dermatitis. J Clin Invest. 1999;103:1103–1111. doi: 10.1172/JCI5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barker JNWN, Allen MH, MacDonald DM. Alterations induced in normal human skin by in vivo interferon-γ. Br J Dermatol. 1990;122:451–458. doi: 10.1111/j.1365-2133.1990.tb14721.x. [DOI] [PubMed] [Google Scholar]
- 20.Carroll JM, Crompton T, Seery JP, Watt FM. Transgenic mice expressing IFN-γ in the epidermis have eczema, hair hypopigmentation and hair loss. J Invest Dermatol. 1997;108:412–422. doi: 10.1111/1523-1747.ep12289702. [DOI] [PubMed] [Google Scholar]
- 21.Hanifin JM. Atopic dermatitis. J Am Acad Dermatol. 1982;6:1–13. doi: 10.1016/s0190-9622(82)70001-5. [DOI] [PubMed] [Google Scholar]
- 22.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;272:104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 23.Vermes I, Haanen C, Steffens-Nakken H, Reutlingsberger CDM. A novel assay for apoptosis: flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labeled Annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 24.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 25.Gavrieli Y, Sherman Y, BenSasson SA. Identification of programmed cell death by in situ specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Norris DA, et al. Human keratinocytes maintain reversible anti-apoptotic defenses in vivo and in vitro. Apoptosis. 1997;2:136–148. doi: 10.1023/a:1026456229688. [DOI] [PubMed] [Google Scholar]
- 27.Noll M, et al. Reconstructed human skin (AST-2000) as a tool for pharmaco-toxicology. Alternatives to Laboratory Animals. 1999;27:302a. (Abstr.) [Google Scholar]
- 28.Wolff, K., Kibbi, A.G., and Mihm, M.C. 1993. Basic pathologic reactions of the skin. In Dermatology in general medicine. T.B. Fitzpatrick, editor. McGraw-Hill. New York, New York, USA. 66–84.
- 29.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 30.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 31.Saunders NA, Jetten AM. Control of growth regulatory and differentiation specific genes in human epidermal keratinocytes by IFN-γ. Antagonism by retinoic acid and transforming growth factor β1. J Biol Chem. 1994;269:2016–2022. [PubMed] [Google Scholar]
- 32.Foster CA, et al. Human epidermal T cells predominantly belong to the lineage expressing α/β T cell receptor. J Exp Med. 1990;171:997–1013. doi: 10.1084/jem.171.4.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bell EB, Sparshott SM, Bunce C. CD4+ T cell memory, CD45R subsets and the persistence of antigen-a unifying concept. Immunol Today. 1998;19:60–64. doi: 10.1016/s0167-5699(97)01211-5. [DOI] [PubMed] [Google Scholar]
- 34.Crabtree GR. Contingent genetic regulatory events in T lymphocyte activation. Science. 1989;270:1189–1191. doi: 10.1126/science.2783497. [DOI] [PubMed] [Google Scholar]
- 35.Dorf ME, Benacerraf B. Suppressor cells and immunoregulation. Annu Rev Immunol. 1984;2:127–157. doi: 10.1146/annurev.iy.02.040184.001015. [DOI] [PubMed] [Google Scholar]
- 36.Lynch DH, Ramsdell F, Alderson MR. Fas and Fas ligand in the homeostatic regulation of immune responses. Immunol Today. 1995;16:569–572. doi: 10.1016/0167-5699(95)80079-4. [DOI] [PubMed] [Google Scholar]
- 37.Pronk GJ, Ramer K, Amiri P, Williams LT. Requirement of an ICE-like protease for induction of apoptosis and ceramide generation by REAPER. Science. 1996;271:808–810. doi: 10.1126/science.271.5250.808. [DOI] [PubMed] [Google Scholar]
- 38.Ramsdell F, Seaman MS, Miller RE, Tough TW, Alderson MR. gld/gld mice are unable to express a functional ligand for Fas. Eur J Immunol. 1994;24:928–933. doi: 10.1002/eji.1830240422. [DOI] [PubMed] [Google Scholar]
- 39.Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science. 1997;277:2005–2007. doi: 10.1126/science.277.5334.2005. [DOI] [PubMed] [Google Scholar]
- 40.Trinchieri G. Interleukin-12: a cytokine produced by antigen-presenting cells with immunoregulatory functions in the generation of T helper cells type 1 and cytotoxic lymphocytes. Blood. 1994;84:4008–4027. [PubMed] [Google Scholar]
- 41.Scheynius A, et al. Expression of interferon-γ receptors in normal and psoriatic skin. J Invest Dermatol. 1992;98:255–258. doi: 10.1111/1523-1747.ep12556086. [DOI] [PubMed] [Google Scholar]
- 42.DeBruin-Weller MS, Knol EF, Bruijnzeel-Koomen CAFM. Atopy patch testing—a diagnostic tool? Allergy. 1999;54:784–791. doi: 10.1034/j.1398-9995.1999.00097.x. [DOI] [PubMed] [Google Scholar]
- 43.Wrone-Smith T, et al. Keratinocytes derived from psoriatic plaques are resistant to apoptosis compared with normal skin. Am J Pathol. 1997;151:1321–1329. [PMC free article] [PubMed] [Google Scholar]
- 44.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 45.Watanabe-Fukunaga R, et al. The cDNA structure, expression, and chromosomal assignment of the mouse Fas antigen. J Immunol. 1992;148:1274–1280. [PubMed] [Google Scholar]
- 46.Tagawa YI, Kakuta S, Iwakura Y. Involvement of Fas/Fas ligand system-mediated apoptosis in the development of concanavalin A-induced hepatitis. Eur J Immunol. 1998;28:4105–4113. doi: 10.1002/(SICI)1521-4141(199812)28:12<4105::AID-IMMU4105>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 47.Sayama K, Yonehara S, Watanabe Y, Miki Y. Expression of Fas antigen on keratinocytes in vivo and induction of apoptosis in cultured keratinocytes. J Invest Dermatol. 1994;103:330–334. doi: 10.1111/1523-1747.ep12394858. [DOI] [PubMed] [Google Scholar]
- 48.Matsue H, Kobayashi H, Hosokawa T, Akitaya T, Ohkawara A. Keratinocytes constitutively express the Fas antigen that mediates apoptosis in IFN-γ cultured keratinocytes. Arch Dermatol Res. 1995;287:315–320. doi: 10.1007/BF01105085. [DOI] [PubMed] [Google Scholar]
- 49.Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privileged sites. Science. 1995;270:1189–1191. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- 50.Barker JNWN, Mitra RS, Griffiths CEM, Dixit VM, Nickoloff BJ. Keratinocytes as initiators of inflammation. Lancet. 1991;337:211–214. doi: 10.1016/0140-6736(91)92168-2. [DOI] [PubMed] [Google Scholar]
- 51.Nickoloff BJ. The role of gamma interferon in epidermal trafficking of lymphocytes with emphasis on molecular and cellular adhesion events. Arch Dermatol. 1988;124:1835–1843. [PubMed] [Google Scholar]
- 52.Viard I, et al. Inhibition of toxic epidermal necrolysis by blockade of CD95 with human intravenous immunoglobulin. Science. 1998;282:490–493. doi: 10.1126/science.282.5388.490. [DOI] [PubMed] [Google Scholar]
- 53.Liu C, Walsh CM, Young JDE. Perforin: structure and function. Immunol Today. 1995;16:194–201. doi: 10.1016/0167-5699(95)80121-9. [DOI] [PubMed] [Google Scholar]
- 54.Smyth MJ, Trapani JA. Granzymes: exogeneous proteinases that induce target cell apoptosis. Immunol Today. 1995;16:202–206. doi: 10.1016/0167-5699(95)80122-7. [DOI] [PubMed] [Google Scholar]
- 55.Henkart PA. Lymphocyte-mediated cytotoxicity: two pathways and multiple effector molecules. Immunity. 1994;1:343–350. doi: 10.1016/1074-7613(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 56.Kemeny DM. CD8+ T cells in atopic disease. Curr Opin Immunol. 1998;10:628–633. doi: 10.1016/s0952-7915(98)80080-0. [DOI] [PubMed] [Google Scholar]
- 57.Hamid Q, Boguniewicz M, Leung DY. Differential in situ gene expression in acute versus chronic atopic dermatitis. J Clin Invest. 1994;94:870–876. doi: 10.1172/JCI117408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hamid Q, et al. In vivo expression of IL-12 and IL-13 in atopic dermatitis. J Allergy Clin Immunol. 1996;98:225–231. doi: 10.1016/s0091-6749(96)70246-4. [DOI] [PubMed] [Google Scholar]
- 59.Fujimura T, et al. Conversion of the CD4+ T cell profile from T(H2)-dominant type to T(H1)-dominant type after varicella-zoster virus infection in atopic dermatitis. J Allergy Clin Immunol. 1997;100:274–282. doi: 10.1016/s0091-6749(97)70236-7. [DOI] [PubMed] [Google Scholar]
- 60.Haake AR, Polakowska RR. Cell death by apoptosis in epidermal biology. J Invest Dermatol. 1993;101:107–112. doi: 10.1111/1523-1747.ep12363594. [DOI] [PubMed] [Google Scholar]
- 61.Weedon D, Searle J, Kerr JFR. Apoptosis. Am J Dermatopathol. 1979;1:133–144. [PubMed] [Google Scholar]
- 62.Stark HJ, Baur M, Breitkreuz D, Mirancea N, Fusenig NE. Organotypic keratinocyte cocultures in defined medium with regular epidermal morphogenesis and differentiation. J Invest Dermatol. 1999;112:681–691. doi: 10.1046/j.1523-1747.1999.00573.x. [DOI] [PubMed] [Google Scholar]