

Abstract
Fabry disease is an X-linked lysosomal storage disorder. Female carriers were long thought to be asymptomatic; however, research has revealed the opposite. Cardiac conditions are the chief causes of death in women with Fabry disease. Although ventricular tachycardia has been reported in male patients with Fabry disease, it is not thought to be a frequent finding in females. We describe the case of a 50-year-old woman in whom we used 14-day continuous electrocardiographic monitoring to identify nonsustained ventricular tachycardia, after electrocardiograms and 24-hour Holter monitoring failed to detect the arrhythmia. A permanent implantable cardioverter-defibrillator relieved the patient's symptoms. We discuss why this case supports the need for more extensive electrophysiologic evaluation in women who have Fabry disease.
Keywords: Alpha-galactosidase/blood; arrhythmias, cardiac/etiology; defibrillators, implantable; Fabry disease/complications/epidemiology; female; prevalence; tachycardia, ventricular/diagnosis/etiology/prevention & control; treatment outcome
Fabry disease, an X-linked lysosomal storage disorder, is caused by deficient α-galactosidase activity that leads to the deposition of globotriaosylceramide in cells and tissues. It classically presents with acroparesthesias, angiokeratomas, gastrointestinal symptoms, cataracts, and dyshydrosis during childhood and adolescence. Later progression includes renal, cerebrovascular, and cardiac disease.1 As with other X-linked disorders, it was once thought that females with Fabry disease were mostly asymptomatic and did not need further investigation or treatment; however, cohort studies have revealed that most heterozygous females have clinical manifestations.2 Cardiac disease is the chief cause of early death in these women. Many have palpitations, and 20% have arrhythmias.2,3 The most prevalent arrhythmias are supraventricular tachycardias, atrial fibrillation, and atrial flutter. Although ventricular tachycardia (VT) has occasionally been reported in men with Fabry disease, VT has not been thought to be a typical finding in women. Indeed, we found just one report of a heterozygous woman with nonsustained VT (NSVT) that was detected during 24-hour Holter monitoring.4
We present the case of a woman with Fabry disease who reported presyncope and fatigue and in whom we detected NSVT by unconventional means. In addition, we discuss why this case supports more extensive electrophysiologic evaluation in women with Fabry disease.
Case Report
A 50-year-old woman with Fabry disease presented at our cardiomyopathy clinic for evaluation of episodes of dizziness occurring every other day, palpitations, and progressive fatigue.
The patient had been diagnosed with Fabry disease at age 26 years by means of eye examination after her father had been thus diagnosed. At age 47 years, she had undergone evaluation at our genetics clinic. She had then reported stable angina, dyspnea on exertion, periodic burning sensations in the hands and feet, daily nausea, diarrhea, abdominal pain, chronic pain in all extremities, fatigue, memory loss, depression, and anxiety. She had been symptomatic for many years; her illness was being managed by her primary care physician and nephrologist. She had not yet seen a Fabry specialist or undergone enzyme replacement therapy. Her medical history was otherwise complicated by systemic hypertension, hyperlipidemia, chronic kidney disease with proteinuria, obesity, poorly controlled diabetes mellitus type 2, and transient ischemic attacks.
On physical examination, the patient's blood pressure was 124/62 mmHg, and her heart rate was 80 beats/min. She had angiokeratomas on her lips and abdomen. Her cardiac examination revealed nothing notable.
Mutation analysis for the Fabry disease α-galactosidase gene (Xq22) confirmed her heterozygous status upon the identification of galactosidase alpha mutation 274 G>A (D92N), the same mutation associated with Fabry disease in her family. She started enzyme replacement therapy involving α-galactosidase infusions, adhered to these reasonably well, and visited our genetics clinic every 6 to 12 months.
At that time, she had also been referred to our cardiology clinic. Treadmill exercise testing had revealed sinus rhythm with frequent, normally conducted premature atrial contractions and first-degree atrioventricular block throughout. Cardiac magnetic resonance images (MRI) with gadolinium contrast medium showed mid-myocardial and near-transmural late gadolinium enhancement in the basal and midventricular anterolateral and inferolateral segments. In addition, her left ventricular systolic function was mildly depressed (ejection fraction, 0.50), with mild concentric left ventricular hypertrophy and mild aortic insufficiency.
At the current presentation, she reported a 6-month history of new-onset palpitations, presyncope, and feeling “exhausted, nervous, cold, and tired.” A 24-hour Holter monitor captured predominantly sinus rhythm. Cardiac MRI results were unchanged from those of 3 years earlier, and electrocardiograms (ECGs) revealed only left-axis deviation, nonspecific ST- and T-wave abnormalities, intermittent left bundle branch block, and intermittent ventricular and atrial ectopy.
We decided to use 14-day ECG monitoring to further evaluate the cause of her symptoms. This study captured 4 episodes of NSVT (up to a rate of 170 beats/min and lasting as long as 14 beats at a time), detected on days 2, 3, 4, and 6 (Fig. 1). The patient reported no syncope or near-syncope during these events. Repeat ECG results shortly thereafter remained unchanged from those prior.
Fig. 1.
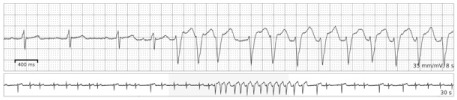
Electrocardiographic tracing shows a 14-beat episode of nonsustained ventricular tachycardia, recorded during 14-day continuous cardiac monitoring.
We were concerned that the near-syncopal episodes were secondary to symptomatic VT. Because of these and the newly documented episodes of NSVT, we thought that the patient was at risk for sudden cardiac arrest, so we placed a dual-chamber ICD for primary prevention.5 Her symptoms improved thereafter, with concurrent medical management involving genetic and cardiologic follow-up evaluation. An ICD interrogation 2 months after placement revealed 3 episodes of nonsustained ventricular arrhythmias with no ICD shocks.
Discussion
To our knowledge, ours is the first report of using extended continuous cardiac monitoring to capture ventricular arrhythmic events in a patient with Fabry disease. Our patient's case underscores the importance of comprehensive cardiac management in Fabry disease and supports the use of newer diagnostic methods to deliver optimal diagnostic care and management.
The estimated prevalence of Fabry disease ranges from 1 in 40,000 to 1 in 117,000 live male births. This enzyme deficit leads to the lysosomal accumulation of glycosphingolypids, especially globotriaosylceramide. The excessive lysosomal storage causes many clinical manifestations, typically skin lesions, neuropathy, stroke, renal failure, and cardiovascular disease.1
Until early in the 21st century, it was thought that Fabry disease, like most X-linked recessive disorders, was mostly asymptomatic in heterozygous females. To the contrary, results of cohort studies such as the Fabry Outcome Survey (of 303 women) have shown that most female carriers report symptoms and that severe clinical manifestations are prevalent.6,7 Among female Fabry disease patients, as many as 77% have neurologic involvement, 50% have cardiac involvement, and 40% have renal involvement.8 Reduction in life expectancy (15 yr) is similar to that in men (20 yr).6,9 These discoveries have engendered a new emphasis on early detection and treatment of women who have this genetic disorder.
Unlike men with Fabry disease, in whom the leading cause of death is renal failure, heterozygous women typically die of cardiac conditions. Chief among these secondary disorders are hypertrophy, left ventricular dysfunction, ischemia, valvular disease, and arrhythmias. The arrhythmias described most often are supraventricular tachycardias, atrial fibrillation, and atrial flutter. However, NSVT has been reported in men after 24-hour Holter monitoring.1,10 In large cohorts, 20% of women and 14% of men have had ventricular arrhythmias.3 Of note are multiple reports of fatal arrhythmias, even arrhythmias resistant to ICD therapy.11
Although cardiac symptoms can be present in Fabry disease, cardiac evaluation is often slight. In a 60-woman cohort, 52% had chest pain and palpitations, but only one third underwent further evaluation.9 In addition, capturing arrhythmic episodes during brief screenings is often difficult because patients might not have symptoms during those windows.
Because of late genetic diagnosis, late initiation of enzyme replacement therapy, and the lingering misconception that women with Fabry disease are mostly asymptomatic, these women might not undergo comprehensive cardiac screening over time.12 We and others support long-term cardiac screening in this population.
We found only one report of a heterozygous woman whose NSVT was detected during 24-hour Holter monitoring.4 Routine ECG and annual Holter monitoring might be reasonable in women with Fabry disease; however, if patients have symptoms that are not captured during these studies, alternative rhythm-monitoring, such as using implantable loop recorders, might viably expand the diagnostic opportunities. Because arrhythmic events were recorded in our patient on days 2, 3, 4, and 6 of screening, we think that 14-day continuous cardiac monitoring can be fruitful in diagnosing infrequent-yet-dangerous arrhythmias in patients with Fabry disease.
References
- 1. Mehta A, Beck M, Linhart A, Sunder-Plassmann, Widmer U.. History of lysosomal storage diseases: an overview. : Mehta A, Beck M, Sunder-Plassmann G, . Fabry disease: perspectives from 5 years of FOS. Oxford (UK): Oxford PharmaGenesis; 2006. Available from: http://www.ncbi.nlm.nih.gov/books/NBK11615/. [PubMed] [Google Scholar]
- 2. Deegan PB, Bähner F, Barba M, Hughes DA, Beck M.. Fabry disease in females: clinical characteristics and effects of enzyme replacement therapy. : Mehta A, Beck M, Sunder-Plassmann G, . Fabry disease: perspectives from 5 years of FOS. Oxford (UK): Oxford PharmaGenesis; 2006. Available from: http://www.ncbi.nlm.nih.gov/books/NBK11591/. [PubMed] [Google Scholar]
- 3. Acharya D, Robertson P, Kay GN, Jackson L, Warnock DG, Plumb VJ, Tallaj JA.. Arrhythmias in Fabry cardiomyopathy. Clin Cardiol 2012; 35( 12): 738– 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Igawa O, Miake J, Hisatome I.. Ventricular tachycardias and dilated cardiomyopathy caused by Fabry disease. Pacing Clin Electrophysiol 2005; 28( 10): 1142– 3. [DOI] [PubMed] [Google Scholar]
- 5. Acharya D, Doppalapudi H, Tallaj JA.. Arrhythmias in Fabry cardiomyopathy. Card Electrophysiol Clin 2015; 7( 2): 283– 91. [DOI] [PubMed] [Google Scholar]
- 6. Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, . et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest 2004; 34( 3): 236– 42. [DOI] [PubMed] [Google Scholar]
- 7. Galanos J, Nicholls K, Grigg L, Kiers L, Crawford A, Becker G.. Clinical features of Fabry's disease in Australian patients. Intern Med J 2002; 32( 12): 575– 84. [DOI] [PubMed] [Google Scholar]
- 8. Sheppard MN. The heart in Fabry's disease. Cardiovasc Pathol 2011; 20( 1): 8– 14. [DOI] [PubMed] [Google Scholar]
- 9. MacDermot KD, Holmes A, Miners AH.. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet 2001; 38( 11): 769– 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shah JS, Hughes DA, Sachdey B, Tome M, Ward D, Lee P, . et al. Prevalence and clinical significance of cardiac arrhythmia in Anderson-Fabry disease. Am J Cardiol 2005; 96( 6): 842– 6. [DOI] [PubMed] [Google Scholar]
- 11. Linhart A. The heart in Fabry disease. : Mehta A, Beck M, Sunder-Plassmann G, . Fabry disease: perspectives from 5 years of FOS. Oxford (UK): Oxford PharmaGenesis; 2006. Available from: http://www.ncbi.nlm.nih.gov/books/NBK11576/. [PubMed] [Google Scholar]
- 12. Wang RY, Lelis A, Mirocha J, Wilcox WR.. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet Med 2007; 9( 1): 34– 45. [DOI] [PubMed] [Google Scholar]