

Paromomycin analogue activity
Novel amphiphilic aminoglycosides are shown to inhibit clinically relevant deactivating enzymes, without undergoing significant deactivation themselves.
Keywords: aminoglycosides, antibacterial agents, antibiotics, APH(3′)-IIIa, drug discovery, paromomycin
Aminoglycoside antibiotics have been in clinical use for the last sixty years. Following the discovery of streptomycin in 1944, many other broad-spectrum aminoglycosides have been discovered (Figure 1).[1,2] Typically, aminoglycosides are classified according to the substitution pattern of the deoxystreptamine unit that forms the core of these antimicrobial compounds. 4,5-Disubstituted deoxystreptamine compounds are comprised in class A aminoglycosides (Figure 1a), whereas 4,6-disubstituted derivatives are in class B (Figure 1b). The isolation of new aminoglycosides declined rapidly in the early seventies, when efforts were diverted to the preparation of semi-synthetic analogues intended to counteract increasing bacterial resistance to these useful drugs.[3] This led to dibekacin (11),[3a] a deoxygenated analogue of kanamycin B (7), and to ar-bekacin (12),[3c] by modifying the N1 group of dibekacin with the 2S-4-amino-2-hydroxybutanoyl moiety originally found in butirosin (2). Although it has been known for decades that aminoglycosides interfere with protein biosynthesis by binding to the prokaryotic ribosome,[4] the lack of precise structural information hampered the identification of beneficial drug modification until the late nineties. A better understanding of the mode of action of aminoglycosides, exemplified by paromomycin (3), was obtained from biochemical[5] and spectroscopic[6] approaches, as well as by mass spectrometry[7] and nuclear magnetic resonance.[8] Definitive confirmation was provided by X-ray structures of the 30S ribosomal subunit bound to amino-glycosides, as well as kinetic studies of protein biosynthesis.[9,10] This long-awaited information led to an increase in structure-based modifications of aminoglycosides, leading to many of semisynthetic analogues from our laboratory and elsewhere.[11,12]
Figure 1.

a) Representative class A aminoglycosides; b) representative class B aminoglycosides.
Aminoglycoside therapy is usually limited to a clinical environment since parenteral injection of these highly hydrophilic drugs is required to obtain the desired plasma concentration in a patient.[2] Their use is also limited by their oto- and nephrotoxicity. Since well-studied dosage strategies are used to maximize their antibiotic potential while minimizing their toxicity,[13] the future of these antibiotics will eventually be compromised by the emergence of bacterial resistance. In order to overcome this threat to human health, the structures of some other classes of antibiotics have also been substantially modified. For example, the β-lactam family has “evolved” remarkably since the first report of penicillin resistance.[1,14] However, clinically effective aminoglycosides have been only minimally modified since their first use.[1,2] Bacteria have developed two general strategies to resist aminoglycosides: 1) diminution of intracellular concentration of the antibiotic, mainly by efflux; and 2) chemical modification of the drug itself or its biological target.[15] Fortunately, bacterial responses influencing aminoglycoside intracellular concentration, as well as the chemical modification of the ribosomal A-site, are still not widespread. However, modification of aminoglycosides by deactivating enzymes is a major threat to the continued clinical efficacy of these antibiotics.[15]
Aminoglycoside deactivating enzymes can be divided into three categories: nucleotidyltransferases (ANTs), acetyltransferases (AACs), and phosphotransferases (APHs).[15] Once adenylated, acetylated or phosphorylated, the affinity of an aminoglycoside for its biological target is drastically attenuated. There are multiple ANTs, AACs and APHs, that can each target different amino or hydroxy groups on the various aminoglycosides. APH(3′)-IIIa mediates the phosphorylation of aminoglycosides at their 3′-OH position by a sequential mechanism where ATP binds first and ADP is the last species to leave the active site.[16] X-ray structures of APH(3′ )-IIIa bound to kanamycin A or neomycin B are available.[17] The Enterococcus faecium enzyme AAC(6)-Ii ′ catalyzes the acetylation of most aminoglycosides at the 6′-N position. This isoform proceeds via an ordered bi bi mechanism, with acetyl coenzyme A (AcCoA) binding first.[18] Crystal structures have been reported for AAC(6′)-Ii in complex with AcCoA,[19] CoA,[20] and some inhibitors.[21e] A number of inhibitors of AAC(6′ )-Ii have been reported.[21]
Based on mechanistic and structural information regarding the mode of action of aminoglycosides as well as the enzymes that deactivate them,[15] aminoglycoside analogues have recently been prepared in an attempt to overcome bacterial resistance.[22] In this regard, we reported the preparation of paromomycin analogues with hydrophobic substituents at the O2″ position.[11a,23] The persistent antimicrobial activities of some of these amphiphilic O2″ analogues compared to the parent paromomycin prompted us to further investigate their properties. Indeed, of paramount interest was the realization that these O2″-ether analogues have a unique and altered mode of binding to the bacterial ribosomal A-site, whereby rings I and II maintain their original position, but rings III and IV assume a 90° twist around the glycosidic bond, as evidenced by X-ray cocrystal structure analysis.[11a,c,23] Herein, we describe our results relating to the in vitro inhibition of two aminoglycoside-deactivating enzymes, APH(3′)-IIIa and AAC(6)-Ii, ′ by O2″-ether analogues of paromomycin.
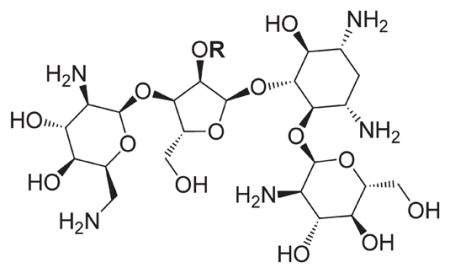
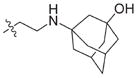
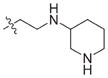
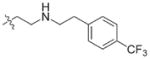
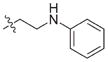
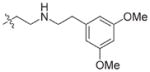
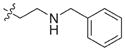
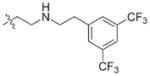
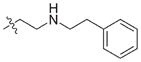
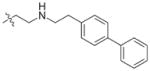
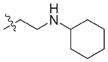
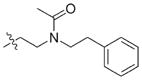
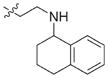
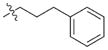
In view of their altered binding mode to the ribosomal A-site, we were interested to see whether the O2″-ether analogues are substrates of APH(3′)-IIIa, a promiscuous aminoglycoside-deactivating enzyme. It is known from the available crystal structures of APH(3′ )-IIIa bound to neomycin that the enzyme interacts with the aminoglycoside in a manner that is different to the ribosomal A-site.[17] We were pleased to observe that the initial phosphorylation rates (v0) of all the O2″-ether analogues tested (compounds 14–38) were significantly slower than that of paromomycin and even undetectable in some cases (compounds 18, 20, 24 and 35; Table 1).
Table 1.
APH(3′)-IIIa-catalyzed phosphorylation rates determined at the same concentration for all paromomycin analogues.
| |||||
|---|---|---|---|---|---|
| Compd | R | v0[a] [nM s−1] | Compd | R | v0[a] [nM s−1] |
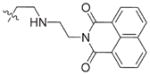
| 3 | H | 54.9 | 26 |
|
7.6 |
| 14 |
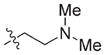
|
9.1 | 27 |
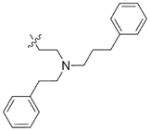
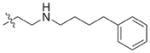
|
8.7 |
| 15 |
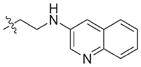
|
12.2 | 28 |
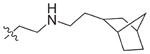
|
9.5 |
| 16 |
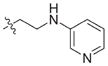
|
8.3 | 29 |
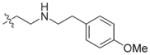
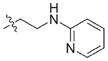
|
11.1 |
| 17 |
|
9.8 | 30 |
|
10.9 |
| 18 |
|
ND[b] | 31 |
|
6.8 |
| 19 |
|
6.4 | 32 |
|
5.6 |
| 20 |
|
ND[b] | 33 |
|
ND[b] |
| 21 |
|
7.0 | 34 |
|
5.4 |
| 22 |
|
8.3 | 35 |
|
ND[b] |
| 23 |
|
6.2 | 36 |
|
6.6 |
| 24 |
|
7.2 | 37 |
|
11.7 |
| 25 |
|
5.4 | 38 |
|
5.2 |
v0 : initial phosphorylation rate;
ND: not detectable.
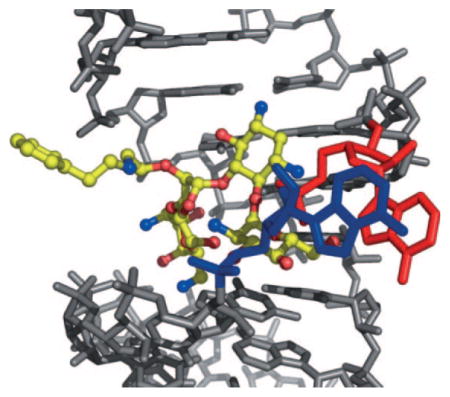
X-ray structural analysis of several O2″-paromomycin analogues revealed that the O2″ substituents are oriented outside of the deep major groove of the ribosomal A-site.[11a,c,23] This observation was also confirmed by the X-ray structure of analogue 20, a key compound in the present study, bound in the A-site of rRNA (Figure 2). The phenylethylamino side chain of compound 20 is outside of the rRNA major groove. A1492 and A1493 are bulged out, as typically observed when an amino-glycoside is bound in the A-site. These observations inspired us to introduce large ether-linked groups at the O2″ position in paromomycin, hoping that these groups could potentially reduce the affinity of the compound for the aminoglycoside-deactivating enzymes. Consistent with our design hypothesis, slower initial phosphorylation rates were observed for analogues 26, 27, 28, 33 and 34, bearing large groups. However, the observation that some analogues with relatively smaller groups, such as 14, 18 and 20, were also phosphorylated significantly more slowly than paromomycin required further investigation.
Figure 2.

X-ray structure of analogue 20 (yellow) bound in the A-site of rRNA (grey; A1492 (blue) and A1493 (red) are highlighted).
To understand why APH(3′)-IIIa was not efficient at catalyzing the phosphorylation of analogues such as 18, 20, 33 and 35, we first verified whether these compounds were indeed bound in the active site of the enzyme by evaluating their ability to inhibit the reaction of APH(3′)III a with its known substrate, amikacin.[24,25] The APH(3′)-IIIa-catalyzed phosphorylation of amikacin was significantly inhibited by low concentrations of the O2″-paromomycin analogues 18 and 20 (Figure 3). From the linear regression (see Figure S1 in Supporting Information), the Ki values determined for compounds 18 and 20 were 3.9 μM and 1.0 μM, respectively (Table 2). The mode of inhibition was shown to be predominantly competitive (Ki ≪Ki′) with respect to amikacin. Furthermore, Km(app) was found to increase with increasing inhibitor concentration, whereas Vmax (app) was relatively insensitive. The observed competitive inhibition suggests that compounds 18 and 20 are bound in the active site of APH(3′)-IIIa.
Figure 3.

Inhibition of amikacin phosphorylation rates (v) by a) analogue 18 (■=0 μM, ●=1 μM, ▲=2.5 μM, ▼=5 μM, ◆ =10 μM, ◀ =15 μM) and b) analogue 20 (■=0 μM, ●=1 μM, ▲=5 μM, ▼=10 μM, ◆ =15 μM).
Table 2.
Ki and Ki′ values of compounds 18 and 20 for inhibition of APH(3′)-IIIa.
| Compd | Ki [μM] | Ki′ [μM] |
|---|---|---|
| 18 | 3.9 | 69.4 |
| 20 | 1.0 | 14.2 |
IC50 values were then determined for a variety of O2″ paromomycin analogues as inhibitors of APH(3′ )-IIIa (Table 3). Considering the steric, electronic and hydrophobic variability of the analogues studied, the similarity of the IC50 values indicates that the O2″-ether groups have a relatively small effect on the inhibitory potency of these paromomycin analogues, suggesting they do not contribute greatly to active site affinity, relative to the aminoglycoside portion.
Table 3.
Inhibition (IC50) of APH (3′)-IIIa by paromomycin analogues functionalized at the O2″ position.[a]
| Compd | IC50 [μM] | Compd | IC50 [μM] |
|---|---|---|---|
| 14 | 17.2 ± 2.3 | 27 | 30.3 ± 5.6 |
| 15 | 30.6 ± 5.7 | 28 | 37.1 ± 3.5 |
| 16 | 2.0 ± 0.2 | 29 | 6.0 ± 0.7 |
| 17 | 4.3 ± 0.5 | 30 | 2.3 ± 0.3 |
| 18 | 12.3 ± 1.7 | 31 | 12.4 ± 1.9 |
| 19 | 16.1 ± 2.5 | 32 | 21.1 ± 1.6 |
| 20 | 9.5 ± 0.7 | 33 | 2.8 ± 0.3 |
| 21 | 18.5 ± 1.8 | 34 | 39.2 ± 7.3 |
| 22 | 24.2 ± 4.5 | 35 | 5.5 ± 0.1 |
| 23 | 11.7 ± 0.1 | 36 | 4.3 ± 0.5 |
| 24 | 3.1 ± 0.4 | 37 | 12.7 ± 1.6 |
| 25 | 16.0 ± 2.0 | 38 | 2.2 ± 0.3 |
| 26 | 5.0 ± 0.7 |
Values represent the mean ± SD of n =3 experiments.
In view of our observation that the APH(3′)-IIIa-catalyz ed phosphorylation of compounds 18, 20, 33 and 35 was experimentally undetectable, we suspected that the O2″-side chain of the inhibitors may obstruct binding of the ATP co-factor to APH(3′)-IIIa. The mode of inhibition of compound 20 with respect to ATP was therefore determined (Figure 4). At low inhibitor concentrations of compound 20 (<5 μM), a Ki value of 2.7 μM and a Ki′ of 0.8 μM were obtained from the linear regression (see Figures S2 and S3 in Supporting Information). Also at low inhibitor concentration, inhibition was observed to be mixed uncompetitive with respect to ATP, indicating that 20 cannot be bound in the ATP site before the binding of ATP. At higher concentrations, the inhibitor might also be bound to the E·ADP complex (Scheme 1), as observed with other APH(3′)-IIIa substrates.[16] According to this model, compounds 18 and 20 are in competition with the aminoglycoside substrate for each form of the enzyme known to bind an aminoglycoside.[16] Inhibition could be due to an altered orientation of the pseudotetrasaccharide harboring the O2″-ether chain in the active site, which causes a 90° twist of the glycosidic bond linking rings III–IV to I–II. Several attempts to obtain X-ray quality crystals of 20 with APH(3′)-IIIa were unsuccessful.
Figure 4.

Inhibition of APH(3′)-IIIa-catalyzed amikacin phosphorylation by analogue 20 as a function of ATP concentration. v=phosphorylation rates, ◆ =0 μM, × =1 μM, ▲=2.5 μM, ●=5 μM, ■=10 μM.
Scheme 1.

Proposed mechanism for the inhibition of APH(3′)-IIIa by compounds 18 and 20. E =APH(3′)-IIIa, S =aminoglycoside substrate of APH(3′)-IIIa, I =inhibitor, P =aminoglycoside product after modification by APH(3′)-IIIa.
Compound 20 was also found to be an inhibitor of AAC(6′)-Ii. A Ki value of 8 ± 3 μM was determined using a known protocol.[21e] This result was expected since AAC(6′)-Ii is known not to modify aminoglycosides lacking a 6′-amino group.[18] Moreover, the Ki value of compound 20 (8 μM) is within error of the reported Km for neomycin C (5 μM),[18] which is the 6′-NH2 analogue of paromomycin. This suggests that the O2″-substituent of compound 20 on its own is not contributing significantly to the inhibition observed.
The promising results in reducing the rate of in vitro O3′ phosphorylation in amikacin in the presence of 20 prompted us to test the antimicrobial activity of compound 20 against a variety of resistant bacterial strains expressing efflux pumps and aminoglycoside-deactivating enzymes. Compound 20 was not effective against most of these strains (Table 4), much like paromomycin and neomycin B. These disappointing results, albeit against some of the most recalcitrant strains, are probably due to the existence of different isoforms of APH(3′)-IIIa and AAC(6′)-Ii and also efflux. It is also possible that these strains express aminoglycoside-deactivating enzymes other than APH(3′)-III and AAC(6′ )-I that can deactivate compound 20. Nevertheless, it is of interest that amphiphilic analogues of paromomycin provided insights into a new mode of binding to the ribosomal A-site while maintaining activity against sensitive strains of Staphylococcus aureus, Escherichia coli, Klebsiella pneumoniae and Acinetobacter baumannii. No synergistic effect was observed for varying concentrations (up to 64 μg mL−1) of compound 20 in the presence of amikacin and paromomycin.
Table 4.
Antibacterial activities of compound 20 and other aminoglycosides (3, 4, 9, 10 and 13) against a variety of different bacterial strains.
| Species | Phenotype | 20 | 3[a] | 4[a] | 9 | 10 | 13 |
|---|---|---|---|---|---|---|---|
| MIC [μg mL−1] | |||||||
| Staphylococcus aureus | ATCC 29213 | 0.5 | 2 | 0.5 | 0.5 | 0.5 | 4 |
| Escherichia coli | ATCC 25922 | 4 | 2 | 1 | 0.5 | 1 | 2 |
| Klebsiella pneumoniae | ATCC 10031 | 2 | 2 | 0.25 | 0.25 | 0.5 | 0.25 |
| Pseudomonas aeruginosa | ATCC 27853 | >64 | >64 | 64 | 0.5 | 0.5 | 2 |
| P. aeruginosa | Wild-type pump | >64 | >64 | 8 | 1 | 0.5 | 2 |
| Acinetobacter baumannii | Susceptible | 2 | 2 | 1 | 0.5 | 0.5 | 2 |
| P. aeruginosa | MexXY up | >64 | >64 | 16 | 4 | 0.5 | 4 |
| A. baumannii | ATCC 19606 | >64 | 16 | 4 | 8 | 4 | 16 |
| P. aeruginosa | Efflux | >64 | >64 | > 64 | 4 | 2 | 16 |
| P. aeruginosa | Efflux | >64 | >64 | > 64 | 8 | 2 | 32 |
| Providencia stuartii | AAC(2′)-I | >64 | >64 | 32 | 64 | 32 | 4 |
| E. coli | APH(3′)-Ib | >64 | >64 | > 64 | 0.25 | 1 | 0.5 |
| S. aureus | APH(3′)-III | >64 | >64 | > 64 | 0.5 | 1 | 8 |
| A. baumannii | ANT(2″)-I, APH(3′)-VI, Perm/Efflux | >64 | >64 | > 64 | >64 | >64 | > 64 |
| Acinetobacter spp. | APH(3′)-VI | >64 | >64 | 16 | 0.5 | 1 | > 64 |
| S. aureus | ANT(4′)-I | >64 | >64 | > 64 | 0.5 | >64 | > 64 |
| P. aeruginosa | ANT(4′)-II | >64 | >64 | 8 | 1 | 64 | 64 |
| Serratia marcescens | AAC(6′)-I | 8 | 4 | 4 | 2 | 64 | 64 |
| P. aeruginosa | AAC(6′)-I | >64 | >64 | > 64 | 64 | >64 | > 64 |
| P. aeruginosa | AAC(6′)-II | >64 | >64 | 8 | 64 | 32 | 4 |
| E. coli | ANT(2″)-I | 8 | 8 | 2 | >64 | >64 | 4 |
| Enterobacter cloacae | AAC(6′)-I, ANT(2″)-I, APH(3′)-I | >64 | >64 | > 64 | >64 | >64 | 64 |
| S. aureus | AAC(6′)-I, APH(2″) | >64 | >64 | > 64 | >64 | >64 | > 64 |
| P. aeruginosa | AAC(3)-I | >64 | >64 | 8 | 64 | 0.5 | 4 |
| E. coli | AAC(3)-IV | 16 | 16 | 8 | 32 | >64 | 2 |
Paromomycin (3); neomycin B (4).
Amphiphilic O2″-ether analogues of paromomycin such as compound 20, demonstrating a new mode of binding to the ribosomal A-site and showing inhibitory activity similar to the parent aminoglycoside, were used as a model to understand the impact of the O2″-modification on two aminoglycoside-deactivating enzymes, APH(3)-IIIa ′ and AAC(6′)-Ii. Compound 20 was not a substrate for these clinically relevant enzymes, but was capable of inhibiting both with Ki values in the low micromolar range. Competitive inhibition was observed for APH(3′)- IIIa relative to the natural substrate amikacin, and uncompetitive inhibition was observed for APH(3′ )-IIIa with respect to ATP at low inhibitor concentration. The IC50 values determined for a series of paromomycin O2″ analogues indicate that the ether moiety introduced at the O2″ position is not a determining factor for APH(3′)-IIIa affinity. Further work will involve incorporation of deoxygenation and N1-substitution with the 2S-4-amino-2-hydroxybutanoyl moiety, known to impart activity against resistant strains as in the case of amikacin.[3b]
Experimental Section
Experimental details are given in the Supporting Information.
Supplementary Material
Acknowledgments
We thank the Natural Sciences and Engineering Research Council of Canada (NSERC), the Fonds Québécois de la Recherche sur la Nature et les Technologies (FQRNT), and the Fonds de Recherche en Santé du Québec (FRSQ) for fellowships, and Achaogen Inc. (San Francisco, USA) for financial assistance and antibacterial testing.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cmdc.201100346.
References
- 1.Walsh C. Antibiotics: Actions, Origins, Resistance. Chapters 1–4 ASM Press; Washington DC: 2003. [Google Scholar]
- 2.Bryskier A, editor. Antimicrobial Agents: Antibacterials and Antifungals. Chapters 2 and 16 ASM Press; Washington DC: 2005. [Google Scholar]
- 3.For example see: Umezawa H, Umezawa S, Tsuchiya T, Okazaki Y. J Antibiot. 1971;24:485–487. doi: 10.7164/antibiotics.24.485.Kawaguchi H, Naito T, Nakagawa S, Fujisawa K. J Antibiot. 1972;25:695–708. doi: 10.7164/antibiotics.25.695.Kondo S, Linuma K, Yamamoto H, Ikeda Y, Maeda K, Umezawa H. J Antibiot. 1973;26:705–707. doi: 10.7164/antibiotics.26.705.Hanessian S, Takamoto T, Masse R, Patil G. Can J Chem. 1978;56:1482–1491.
- 4.Moazed D, Noller HF. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- 5.For example see: Recht MI, Fourmy D, Blanchard KD, Dahlquist D, Puglisi JD. J Mol Biol. 1996;262:421–436. doi: 10.1006/jmbi.1996.0526.
- 6.Fourmy D, Recht MI, Blanchard SC, Puglisi JD. Science. 1996;274:1367–1371. doi: 10.1126/science.274.5291.1367. [DOI] [PubMed] [Google Scholar]
- 7.a) Hofstadler SA, Griffey RH. Curr Opin Drug Discovery Dev. 2000;3:423–431. [PubMed] [Google Scholar]; b) Griffey RH, Hofstadler SA, Sannes-Lowery KA, Ecker DJ, Crooke ST. Proc Natl Acad Sci USA. 1999;96:10129–10133. doi: 10.1073/pnas.96.18.10129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Fourmy D, Yoshizawa S, Puglisi JD. J Mol Biol. 1998;277:333–345. doi: 10.1006/jmbi.1997.1551. [DOI] [PubMed] [Google Scholar]; b) Fourmy D, Recht MI, Puglisi JD. J Mol Biol. 1998;277:347–362. doi: 10.1006/jmbi.1997.1552. [DOI] [PubMed] [Google Scholar]; c) Lynch SR, Gonzalez RL, Jr, Puglisi JD. Structure. 2003;11:43–53. doi: 10.1016/s0969-2126(02)00934-6. [DOI] [PubMed] [Google Scholar]
- 9.Carter AP, Clemons WM, Bordersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Nature. 2000;407:340–348. doi: 10.1038/35030019.Also see: Vicens Q, Westhof E. Structure. 2001;9:647–658. doi: 10.1016/s0969-2126(01)00629-3.
- 10.a) Pape T, Wintermeyer W, Rodnina MV. EMBO J. 1998;17:7490–7497. doi: 10.1093/emboj/17.24.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pape T, Wintermeyer W, Rodnina MV. EMBO J. 1999;18:3800–3807. doi: 10.1093/emboj/18.13.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Pape T, Wintermeyer W, Rodnina MV. Nat Struct Biol. 2000;7:104–107. doi: 10.1038/72364. [DOI] [PubMed] [Google Scholar]
- 11.a) Hanessian S, Szychowski J, Adhikari SS, Vasquez G, Pachamuthu K, Swayze EE, Migawa MT, Ranken R, François B, Wirmer-Bartoschek J, Kondo J, Westhof E. J Med Chem. 2007;50:2352–2369. doi: 10.1021/jm061200+. [DOI] [PubMed] [Google Scholar]; b) Hanessian S, Adhikari SS, Szychowski J, Pachamuthu K, Wang X, Migawa MT, Griffey RH, Swayze EE. Tetrahedron. 2007;63:827–846. [Google Scholar]; c) Kondo J, Pachamuthu K, François B, Szychowski J, Hanessian S, Westhof E. Chem-MedChem. 2007;2:1631–1638. doi: 10.1002/cmdc.200700113. [DOI] [PubMed] [Google Scholar]; d) Moitessier N, Westhof E, Hanessian S. J Med Chem. 2006;49:1023–1033. doi: 10.1021/jm0508437. [DOI] [PubMed] [Google Scholar]; e) François B, Szychowski J, Adhikari SS, Pachamuthu K, Swayze EE, Griffey RH, Migawa MT, Westhof E, Hanessian S. Angew Chem. 2004;116:6903–6906. doi: 10.1002/anie.200462092. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:6735–6738. doi: 10.1002/anie.200462092. [DOI] [PubMed] [Google Scholar]; f) Hanessian S, Tremblay M, Kornienko A, Moitessier N. Tetrahedron. 2001;57:3255–3265. [Google Scholar]
- 12.a) Kudyba I, Fernandez DP, Boettgerb EC, Vasella A. Carbohydr Res. 2007;342:499–519. doi: 10.1016/j.carres.2006.09.014. [DOI] [PubMed] [Google Scholar]; b) Blount KF, Tor Y. ChemBioChem. 2006;7:1612–1621. doi: 10.1002/cbic.200600109. [DOI] [PubMed] [Google Scholar]; c) Ding D, Hofstadler SA, Swayze EE, Risen L, Griffey RH. Angew Chem. 2003;115:3531–3534. doi: 10.1002/anie.200351354. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2003;42:3409–3412. doi: 10.1002/anie.200351354. [DOI] [PubMed] [Google Scholar]; d) Wang J, Li J, Tuttle D, Takemoto JY, Chang CT. Org Lett. 2002;4:3997–4000. doi: 10.1021/ol026588r. [DOI] [PubMed] [Google Scholar]; e) Greenberg WA, Priestley ES, Sears PS, Alper PB, Rosenbohm C, Hendrix M, Hung S, Wong C. J Am Chem Soc. 1999;121:6527–6541. [Google Scholar]; f) Alper PB, Hendrix M, Sears P, Wong C. J Am Chem Soc. 1998;120:1965–1978. [Google Scholar]
- 13.a) Rougier F, Ducher M, Maurin M, Corvaisier S, Claude D, Jelliffe R, Maire P. Clinic Pharmacokinetics. 2003;42:493–500. doi: 10.2165/00003088-200342050-00007. [DOI] [PubMed] [Google Scholar]; b) Bartal C, Danon A, Schlaeffer F, Reisenberg K, Alkan M, Smoliakov R, Sidi A, Almog Y. Am J Med. 2003;114:194–198. doi: 10.1016/s0002-9343(02)01476-6. [DOI] [PubMed] [Google Scholar]
- 14.Fisher JF, Meroueh SO, Mobashery S. Chem Rev. 2005;105:395–424. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 15.Magnet S, Blanchard JS. Chem Rev. 2005;105:477–497. doi: 10.1021/cr0301088. [DOI] [PubMed] [Google Scholar]
- 16.a) Hon WC, McKay GA, Thompson PR, Sweet RM, Yang DSC, Wright GD, Berghuis AM. Cell. 1997;89:887–895. doi: 10.1016/s0092-8674(00)80274-3. [DOI] [PubMed] [Google Scholar]; b) McKay GA, Wright GD. J Biol Chem. 1995;270:24 686–24 692. doi: 10.1074/jbc.270.42.24686. [DOI] [PubMed] [Google Scholar]; c) McKay GA, Wright GD. Biochemistry. 1996;35:8680–8685. doi: 10.1021/bi9603884. [DOI] [PubMed] [Google Scholar]; d) Burk DL, Hon WC, Leung AK, Berghuis AM. Biochemistry. 2001;40:8756–8764. doi: 10.1021/bi010504p. [DOI] [PubMed] [Google Scholar]; e) Thompson PR, Boehr DD, Berghuis AM, Wright GD. Biochemistry. 2002;41:7001–7007. doi: 10.1021/bi0256680. [DOI] [PubMed] [Google Scholar]
- 17.Fong DH, Berghuis AM. EMBO J. 2002;21:2323–2331. doi: 10.1093/emboj/21.10.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Wright GD, Ladak P. Antimicrob Agents Chemother. 1997;41:956–960. doi: 10.1128/aac.41.5.956. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Draker KA, Wright GD. Biochemistry. 2004;43:446–454. doi: 10.1021/bi035667n. [DOI] [PubMed] [Google Scholar]
- 19.Wybenga-Groot LE, Draker K-A, Wright GD, Berghuis AM. Structure. 1999;7:497–507. doi: 10.1016/s0969-2126(99)80066-5. [DOI] [PubMed] [Google Scholar]
- 20.Burk DL, Ghuman N, Wybenga-Groot LE, Berghuis AM. Prot Sci. 2003;12:426–437. doi: 10.1110/ps.0233503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a) Freiburger LA, Baettig OM, Berghuis AM, Auclair K, Mittermaier AK. Nat Struct Mol Biol. 2011;18:288–294. doi: 10.1038/nsmb.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gao F, Yan X, Vong K, Auclair K. Chem Eur J. 2009;15:2064–2070. doi: 10.1002/chem.200802172. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gao F, Yan X, Zahr O, Larsen A, Vong K, Auclair K. Bioorg Med Chem Lett. 2008;18:5518–5522. doi: 10.1016/j.bmcl.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gao F, Yan X, Shakya T, Baettig OM, Ait-Mohand-Brunet S, Berghuis AM, Wright GD, Auclair K. J Med Chem. 2006;49:5273–5281. doi: 10.1021/jm060732n. [DOI] [PubMed] [Google Scholar]; e) Gao F, Yan X, Baettig OM, BerghuisK AM. Auclair, Angew Chem. 2005;117:7019–7022. doi: 10.1002/anie.200501399. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:6859–6862. doi: 10.1002/anie.200501399. [DOI] [PubMed] [Google Scholar]
- 22.a) Bera S, Zhanel GG, Schweizer F. J Med Chem. 2008;51:6160–6164. doi: 10.1021/jm800345u. [DOI] [PubMed] [Google Scholar]; b) Bera S, Zhanel GG, Schweizer F. J Med Chem. 2010;53:3626–3631. doi: 10.1021/jm1000437. [DOI] [PubMed] [Google Scholar]; c) Bastida A, Hidalgo A, Chiara JL, Torrado M, Corzana F, Pérez-Canadillas JM, Groves P, Garcia-Junceda E, Gonzalez C, Jimenez-Barbero J, Asensio JL. J Am Chem Soc. 2006;128:100–116. doi: 10.1021/ja0543144. [DOI] [PubMed] [Google Scholar]; d) Blount KF, Zhao F, Hermann T, Tor Y. J Am Chem Soc. 2005;127:9818–9829. doi: 10.1021/ja050918w. [DOI] [PubMed] [Google Scholar]; e) Haddad J, Vakulenko S, Mobashery S. J Am Chem Soc. 1999;121:11922–11923. [Google Scholar]; f) Roestamadji J, Grapsas I, Mobashery S. J Am Chem Soc. 1995;117:80–84. [Google Scholar]; g) Hanessian S, Szychowski J, Campos-Reales Pineda NB, Furtos A, Keillor JW. Bioorg Med Chem Lett. 2007;17:3221–3225. doi: 10.1016/j.bmcl.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 23.Hanessian S, Pachamuthu K, Szychowski J, Giguère A, Swayze EE, Migawa MT, François B, Kondo J, Westhof E. Bioorg Med Chem Lett. 2010;20:7097–7101. doi: 10.1016/j.bmcl.2010.09.084. [DOI] [PubMed] [Google Scholar]
- 24.McKay GA, Thompson PR, Wright GD. Biochemistry. 1994;33:6936–6944. doi: 10.1021/bi00188a024. [DOI] [PubMed] [Google Scholar]
- 25.For amikacin, the Km value of 245 μM reported by McKay et al. in Reference [24] (we observed Km = 150 μM) is higher than for other commercially available aminoglycosides and facilitates the detection of the phosphorylation at the concentrations used in the assay. Also, amikacin is only singly phosphorylated whereas aminoglycosides, such as paromomycin, are phosphorylated twice, and their biphasic phosphorylation curves make analysis more complicated.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.