

Eisenmenger syndrome (ES) is a complex and disastrous medical problem with profound cyanosis and clinical deterioration by significant right to left shunting. This syndrome is the most advanced form of pulmonary arterial hypertension (PAH) associated with congenital heart disease (PAH-CHD). Inverted shunt changes structures of pulmonary vasculature like all forms of PAH in ES (1). In the past, the management of patients with ES was limited to conventional therapy with an emphasis on regular informed cardiovascular follow-up. Subsequent clinical studies have made it possible to improve to patient survival and functional capacity. There are major therapeutic targets in PAH treatment using endothelin-receptor antagonists, phosphodiesterase type-5 inhibitors, and prostacyclin derivatives.
In the REVEAL registry, patient’s survival of advanced PAH remains poor despite advanced targeted therapy. Survival rates in newly diagnosed PAH patients after the development of PAH-specific therapies were 86.3% and 61.2% at 1 and 5 years, respectively (2). This result is still disappointing although PAH-CHD shows better outcomes compare to other etiologies of PAH. Nevertheless, many studies suggest advanced PAH therapies should be needed to improve the survival of PAH patients. The BREATHE-5 trial, first placebo-controlled trial in patients with ES, demonstrated a significant improvement of hemodynamics and exercise capacity without adversely affecting systemic arterial oxygen saturation on bosentan-treated patients (3). Other recent randomized controlled trials in ES with phosphodiesterase type-5 inhibitors have shown improvements in exercise capacity and hemodynamics (4,5). In common with other world registries, our group also reports the importance of specialized therapies to improve the survival of PAH patients in Korean Registry of pulmonary arterial hypertension (KORPAH) (Table 1) (6). The demographics of the KORPAH registry participants were similar to those of western registry including 159 patients of PAH-CHD (26.9%) of all patients (n=625) (Figure 1) (6).
Table 1. PAH-specific medications of KORPAH in all patients and incident patients.
| Medications | Treatments in all patients (n=625) (%) | Treatments in incidence cases (n=297) (%) |
|---|---|---|
| No. of patients receiving PAH-specific medications in all treatments | 380 (60.8) | 182 (61.3) |
| No. of medications in all PAH-specific treatments | 155 (40.9) | 93 (51.1) |
| Single sildenafil | 45 (11.8) | 21 (11.6) |
| Single inhaled iloprost | 22 (5.8) | 17 (9.3) |
| Single beraprost | 86 (22.6) | 23 (12.6) |
| Combinations of above single medications | 72 (18.9) | 28 (15.4) |
Figure 1.
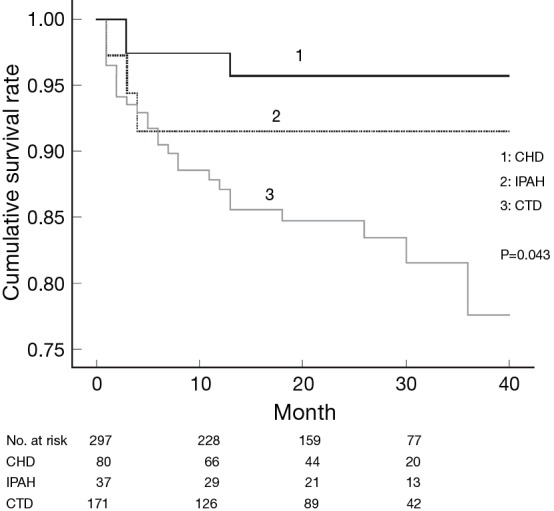
Comparison of survival according to the etiologies of PAH of the incident cases in the KORPAH (n=297). This figure presents a comparison of prognoses according to the etiologies of PAH. PAH with CTD corresponded to the highest mortality (18.8%), followed by idiopathic PAH (IPAH) (8.1%) and PAH with congenital heart disease CHD (3.9%) (P=0.043). CHD, congenital heart disease; CTD, connective tissue disease. [Reproduced by permission of the Korean Academy of Medical Sciences (6)].
The World Health Organization (WHO) functional classification at the time of diagnosis has major implications as a clinical predictor of mortality and survival for patients with PAH (7). We reported the same results already in the retrospective cohort (Figure 2) (8). The EARLY study, randomized controlled trial of a PAH-targeted therapy in WHO functional class (FC) II patients, suggested that patients with WHO FC II PAH have a severe and often fatally progressive disease (9). These findings demonstrated the importance of the advanced targeted therapy in a timely manner.
Figure 2.
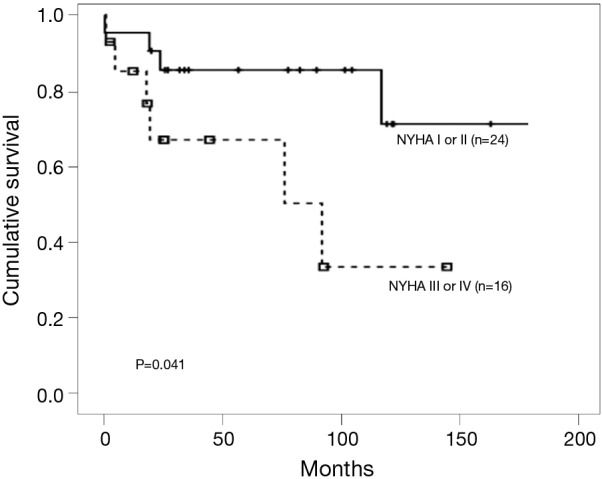
Median overall survival time of patients by NYHA functional classification. Patients with NYHA class I or II at the time of diagnosis showed significantly better survival than those with more severe functional class. [Reprinted with permission (8)].
Several studies have reported beneficial effects of inhaled iloprost, resulting in improved WHO functional class, hemodynamics, an increased 6-minute walking distance, and quality of life (10,11). A retrospective study of adult patients with PAH to CHD in Korea also suggested that inhaled iloprost as a perioperative medical intervention for patients with PAH-CHD is safe and effective in improving the systemic oxygen saturation and for early recovery in the postoperative course (Table 2) (12). Also, a prospective single-arm study which 18 patients with ES and exertional dyspnea according to WHO functional class III or IV were prospectively recruited had showed that 24 weeks of inhaled iloprost therapy in patients with ES led to significant improvements in exercise capacity, quality of life, and right ventricular function. These results likely explain the symptomatic relief reported by patients with ES receiving iloprost therapy (13).
Table 2. Clinical course of the iloprost group vs. the control group.
| Medications | Iloprost group (n=28) | Control group (n=17) | P value |
|---|---|---|---|
| Mortality | |||
| Use of iNOa (%) | 4 (17.9) | 8 (52.9) | 0.021 |
| Use of iNOa (ppm) | 24 vs. 8 | 31 vs. 7 | 0.064 |
| Use of iNOa (h) | 11.2 vs. 4.5 | 25 vs. 6.8 | 0.031 |
| Plasma BNPb (pg/mL) | 98 vs. 46 | 265 vs. 92 | 0.008 |
| Mechanical ventilation time (h) | 10.1 vs. 12.5 | 41.1 vs. 46.1 | 0.018 |
| ICU stay (h) | 39.4 vs. 26.4 | 90.3 vs. 60.8 | 0.005 |
| Chest tube use (h) | 63.9 vs. 22.7 | 89.3 vs. 42.8 | 0.039 |
| Inotropic support (h) | 103.8 vs. 88.2 | 74.5 vs. 56.0 | 0.246 |
| Drug used (ìg/kg/h) | |||
| Milrinone | 0.375–0.5 | 0.375–0.5 | |
| Dopamine | 5–10 | 5–10 |
a, administered via an endotracheal tube before weaning when clinically necessary for the immediate postoperative period; b, Checked on the 7th postoperative day.
The recent study of the German National Resister for congenital heart defects (GNR-CHD) contains a nation-wide data with a large population of ES patients in the community (14). Among 153 patients with ES, 57.5% of patients were treated with PAH-specific medical therapies. Of those, 17.6% of patients received combination therapy; 76.1% of patients on monotherapy were on bosentan and 44.4% of patients treated primarily with Sildenafil were also on this drug as a second line. The GNR-CHD is a well-designed study and recruited a large number of patients representing the community-based population. The result of this study may be valuable data on advanced targeted therapy. However, as the intrinsic drawback of registry, the lack of uniform treatment strategy couldn’t explain effective and timely treatments in PAH-targeted therapy. This limitation was shown as no outcome difference between monotherapy and dual targeted therapy because of the long escalation time. Nevertheless, the strongest message from GNR-CHD is the better clinical outcome in the large volume centers than remaining centers that means the need of expert care from centers of excellence.
In summary, the GNR-CHD demonstrated better survival of advanced targeted therapy based on the real world as well as tertiary referrals in the Germany. This modern real world registry data reinforce the reason why specialized medical therapies in PAH expert center should be considered in ES patients.
Acknowledgements
Funding: This research was partly supported by the Gachon University Gil Medical Center (Grant number: 2015-02) and the Next-generation Medical Device Development Program for Newly-Created Market of the National Research Foundation (NRF) funded by the Korean government, MSIP (No. 2015M3D5A1066043).
Footnotes
Provenance: This is an invited Editorial commissioned by the Section Editor Haiyun Yuan (Department of Cardiovascular Surgery, Guangdong Provincial Cardiovascular Institute, Guangdong General Hospital, Guangzhou, China).
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Pietra GG, Capron F, Stewart S, et al. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol 2004;43:25S-32S. 10.1016/j.jacc.2004.02.033 [DOI] [PubMed] [Google Scholar]
- 2.Farber HW, Miller DP, Poms AD, et al. Five-year outcomes of patients enrolled in the REVEAL Registry. Chest 2015;148:1043-54. 10.1378/chest.15-0300 [DOI] [PubMed] [Google Scholar]
- 3.Galiè N, Beghetti M, Gatzoulis MA, et al. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study. Circulation 2006;114:48-54. 10.1161/CIRCULATIONAHA.106.630715 [DOI] [PubMed] [Google Scholar]
- 4.Mukhopadhyay S, Nathani S, Yusuf J, et al. Clinical efficacy of phosphodiesterase-5 inhibitor tadalafil in Eisenmenger syndrome--a randomized, placebo-controlled, double-blind crossover study. Congenit Heart Dis 2011;6:424-31. 10.1111/j.1747-0803.2011.00561.x [DOI] [PubMed] [Google Scholar]
- 5.D'Alto M, Romeo E, Argiento P, et al. Bosentan-sildenafil association in patients with congenital heart disease-related pulmonary arterial hypertension and Eisenmenger physiology. Int J Cardiol 2012;155:378-82. 10.1016/j.ijcard.2010.10.051 [DOI] [PubMed] [Google Scholar]
- 6.Chung WJ, Park YB, Jeon CH, et al. Baseline characteristics of the Korean Registry of Pulmonary Arterial Hypertension. J Korean Med Sci 2015;30:1429-38. 10.3346/jkms.2015.30.10.1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sitbon O, Galiè N. Treat-to-target strategies in pulmonary arterial hypertension: the importance of using multiple goals. Eur Respir Rev 2010;19:272-8. 10.1183/09059180.00008210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park YM, Chung WJ, Choi DY, et al. Functional class and targeted therapy are related to the survival in patients with pulmonary arterial hypertension. Yonsei Med J 2014;55:1526-32. 10.3349/ymj.2014.55.6.1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simonneau G, Galiè N, Jansa P, et al. Long-term results from the EARLY study of bosentan in WHO functional class II pulmonary arterial hypertension patients. Int J Cardiol 2014;172:332-9. 10.1016/j.ijcard.2013.12.179 [DOI] [PubMed] [Google Scholar]
- 10.Ivy DD, Doran AK, Smith KJ, et al. Short- and long-term effects of inhaled iloprost therapy in children with pulmonary arterial hypertension. J Am Coll Cardiol 2008;51:161-9. 10.1016/j.jacc.2007.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Opitz CF, Wensel R, Winkler J, et al. Clinical efficacy and survival with first-line inhaled iloprost therapy in patients with idiopathic pulmonary arterial hypertension. Eur Heart J 2005;26:1895-902. 10.1093/eurheartj/ehi283 [DOI] [PubMed] [Google Scholar]
- 12.Sung KW, Jeon YB, Kim NY, et al. The effects of perioperative inhaled iloprost on pulmonary hypertension with congenital heart disease. Cardiology 2013;126:224-9. 10.1159/000354248 [DOI] [PubMed] [Google Scholar]
- 13.Cha KS, Cho KI, Seo JS, et al. Effects of inhaled iloprost on exercise capacity, quality of life, and cardiac function in patients with pulmonary arterial hypertension secondary to congenital heart disease (the Eisenmenger syndrome) (from the EIGER Study). Am J Cardiol 2013;112:1834-9. 10.1016/j.amjcard.2013.08.003 [DOI] [PubMed] [Google Scholar]
- 14.Diller GP, Körten MA, Bauer UM, et al. Current therapy and outcome of Eisenmenger syndrome: data of the German National Register for congenital heart defects. Eur Heart J 2016;37:1449-55. 10.1093/eurheartj/ehv743 [DOI] [PMC free article] [PubMed] [Google Scholar]