

Summary
Human cancers are characterized by the presence of oncogene-induced DNA replication stress (DRS), making them dependent on repair pathways such as break-induced replication (BIR) for damaged DNA replication forks. To better understand BIR, we performed a targeted siRNA screen for genes whose depletion inhibited G1 to S phase progression when oncogenic cyclin E was overexpressed. RAD52, a gene dispensable for normal development in mice, was among the top hits. In cells in which fork collapse was induced by oncogenes or chemicals, the Rad52 protein localized to DRS foci. Depletion of Rad52 by siRNA or knockout of the gene by CRISPR/Cas9 compromised restart of collapsed forks and led to DNA damage in cells experiencing DRS. Furthermore, in cancer-prone, heterozygous APC mutant mice, homozygous deletion of the Rad52 gene suppressed tumor growth and prolonged lifespan. We therefore propose that mammalian RAD52 facilitates repair of collapsed DNA replication forks in cancer cells.
Keywords: RAD52, DNA replication stress, break-induced replication, DNA recombination, cancer
Graphical Abstract
Highlights
-
•
Mammalian RAD52 is involved in the oncogene-induced DNA replication stress response
-
•
Mammalian RAD52 functions in the repair of collapsed DNA replication forks
-
•
Rad52 deficiency prolongs the lifespan of Apcmin/+ mice
RAD52 is dispensable for life in mammals but is required for double-stranded break (DSB) repair in yeast. Sotiriou et al. now show that mammalian RAD52 plays an important role in response to DNA replication stress by mediating the repair of collapsed DNA replication forks.
Introduction
Genomic instability, a key hallmark of cancer, is driven in part by oncogene-induced DNA replication stress (DRS). Specifically, in cancer cells, activated oncogenes induce dissociation of the replication machinery from the DNA fork (fork collapse), formation of DNA double-strand breaks (DSBs), and genomic instability (Gorgoulis et al., 2005, Bartkova et al., 2005, Bartkova et al., 2006, Bonner et al., 2008, Halazonetis et al., 2008, Negrini et al., 2010, Arlt et al., 2012, Hills and Diffley, 2014, Macheret and Halazonetis, 2015). Prolonged exposure to chemical agents that interfere with DNA replication can also lead to fork collapse (Branzei and Foiani, 2010, Petermann et al., 2010, Yeeles et al., 2013).
Following fork collapse, DNA replication can be completed by repair of the collapsed forks, by incoming replication forks, or by dormant origin firing (Branzei and Foiani, 2010, Blow et al., 2011, Yeeles et al., 2013, Mayle et al., 2015). We previously described break-induced replication (BIR) as a repair pathway for collapsed DNA replication forks in cancer cells (Costantino et al., 2014) and, more recently, the scope of BIR in human cells was expanded to include DNA replication repair in prophase and alternative lengthening of telomeres (Minocherhomji et al., 2015, Dilley et al., 2016, Roumelioti et al., 2016).
BIR has been studied extensively in budding yeast, as a homologous recombination (HR)-based repair pathway for one-ended DNA DSBs (Llorente et al., 2008, Malkova and Ira, 2013, Anand et al., 2013). In BIR, formation of a D loop is followed by establishment of a replication fork. Notably, the D loop moves together with the replication fork, and DNA replication is conservative (Donnianni and Symington, 2013, Saini et al., 2013, Wilson et al., 2013). These unique properties distinguish BIR-initiated forks from origin-initiated forks and suggest the involvement of different proteins at these two types of forks. Indeed, Pol32, a nonessential subunit of budding yeast DNA polymerase delta, is required for BIR, but not for origin-initiated replication (Lydeard et al., 2007). Mammalian PolD3, the ortholog of budding yeast Pol32, is also required for BIR, as is PolD4, another subunit of mammalian DNA polymerase delta that has no apparent ortholog in budding yeast (Costantino et al., 2014, Murga et al., 2016).
In addition to Pol32, BIR in yeast requires Rad52 (Llorente et al., 2008, Payen et al., 2008). However, the role of Rad52 in yeast is not specific to BIR; DNA DSB repair by gene conversion (a.k.a. synthesis-dependent strand annealing) and single-strand annealing also require Rad52, and yeast mutants lacking Rad52 are very sensitive to DNA damaging agents (Symington, 2002, Sugawara et al., 2003).
Rad52 is conserved at the amino acid level from yeast to human, but its function is apparently only partially conserved. Thus, human Rad52 retains the strand-annealing activity (Kagawa et al., 2002, Singleton et al., 2002), but gene conversion is mediated primarily by BRCA2 (Prakash et al., 2015). Accordingly, whereas homozygous deletion of the BRCA2 gene in mice leads to embryonic lethality (Sharan et al., 1997), RAD52-knockout mice have a normal lifespan and no major phenotype, raising the question of what the physiological function of mammalian Rad52 is (Rijkers et al., 1998, Yamaguchi-Iwai et al., 1998).
We previously performed an siRNA screen to identify DNA repair genes that are important for cell cycle progression when cyclin E is overexpressed (Costantino et al., 2014). POLD3 was one of the top hits. Here we performed a more focused screen centering on genes that function in HR, and we identified RAD52. Further characterization revealed that RAD52 has a role in BIR, making it a potential target for the development of cancer-specific therapies.
Results
RAD52 Plays a Role in the Response to Oncogene-Induced DNA Replication Stress
In an effort to identify genes that function in BIR in human cells, we performed an siRNA screen targeting about 70 genes that had previously been linked to DNA DSB repair and HR (Table S1). The requirement of these genes in BIR was examined using U2OS cells that overexpress cyclin E in an inducible manner (Bartkova et al., 2005). In these well-characterized cells, cyclin E overexpression leads to DRS, and the damaged replication forks are repaired to a significant degree by BIR. Thus, when BIR is inhibited—for example, by depleting PolD3—progression through the cell cycle is delayed (Costantino et al., 2014).
To enable monitoring of cell cycle progression, the cells were pulsed consecutively with two thymidine analogs (EdU and BrdU; 1 hr pulse each with the two pulses separated by 6 hr) and then examined by flow cytometry (Figure 1A and Figure S1A). Cells that remained in G1 during the 8 hr period would stain negatively for both EdU and BrdU, whereas cells that transitioned from G1 into S phase would stain negatively for EdU and positively for BrdU. As observed before (Costantino et al., 2014), the fraction of cells that remained in G1 over the 8 hr period decreased when cyclin E was overexpressed (Figure 1A, control siRNA; NE, normal cyclin E expression; OE, cyclin E overexpression).
Figure 1.

RAD52 Facilitates S Phase Entry in Cells with Oncogene-Induced DRS
(A) U2OS cells overexpressing cyclin E in a tetracycline (tet)-dependent manner were seeded on plates either in the presence (normal levels of cyclin E, NE) or absence (cyclin E overexpression, OE) of tet. The next day the cells were transfected with siRNA; after 3 days, they were pulse labeled with EdU for 1 hr, and 6 hr later they were pulse labeled with BrdU for 1 hr. Nocodazole (noc) was added between the EdU and BrdU pulses to prevent mitotic cells from proceeding into G1. The percentages of EdU−/BrdU− OE and NE cells were determined by flow cytometry and plotted. Selected siRNAs are indicated: ctl, control; E, EdU; B, BrdU.
(B) Means and standard deviations of EdU−/BrdU− percentages of cells transfected with the indicated siRNAs. Two different siRNAs were used to target RAD52 and PIF1. In this and all other figures, one, two, three, and four asterisks denote statistical significance levels of p < 0.05, p < 0.01, p < 0.001, and p < 0.0001, respectively, and relevant statistical parameters are listed in Table S2.
(C) CRISPR/Cas9-mediated inactivation of the RAD52 gene in three different knockout (KO) clones of U2OS cells inducibly overexpressing cyclin E. Lack of Rad52 protein expression (top) and robust cyclin E induction (bottom) in the three clones were documented by immunoblot analysis.
(D) CRISPR/Cas9-mediated inactivation of the RAD52 gene compromises entry into S phase preferentially in cells overexpressing cyclin E (OE) as compared to cells expressing normal cyclin E (NE) levels. Means and standard deviations of the percentages of EdU−/BrdU− cells were determined using the experimental design shown in (A).
Most of the siRNAs tested did not affect the fraction of NE or OE cells that remained in G1 over the 8 hr period (Figure 1A and Table S1). This included two siRNAs that depleted the helicase Pif1, even though in budding yeast Pif1 is required for BIR (Wilson et al., 2013). A small number of siRNAs preferentially enhanced the fraction of cells that remained in G1 when cyclin E was overexpressed. These were the siRNAs targeting SLX4, MUS81, SMARCAL1, TIPIN, TIMELESS, POLD4, and RAD52 (Figures 1A and 1B). Slx4, an adaptor protein that binds Mus81; Mus81, a nuclease; and SmarcaL1, a helicase, remodel damaged replication forks (Bétous et al., 2012, Pepe and West, 2014, Sarbajna et al., 2014); Tipin and Timeless are part of the replication fork protection complex (Chou and Elledge, 2006, Errico and Costanzo, 2012), while PolD4 functions in BIR (Costantino et al., 2014). We decided to pursue the last hit, Rad52, which is dispensable for normal development in mice, but whose homolog in budding yeast is important for all forms of HR, including BIR.
Using CRISPR/Cas9 (Jinek et al., 2012), we generated RAD52-knockout clones in the context of the U2OS cells, in which cyclin E could be inducibly overexpressed. Three independent clones were obtained. Clone 2G contained two mutant alleles, whereas in clones 3C and 4A only one mutant allele was identified (Figure S1B). Clone 3C retained a wild-type (WT) allele, whereas in clones 2G and 4A no WT alleles could be identified. Consistent with the sequencing data, Rad52 protein was undetectable in clones 2G and 4A and barely detectable in clone 3C (Figure 1C). Importantly, all clones retained the capacity to regulate cyclin E levels in a tetracycline (tet)-dependent manner (Figure 1C), and all of them displayed decreased progression from G1 into S phase when cyclin E was overexpressed (Figures 1D and S1C–S1F).
RAD52 Is Recruited to Sites of DRS
We next examined the intracellular localization of Rad52 in cells that were either overexpressing cyclin E or exposed to chemical agents, such as hydroxyurea (HU) and camptothecin (CPT), that induce DRS. In about 20% of cells overexpressing cyclin E for 4 days, endogenous Rad52 localized to DRS foci, as marked by staining for RPA and ATRIP (Figures 2A, 2B, and S2A). Recruitment of Rad52 to DRS foci was also observed in about 50% of the cells exposed to HU for 24 hr but was mostly absent in cells exposed to HU for 2 hr (Figures 2C, 2D, and S2B). These kinetics parallel the known effects of HU on DNA replication forks; short treatment of cells with HU leads to a reduction in the ribonucleotide pools and fork stalling, whereas forks collapse after prolonged exposure to HU (Petermann et al., 2010).
Figure 2.

Rad52 Is Recruited to Sites of DRS
(A) Means and standard deviations of the percentages of cells displaying Rad52, RPA, Atrip, or Rad51 foci in the presence of normal (NE) or high (OE) levels of cyclin E. The results are derived from three independent experiments.
(B) Representative immunofluorescence images showing colocalization of Rad52 and RPA foci in cells overexpressing cyclin E (OE).
(C) Means and standard deviations of the percentages of cells displaying Rad52, RPA, 53BP1, or Rad51 foci following treatment with HU or CPT for 0, 2, or 24 hr. The results are derived from three independent experiments.
(D) Representative immunofluorescence images showing colocalization of Rad52 and RPA foci in cells treated with HU for 24 hr.
(E) Posttranslational modifications of chromatin-bound Rad52 in cells treated with hydroxyurea (HU) for 24 hr or exposed to ionizing radiation (IR). U2OS parental cells (WT) and clone 2G with both alleles of RAD52 inactivated were cultured in the presence of tet to maintain normal levels of cyclin E. The bands corresponding to Rad52 are indicated.
(F) ATR dependence of HU-induced posttranslational modification of chromatin-bound Rad52. U2OS parental cells were cultured in the presence of HU with or without an ATR inhibitor (ATRi) for 24 hr before being harvested. Tet was present in the media to maintain normal levels of cyclin E. Where indicated, the chromatin extracts were treated with lambda phosphatase (λ ph).
Exposure of cells to CPT, which induces covalent bonding of topoisomerase I to DNA and, subsequently, fork collapse (Pommier, 2006), also led to recruitment of Rad52 to DRS foci; this was evident within 2 hr of exposure in some cells but was much more evident at 24 hr (Figures 2C and S2C). Interestingly, in cells overexpressing cyclin E or exposed to either HU or CPT, Rad51 foci were less prevalent than Rad52 foci (Figures 2A and 2C); however, the functional significance, if any, of this apparent difference remains to be investigated.
Exposure of cells to HU for 24 hr was also associated with posttranslational modifications of the chromatin-bound fraction of Rad52, as detected by immunoblotting, whereas a high dose (9 Gy) of ionizing radiation did not elicit similar modifications (Figure 2E). A subset of the Rad52 posttranslational modifications induced in response to HU were ATR dependent, since they were suppressed when the cells were treated with an ATR inhibitor (Figure 2F). Furthermore, the modifications were sensitive to treatment of the chromatin extracts with lambda phosphatase (Figure 2F). Taken together, these results suggest that ATR phosphorylates Rad52 at sites of DRS.
Enhanced DNA Damage Response following Rad52 Depletion in Cells Treated with HU
If Rad52 is important for BIR, then its depletion should compromise the repair of collapsed forks and lead to a stronger DNA damage response. To explore this possibility, we depleted Rad52 by siRNA and monitored H2AX phosphorylation both 2 and 24 hr after adding HU to the cells. At the 24 hr time point, the majority of the replication forks were collapsed, whereas at early time points, a large fraction of the forks are stalled and can resume replication upon HU withdrawal (Figure S3A). The effects of PolD3, Mus81, and Rad51 depletion on H2AX phosphorylation were also examined (Figure S3B).
Two hours after exposure of the cells to HU, γH2AX levels increased modestly and equally in the control, Rad52-, PolD3-, and Mus81-depleted cells (Figures 3A and 3B). After 24 hr exposure to HU, γH2AX phosphorylation levels increased further, but the increase was much stronger in the cells depleted for Rad52 or PolD3 (Figures 3A and 3B). Codepleting Rad52 and PolD3 had the same effect as depleting only Rad52 (Figure 3C). These results are consistent with Rad52 and PolD3 functioning epistatically in repair of collapsed, but not stalled, replication forks. Interestingly, depletion of Rad51 suppressed γH2AX phosphorylation when compared to control siRNA-treated cells at both the 2 and 24 hr time points, an observation that merits further study (Figures 3A and 3B).
Figure 3.

Rad52 Is Required for Fork Restart after Prolonged Exposure of Cells to HU
(A) Rad52 and PolD3 regulate the cellular response to DRS, as ascertained by monitoring histone H2AX phosphorylation (γH2AX) in cells treated with HU for 2 or 24 hr. γH2AX levels were monitored by flow cytometry of cells treated with control (ctl) siRNA or siRNAs targeting RAD52, POLD3, MUS81, or RAD51. PI, propidium iodide.
(B) Means and standard deviations of the percentages of cells assigned to the H2AX phosphorylation gates shown in (A), as determined from three independent experiments. Green, blue, and red indicate background, modest, and high H2AX phosphorylation, respectively.
(C) Rad52 and PolD3 regulate the cellular response to DRS epistatically. siRNA-transfected cells were exposed to HU for 24 hr. Means and standard deviations of the percentages of cells assigned to the high (Hi) H2AX phosphorylation gate were derived from two independent experiments.
(D) Fork restart after prolonged exposure of cells to HU is dependent on Rad52. U2OS parental cells (WT) and clone 2G with both alleles of RAD52 inactivated were cultured in the presence of tet to maintain normal levels of cyclin E. The cells were pulse labeled with CldU for 1 hr, then exposed to HU and a Cdc7 inhibitor for 6 or 24 hr, and finally released into media containing IdU and the Cdc7 inhibitor for 1 hr to allow fork restart. Means and standard deviations of the percentages of restarted forks were derived from three independent DNA fiber experiments.
(E) Effect of depletion of PolD3, PolD4, or Rad52 on repair of DNA DSBs by BIR. Means and standard deviations of the percentages of GFP-positive cells were derived from three independent experiments.
Rad52 Facilitates Restart of Collapsed Replication Forks
To examine whether Rad52 facilitates restart of replication after fork collapse, we performed DNA fiber analysis of cells exposed to HU for 6 or 24 hr. However, we were concerned that new origin firing near a collapsed fork might be misinterpreted as fork restart if replication from the new origin proceeded all the way to the collapsed fork. We therefore performed the assay in the presence of a Cdc7 inhibitor that does not inhibit transcription (Menichincheri et al., 2010, Montagnoli et al., 2010a, Montagnoli et al., 2010b) after demonstrating that this inhibitor effectively suppressed new origin firing in cells released from a 24 hr HU replication block (Figures S3A and S3C).
To monitor fork restart, the cells were incubated with CldU for 1 hr, then incubated with HU for 6 or 24 hr in the presence of the Cdc7 inhibitor; finally, after release from the HU block, the cells were incubated with IdU for 1 hr again in the presence of the Cdc7 inhibitor to prevent new origin firing. The 1 hr IdU incubation period was found to be sufficient to observe restart of replication. In the parental U2OS cells, replication restart was observed after exposure to HU for both 6 and 24 hr, although this was the case to a greater extent for the shorter incubation period (Figure 3D). In the RAD52-knockout clone, replication restart was significantly reduced, especially when the cells were exposed to HU for 24 hr (Figures 3D and S3D). Since the Cdc7 inhibitor prevented new origin firing, these results suggest that repair and restart of collapsed forks was Rad52 dependent.
Rad52 Enhances the Efficiency of BIR
To further monitor the function of Rad52 in BIR, we employed a green fluorescent protein (GFP)-based reporter assay in which BIR-mediated repair of DNA DSBs induced by the endonuclease I-SceI leads to GFP fluorescence (Costantino et al., 2014). Since the initial description of this assay, we have generated a new, stably transfected cell clone expressing the GFP reporter plasmid that provides a better signal-to-noise ratio than the original clone. Depletion of Rad52 by siRNA in the new clone led to a significant suppression of GFP fluorescence, consistent with Rad52 functioning in BIR (Figure 3E). Depletion of PolD3 and PolD4 also suppressed BIR in this system, although not as efficiently as Rad52. Interestingly, codepletion of Rad52 and PolD3 or Rad52 and PolD4 suppressed BIR, as efficiently as depletion of Rad52 alone, further suggesting that PolD3, PolD4 and Rad52 function epistatically (Figure 3E).
Rad52 Deletion Restrains Oncogenic Progression in Mice
Since BIR repairs collapsed forks in cells with oncogene-induced DRS, its deletion should curtail cancer development and/or progression. First, we examined tumor formation in APCflox/+ mice rendered heterozygous for APC by constitutively expressing the Cre recombinase under the control of a CMV promoter and enhancer. In these mice, the entire coding sequence of the mutant APC allele is deleted by the Cre recombinase (Cheung et al., 2010). In regard to Rad52, the mice either had two WT Rad52 alleles (Rad52+/+) or were homozygous for deletion of the Rad52 gene (Rad52−/−). At 8 months of age, the mice, while still having no overt signs of disease, were sacrificed, and the presence of tumors in the entire small intestine was scored by histology. Deletion of Rad52 did not affect the number of observed tumors (a total of 46 versus 50 tumors in six Rad52+/+ and six Rad52−/− mice, respectively), but resulted in smaller tumor sizes (Figure 4A). Deletion of Rad52 also resulted in an increase in the fraction of tumor cells scoring positive for phosphorylated H2AX, consistent with an inability to repair collapsed replication forks; however, this difference was only evident in the early stages of tumor development, when tumors were less than 1.5 mm in diameter (Figures 4B and S4).
Figure 4.

Rad52 Deficiency Restrains Tumor Growth and Prolongs Survival of Mice with APC Mutations
(A) Comparison of tumors present in the intestines of Rad52+/+;Apcf/+;CMVcre (N = 6) and Rad52−/−;Apcf/+;CMVcre (N = 6) mice. Tumors were stratified according to size (in mm) or according to histopathological criteria: LD, low-grade dysplasia; HD, high-grade dysplasia; iMc, intramucosal; and sMc, submucosal.
(B) Proliferation (Ki67) and DNA damage (γH2AX) indices of the tumors present in the intestines of the Rad52+/+;Apcf/+;CMVcre and Rad52−/−;Apcf/+;CMVcre mice. Means and standard deviations of the indices were calculated after stratifying the tumors into three groups according to size.
(C) Survival fractions of Rad52+/+;Apcmin/+ (N = 8) and Rad52−/−;Apcmin/+ (N = 8) mice. Three of the Rad52−/−;Apcmin/+ mice, indicated by vertical lines in the graph, had not died at the time the data were recorded and were considered censored for the statistical analysis.
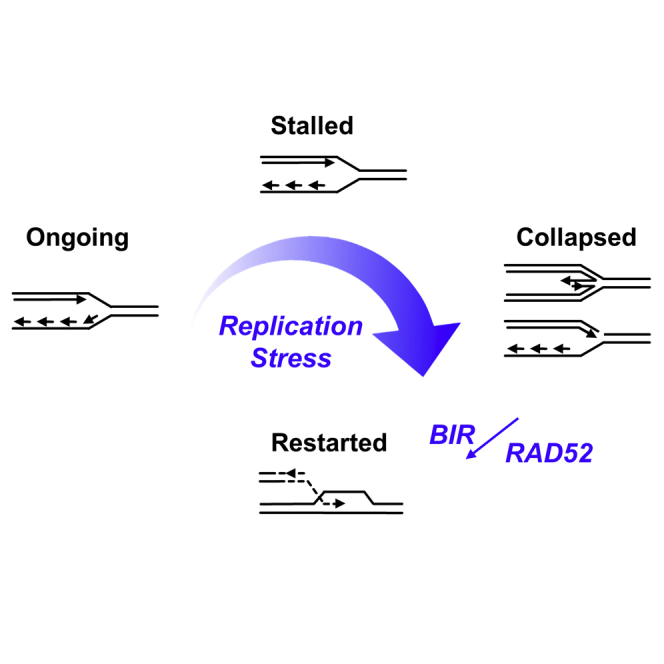
(D) Proposed model for the role of Rad52 in BIR.
The effect of Rad52 on cancer progression was further examined in APCmin heterozygous mice. In a WT Rad52 background, the lifespan of these mice is significantly shorter than that of mice bearing the APCflox allele described above (Moser et al., 1990). We therefore used the APCmin model to ascertain whether loss of Rad52 affected survival. Indeed, lifespan of the APCmin heterozygous mice was significantly extended from a mean of 127 days for the Rad52+/+ mice to 178 days for the Rad52−/− mice (Figure 4C).
Discussion
The function of Rad52 in DNA repair is well established in yeast, but less well defined in higher eukaryotes and mammals (Symington, 2002). In fact, mice with homozygous deletion of the Rad52 gene are viable and have no obvious phenotype (Rijkers et al., 1998). This could indicate that Rad52 serves a backup function that is relevant only when the primary DNA DSB repair pathways are inactivated or overwhelmed by excessive DNA damage. Supporting this model, depletion of Rad52 is synthetic lethal with BRCA2 deficiency in human cell lines (Feng et al., 2011, Lok and Powell, 2012). Also, cells rely on Rad52 for repair when extensive DNA damage overwhelms the repair capacities of BRCA1 and 53BP1 (Ochs et al., 2016). However, the findings reported here suggest that Rad52 has more than a backup role in DNA repair. Specifically, we propose that Rad52 has a key role in BIR repair of collapsed DNA replication forks (Figure 4D).
Several observations support a role of Rad52 in BIR. Endogenous Rad52 localized to sites of DRS in cells with collapsed DNA replication forks, depletion of Rad52 led to increased levels of DNA damage in cells exposed to HU for 24 hr, DNA replication restart from collapsed forks was dependent on Rad52, and Rad52 depletion suppressed repair of DNA DSBs by BIR in a GFP-based reporter assay. Together with the known biochemical function of mammalian Rad52 in strand annealing (Kagawa et al., 2002, Singleton et al., 2002), these results argue that strand invasion by Rad52 leads to DNA structures that are conducive to initiation of DNA replication after fork collapse.
In yeast, Rad52 is important for Rad51-dependent and Rad51-independent forms of HR. Whereas repair of DNA DSBs by gene conversion in yeast is Rad51 dependent, BIR can be Rad51 independent (Ira and Haber, 2002, Payen et al., 2008). In this case, the strand annealing activity of Rad52 is important, and indeed, it is easy to envision the presence of single-stranded template DNA to which the invading strand can anneal at collapsed DNA replication forks. By analogy, mammalian Rad52, with or without Rad51, may be involved in the strand-invasion step of BIR, as is the case for its ortholog in yeast (Llorente et al., 2008, Malkova and Ira, 2013, Anand et al., 2013).
A role for Rad52 in BIR can explain why its depletion in cells with DRS leads to increased DNA damage (Wray et al., 2008, Murfuni et al., 2013, Galanos et al., 2016; Figures 3A and 3B), why the RAD52 gene is amplified in human cancers, and why its inactivation curtails cancer development (Treuner et al., 2004, Cramer-Morales et al., 2013, Lieberman et al., 2016; Figure 4). BIR also mediates DNA repair synthesis in mitosis (Minocherhomji et al., 2015), and it is noteworthy that Rad52 is essential also in this context (Bhowmick et al., 2016).
Experimental Procedures
siRNA Screen and Generation of RAD52-Deficient Clones
For the siRNA screen, U2OS cells engineered to overexpress cyclin E in an inducible (tet-off system) manner (U2OS-CycE cells) were plated in the presence or absence of tet, and the next day they were transfected with siRNA. Then, 3 days later, the cells were pulsed for 1 hr with EdU; 6 hr after that, they were pulsed for 1 hr with BrdU and then processed for flow cytometry as described (Costantino et al., 2014). To generate RAD52-knockout U2OS-CycE cells, two CRISPR/Cas9 constructs targeting exons 3 and 9 of the human RAD52 gene, respectively, were used. After transfection, single clones were expanded and characterized by DNA sequencing of the targeted alleles and by immunoblotting for Rad52 protein.
Immunofluorescence and Flow Cytometry Analysis of the γH2AX Content
Either U2OS-CycE cells grown in the presence or absence of tet for 4 days or U2OS parental cells treated with 2 mM HU or 2 μM CPT were processed for immunofluorescence, as described (Costantino et al., 2014). To monitor γH2AX levels by flow cytometry, U2OS cells transfected with the indicated siRNAs were treated with 2 mM HU for 0, 2, or 24 hr; fixed in 70% ice-cold ethanol; and stained using the FlowCellect Histone H2AX Phosphorylation Assay Kit (Millipore).
DNA Fiber Analysis
U2OS cells were pulse labeled with CldU for 1 hr, then treated with 2 mM HU and Cdc7 inhibitor for 6 or 24 hr, and finally pulse labeled with IdU for 1 hr in the presence of the Cdc7 inhibitor. DNA fibers were spread on APS-coated coverslips and visualized using primary antibodies recognizing CldU or IdU.
See the Supplemental Information for a full list of antibodies as well as a full list and detailed description of the methods used in this study.
Author Contributions
S.K.S. and T.D.H. conceived the study. S.K.S., I.K., N.L., K.E., C.D.-R., F.H., L.P., N.L.N., and S.B. planned and performed the experiments. L.S. planned the chemical synthesis, and S.T. synthesized the Cdc7 inhibitor. S.K.S., I.K., N.L., K.E., C.D.-R., F.H., L.P., S.T., N.L.N., S.B., F.O., C.L., J.L., V.G.G., L.S., and T.D.H. proposed experiments, discussed the results, and contributed to the writing of the manuscript.
Acknowledgments
The authors thank Ian Hickson for sharing unpublished results, Massimo Lopes for advice on the DNA-fiber-spreading protocol, and Samuel Espy for help on the chemical synthesis of the Cdc7 inhibitor. This study was supported by funds from the Swiss National Science Foundation (SNF 160322) and the European Commission (ERC ONIDDAC) to T.D.H., funds from the Swiss National Science Foundation (CRSI33_130016) to L.S., funds from the Novo Nordisk Foundation (NNF 14CC0001) and Danish Cancer Society (R72-A4436) to C.L. and J.L., and funds from the Greek GSRT Program (Aristeia II-3020) to V.G.G., as well as an EMBO long-term postdoctoral fellowship to I.K.
Published: December 15, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, four figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2016.10.038.
Supplemental Information
References
- Anand R.P., Lovett S.T., Haber J.E. Break-induced DNA replication. Cold Spring Harb. Perspect. Biol. 2013;5:a010397. doi: 10.1101/cshperspect.a010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt M.F., Wilson T.E., Glover T.W. Replication stress and mechanisms of CNV formation. Curr. Opin. Genet. Dev. 2012;22:204–210. doi: 10.1016/j.gde.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J., Horejsí Z., Koed K., Krämer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J.M., Lukas C. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bartkova J., Rezaei N., Liontos M., Karakaidos P., Kletsas D., Issaeva N., Vassiliou L.V., Kolettas E., Niforou K., Zoumpourlis V.C. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Bétous R., Mason A.C., Rambo R.P., Bansbach C.E., Badu-Nkansah A., Sirbu B.M., Eichman B.F., Cortez D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26:151–162. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick R., Minocherhomji S., Hickson I.D. RAD52 facilitates mitotic DNA synthesis following replication stress. Mol. Cell. 2016;64:1117–1126. doi: 10.1016/j.molcel.2016.10.037. this issue. [DOI] [PubMed] [Google Scholar]
- Blow J.J., Ge X.Q., Jackson D.A. How dormant origins promote complete genome replication. Trends Biochem. Sci. 2011;36:405–414. doi: 10.1016/j.tibs.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner W.M., Redon C.E., Dickey J.S., Nakamura A.J., Sedelnikova O.A., Solier S., Pommier Y. GammaH2AX and cancer. Nat. Rev. Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D., Foiani M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010;11:208–219. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- Cheung A.F., Carter A.M., Kostova K.K., Woodruff J.F., Crowley D., Bronson R.T., Haigis K.M., Jacks T. Complete deletion of Apc results in severe polyposis in mice. Oncogene. 2010;29:1857–1864. doi: 10.1038/onc.2009.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou D.M., Elledge S.J. Tipin and Timeless form a mutually protective complex required for genotoxic stress resistance and checkpoint function. Proc. Natl. Acad. Sci. USA. 2006;103:18143–18147. doi: 10.1073/pnas.0609251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino L., Sotiriou S.K., Rantala J.K., Magin S., Mladenov E., Helleday T., Haber J.E., Iliakis G., Kallioniemi O.P., Halazonetis T.D. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science. 2014;343:88–91. doi: 10.1126/science.1243211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer-Morales K., Nieborowska-Skorska M., Scheibner K., Padget M., Irvine D.A., Sliwinski T., Haas K., Lee J., Geng H., Roy D. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood. 2013;122:1293–1304. doi: 10.1182/blood-2013-05-501072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilley R.L., Verma P., Cho N.W., Winters H.D., Wondisford A.R., Greenberg R.A. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature. 2016;539:54–58. doi: 10.1038/nature20099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnianni R.A., Symington L.S. Break-induced replication occurs by conservative DNA synthesis. Proc. Natl. Acad. Sci. USA. 2013;110:13475–13480. doi: 10.1073/pnas.1309800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errico A., Costanzo V. Mechanisms of replication fork protection: a safeguard for genome stability. Crit. Rev. Biochem. Mol. Biol. 2012;47:222–235. doi: 10.3109/10409238.2012.655374. [DOI] [PubMed] [Google Scholar]
- Feng Z., Scott S.P., Bussen W., Sharma G.G., Guo G., Pandita T.K., Powell S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA. 2011;108:686–691. doi: 10.1073/pnas.1010959107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanos P., Vougas K., Walter D., Polyzos A., Maya-Mendoza A., Haagensen E.J., Kokkalis A., Roumelioti F.M., Gagos S., Tzetis M. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016;18:777–789. doi: 10.1038/ncb3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis V.G., Vassiliou L.V., Karakaidos P., Zacharatos P., Kotsinas A., Liloglou T., Venere M., Ditullio R.A., Jr., Kastrinakis N.G., Levy B. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Halazonetis T.D., Gorgoulis V.G., Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Hills S.A., Diffley J.F. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014;24:R435–R444. doi: 10.1016/j.cub.2014.04.012. [DOI] [PubMed] [Google Scholar]
- Ira G., Haber J.E. Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol. Cell. Biol. 2002;22:6384–6392. doi: 10.1128/MCB.22.18.6384-6392.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagawa W., Kurumizaka H., Ishitani R., Fukai S., Nureki O., Shibata T., Yokoyama S. Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol. Cell. 2002;10:359–371. doi: 10.1016/s1097-2765(02)00587-7. [DOI] [PubMed] [Google Scholar]
- Lieberman R., Xiong D., James M., Han Y., Amos C.I., Wang L., You M. Functional characterization of RAD52 as a lung cancer susceptibility gene in the 12p13.33 locus. Mol. Carcinog. 2016;55:953–963. doi: 10.1002/mc.22334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente B., Smith C.E., Symington L.S. Break-induced replication: what is it and what is it for? Cell Cycle. 2008;7:859–864. doi: 10.4161/cc.7.7.5613. [DOI] [PubMed] [Google Scholar]
- Lok B.H., Powell S.N. Molecular pathways: understanding the role of Rad52 in homologous recombination for therapeutic advancement. Clin. Cancer Res. 2012;18:6400–6406. doi: 10.1158/1078-0432.CCR-11-3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydeard J.R., Jain S., Yamaguchi M., Haber J.E. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 2007;448:820–823. doi: 10.1038/nature06047. [DOI] [PubMed] [Google Scholar]
- Macheret M., Halazonetis T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015;10:425–448. doi: 10.1146/annurev-pathol-012414-040424. [DOI] [PubMed] [Google Scholar]
- Malkova A., Ira G. Break-induced replication: functions and molecular mechanism. Curr. Opin. Genet. Dev. 2013;23:271–279. doi: 10.1016/j.gde.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayle R., Campbell I.M., Beck C.R., Yu Y., Wilson M., Shaw C.A., Bjergbaek L., Lupski J.R., Ira G. DNA REPAIR. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science. 2015;349:742–747. doi: 10.1126/science.aaa8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichincheri M., Albanese C., Alli C., Ballinari D., Bargiotti A., Caldarelli M., Ciavolella A., Cirla A., Colombo M., Colotta F. Cdc7 kinase inhibitors: 5-heteroaryl-3-carboxamido-2-aryl pyrroles as potential antitumor agents. 1. Lead finding. J. Med. Chem. 2010;53:7296–7315. doi: 10.1021/jm100504d. [DOI] [PubMed] [Google Scholar]
- Minocherhomji S., Ying S., Bjerregaard V.A., Bursomanno S., Aleliunaite A., Wu W., Mankouri H.W., Shen H., Liu Y., Hickson I.D. Replication stress activates DNA repair synthesis in mitosis. Nature. 2015;528:286–290. doi: 10.1038/nature16139. [DOI] [PubMed] [Google Scholar]
- Montagnoli A., Moll J., Colotta F. Targeting cell division cycle 7 kinase: a new approach for cancer therapy. Clin. Cancer Res. 2010;16:4503–4508. doi: 10.1158/1078-0432.CCR-10-0185. [DOI] [PubMed] [Google Scholar]
- Montagnoli A., Ballinari D., Ciavolella A., Rainoldi S., Menichincheri M., Pesenti E., Galvani A., Isacchi A., Moll J. Activity of the Cdc7 inhibitor NMS-1116354 as single agent and in combination in breast cancer models. EJC Suppl. 2010;8:49. [Google Scholar]
- Moser A.R., Pitot H.C., Dove W.F. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- Murfuni I., Basile G., Subramanyam S., Malacaria E., Bignami M., Spies M., Franchitto A., Pichierri P. Survival of the replication checkpoint deficient cells requires MUS81-RAD52 function. PLoS Genet. 2013;9:e1003910. doi: 10.1371/journal.pgen.1003910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga M., Lecona E., Kamileri I., Díaz M., Lugli N., Sotiriou S.K., Anton M.E., Méndez J., Halazonetis T.D., Fernandez-Capetillo O. POLD3 is haploinsufficient for DNA replication in mice. Mol. Cell. 2016;63:877–883. doi: 10.1016/j.molcel.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini S., Gorgoulis V.G., Halazonetis T.D. Genomic instability—an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- Ochs F., Somyajit K., Altmeyer M., Rask M.B., Lukas J., Lukas C. 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol. 2016;23:714–721. doi: 10.1038/nsmb.3251. [DOI] [PubMed] [Google Scholar]
- Payen C., Koszul R., Dujon B., Fischer G. Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet. 2008;4:e1000175. doi: 10.1371/journal.pgen.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepe A., West S.C. MUS81-EME2 promotes replication fork restart. Cell Rep. 2014;7:1048–1055. doi: 10.1016/j.celrep.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E., Orta M.L., Issaeva N., Schultz N., Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- Prakash R., Zhang Y., Feng W., Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015;7:a016600. doi: 10.1101/cshperspect.a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijkers T., Van Den Ouweland J., Morolli B., Rolink A.G., Baarends W.M., Van Sloun P.P., Lohman P.H., Pastink A. Targeted inactivation of mouse RAD52 reduces homologous recombination but not resistance to ionizing radiation. Mol. Cell. Biol. 1998;18:6423–6429. doi: 10.1128/mcb.18.11.6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumelioti F.M., Sotiriou S.K., Katsini V., Chiourea M., Halazonetis T.D., Gagos S. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 2016:e201643169. doi: 10.15252/embr.201643169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N., Ramakrishnan S., Elango R., Ayyar S., Zhang Y., Deem A., Ira G., Haber J.E., Lobachev K.S., Malkova A. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature. 2013;502:389–392. doi: 10.1038/nature12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbajna S., Davies D., West S.C. Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 2014;28:1124–1136. doi: 10.1101/gad.238303.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan S.K., Morimatsu M., Albrecht U., Lim D.S., Regel E., Dinh C., Sands A., Eichele G., Hasty P., Bradley A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 1997;386:804–810. doi: 10.1038/386804a0. [DOI] [PubMed] [Google Scholar]
- Singleton M.R., Wentzell L.M., Liu Y., West S.C., Wigley D.B. Structure of the single-strand annealing domain of human RAD52 protein. Proc. Natl. Acad. Sci. USA. 2002;99:13492–13497. doi: 10.1073/pnas.212449899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N., Wang X., Haber J.E. In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol. Cell. 2003;12:209–219. doi: 10.1016/s1097-2765(03)00269-7. [DOI] [PubMed] [Google Scholar]
- Symington L.S. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 2002;66:630–670. doi: 10.1128/MMBR.66.4.630-670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treuner K., Helton R., Barlow C. Loss of Rad52 partially rescues tumorigenesis and T-cell maturation in Atm-deficient mice. Oncogene. 2004;23:4655–4661. doi: 10.1038/sj.onc.1207604. [DOI] [PubMed] [Google Scholar]
- Wilson M.A., Kwon Y., Xu Y., Chung W.H., Chi P., Niu H., Mayle R., Chen X., Malkova A., Sung P., Ira G. Pif1 helicase and Polδ promote recombination-coupled DNA synthesis via bubble migration. Nature. 2013;502:393–396. doi: 10.1038/nature12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray J., Liu J., Nickoloff J.A., Shen Z. Distinct RAD51 associations with RAD52 and BCCIP in response to DNA damage and replication stress. Cancer Res. 2008;68:2699–2707. doi: 10.1158/0008-5472.CAN-07-6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi-Iwai Y., Sonoda E., Buerstedde J.M., Bezzubova O., Morrison C., Takata M., Shinohara A., Takeda S. Homologous recombination, but not DNA repair, is reduced in vertebrate cells deficient in RAD52. Mol. Cell. Biol. 1998;18:6430–6435. doi: 10.1128/mcb.18.11.6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles J.T., Poli J., Marians K.J., Pasero P. Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 2013;5:a012815. doi: 10.1101/cshperspect.a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.