

Abstract
Meniere’s Disease (MD) is a complex disorder associated with an accumulation of endolymph in the membranous labyrinth in the inner ear. It is characterized by recurrent attacks of spontaneous vertigo associated with sensorineural hearing loss (SNHL) and tinnitus. The SNHL usually starts at low and medium frequencies with a variable progression to high frequencies. We identified a novel missense variant in the PRKCB gene in a Spanish family with MD segregating low-to-middle frequency SNHL. Confocal imaging showed strong PKCB II protein labelling in non-sensory cells, the tectal cells and inner border cells of the rat organ of Corti with a tonotopic expression gradient. The PKCB II signal was more pronounced in the apical turn of the cochlea when compared with the middle and basal turns. It was also much higher in cochlear tissue than in vestibular tissue. Taken together, our findings identify PRKCB gene as a novel candidate gene for familial MD and its expression gradient in supporting cells of the organ of Corti deserves attention, given the role of supporting cells in K+ recycling within the endolymph, and its apical turn location may explain the onset of hearing loss at low frequencies in MD.
Introduction
The auditory system is responsible for sound sensation. The frequency selectivity, known as tonotopy, is based on the structural properties along the cochlear sensory epithelium, the organ of Corti, and the topographic organization of the auditory pathway (1). There are multiple gradients in the gene expression profile from base to apex in the cochlea (2,3), and these gradients may be crucial to understand the mechanisms for low and high frequency sensorineural hearing loss (SNHL).
Meniere’s disease (MD, [MIM 156000]) is a chronic multifactorial disorder affecting the inner ear, characterized by recurrent episodes of vertigo, tinnitus and low-to-medium frequencies SNHL (4). The most disabling symptom of MD is the unpredictable attacks of vertigo, which may last several hours. The vertigo is accompanied by fluctuating aural symptoms (tinnitus, pressure, hearing loss), nausea, vomiting and headache. Initially, the clinical findings are normal between attacks, but as MD progresses, SNHL increases and becomes permanent (5). The disorder may affect both ears in 25-40% of patients (6) and bilateral MD is associated with increased vestibular symptoms and a negative impact on health-related quality of life (7). The hearing loss profile may be limited to low frequencies in most cases, but some patients may show a pan-tonal SNHL (8). Hearing levels fluctuate at the onset of the disease and several types of audiometric plots have been described, including: a) low and high frequency SNHL with preservation at 2,000Hz; b) an upward-sloping curve with low frequencies affected; c) pan-tonal SNHL; or d) a downward-sloping curve with an effect on high frequencies (9,10). These variations in hearing profile may reflect the early involvement of the apical turn of the cochlea and the later involvement of the middle and basal turns as the disease progresses.
Perception of tinnitus varies among patients, but in some cases, it may also become permanent and a major concern for patients. There is a great phenotypic variability in these patients and the symptoms may overlap with other vestibular disorders such as vestibular migraine (11).
In general, MD has a small female preponderance and it is more commonly found in Caucasian than in Asian descendent populations (12). Although its exact aetiology is unknown, the accumulation of endolymph in the scala media of the cochlea and the vestibular organs produces swelling of the membranous labyrinth (endolymphatic hydrops), and suggests that MD is related to abnormal recycling of Na+ and/or K+ in the endolymph (13).
Most MD cases are sporadic, but familial clustering has been reported (14–16), supporting the hypothesis of a genetic susceptibility to the development of certain forms of MD (17). Therefore, familial MD (FMD) is observed in 8-10% among sporadic cases in the European population and it shows different patterns of inheritance (15,16), suggesting some genetic heterogeneity in FMD.
In the present study, we have performed whole-exome sequencing (WES) in a family with three affected men in two generations. We report the identification of a novel missense variant in PRKCB gene, which segregates with hearing loss in the three patients, and we show that PRKCB gene has an expression gradient in the supporting cells of the cochlea.
Results
A novel missense variant in PRKCB segregates with low-frequency sensorineural hearing loss in a Spanish family with Meniere’s disease
The family consisted of two patients with MD (II-2 and III-14) and a third individual presenting an otosclerosis-like phenotype (II-11). Several members of this pedigree reported a history of migraine (II-3, II-5, III-5, III-8), including individuals II-2 and II-11 (Fig. 1A). The proband (III-14) was a 31 year-old man with bilateral MD. He developed a left ear MD at 19 years old and the right ear became affected seven years later. Initially, the hearing loss affected low to medium frequencies (125 to 1,000 Hz) with preservation of high frequencies (2,000 to 8,000 Hz). The hearing loss profile was identical in the right ear. The patient presented a bilateral tinnitus with fluctuating hearing loss in both ears. He was treated with betahistine and torasemide with a reduction in the number of attacks. After 11 years of follow-up, he had a SNHL involving 125-2,000 Hz frequencies with a pure tone average (PTA) of 46 dB HL in the left ear and 23 dB in the right ear (Fig. 1B).
Figure 1.

Spanish family with Meniere’s disease with a novel variant in the PRKCB gene. (A) Pedigree of a family presenting AD FMD with three affected men in two generations. Symbols in the pedigree represent: complete MD phenotype (full black square), numbers at the left of the symbol indicate the age of onset; incomplete phenotype (partial black square or circle, upper-left black: vestibular phenotype; lower-right black: cochlear phenotype). Abbreviations: M, Migraine; MA, Migraine with aura; MO, Migraine without aura. (B) Air-conduction audiograms showing a severe sensorineural hearing loss starting at low frequencies in affected individuals at several ages (age symbols shown below the chart); upper X axis represents frequencies in Hz, and Y axis represents hearing level (HL) in dB. (C) Genomic sequence at PRKCB gene (chr16:23999898G > T) with the novel variant (M) segregating hearing loss (indicated in red).
His father (II-11) was diagnosed with otosclerosis in the right ear and migraine. He has no vestibular symptoms. He underwent stapes surgery at 51 years old and his hearing levels were successfully restored. One year after surgery, he developed a sudden low-frequency SNHL in the right ear that was maintained for 18 months at 60 dB at 250 and 500 Hz with no response to steroids. Three years after the episode of sudden SNHL, hearing levels at low frequencies were at 45 dB. The paternal uncle of the proband (II-2) has a unilateral MD of the left ear that started at 65 years old with a pan-tonal SNHL of 60 dB. He suffered from multiple episodes of vertigo. He was treated with betahistine and torasemide, followed by intratympanic gentamicin with persistence of the attacks. After 3 years of intractable vertigo, the patient underwent a labyrinthectomy, which controlled the symptoms without further relapses.
We used the Lifescope(TM) standard pipeline to analyse our WES data of individuals II-2, II-11 and III-14. Each sample gives an average depth of coverage within targets of 113 reads. We filtered out single nucleotide variants (SNVs) with a GQ < 100 and a read depth (DP) < 20. The pattern of inheritance was considered autosomal dominant (AD) with incomplete penetrance, but according to the pedigree, oligogenic inheritance could not be ruled out.
We generated a list of candidate SNVs, based on the results obtained with our in-house pipeline, which includes different bioinformatics tools to prioritize variants (see Supplementary Material, Fig. S1 and Table S1). After the filtering and prioritization process, we selected a novel missense variant with a high score affecting the PRKCB gene and validated it by Sanger sequencing (Fig. 1C). This variant segregated the hearing loss phenotype in individuals II-2 and III-14 with a diagnosis of MD and in the patient II-11 with an otosclerosis-like phenotype. The novel missense variant (chr16: 23999898 G > T) in the PRKCB gene is located in exon 3 and it generates an amino acid change from glycine to valine (c.G275T:p.G92V) affecting the protein sequence and possibly its function. The presence of the variant was assessed in different populations with several databases: ExAC v0.3, 1000 Genomes, the National Heart, Lung, and Blood Institute Exome Variant Server, and our in-house exome database (including six multi-case families with MD and 500 controls). In total, data from > 71,000 individuals from unrelated populations were reviewed and none of them had the variant.
Protein kinase C beta type is a family of serine- and threonine-specific protein kinases involved in various cellular processes. In humans, the PRKCB gene has six annotated protein coding transcripts (Supplementary Material, Table S2), and the SNV identified affects two of these transcripts, one of them being PRKCB-002 (ENST0000303531), the canonical isoform.
PRKCB gene expression in the human cochlea and semicircular canals
Supplementary Material, Figure S2A shows PRKCB expression in both human adult cochlea and the semicircular canals (SCs). The relative expression was calculated using qPCR (ΔCt cochlea = 2.83 ± 0.40; ΔCt SCs = 2.49 ± 0.38) (Supplementary Material, Fig. S2B). No significant differences were found between the cochlea and vestibular tissues.
PKCB II protein labelling shows a tonotopic gradient in inner border cells and tectal cells
The PKCB II antibody intensely labelled two types of cochlear supporting cells, the tectal cells and inner border cells, in the apical turn of the cochlea (Figs. 2 and 3A). Tectal cells are across the “outer tunnel” from the third row of outer hair cells, adjacent to the Hensen cells and have, in fact, previously been considered the first row of Hensen cells, until it was determined that they had many morphological differences from the other Hensen cells. Inner border cells are immediately adjacent to cochlear inner hair cells. The inner border and tectal cells were not labelled in the middle and basal turns, however, Deiters cells underneath the outer hair cells in these two turns were moderately labelled. Both types of cells were more intensely labelled than any cell type in the vestibular epithelium (Fig. 3). The transitional epithelium in the crista ampullaris was lightly labelled (above background levels observed in the rest of the neuroepithelium). Interestingly, neither the vestibular dark cells, nor the cochlear stria vascularis, were labelled. Although most of the significant differences in label were apparent to the naked eye, we quantified the intensity levels using ImageJ (NIH) to distinguish between differences that were not apparent (Supplementary Material, Table S3). Apical turn tectal cells, inner border cells and afferent boutons innervating inner hair cells, were the most intensely labelled, then middle and basal turn Deiters cells, and finally cochlear hair cells and vestibular hair cells, which were indistinguishable from background levels. Vestibular transitional epithelium cells (another type of supporting cell) were just slightly above background levels.
Figure 2.
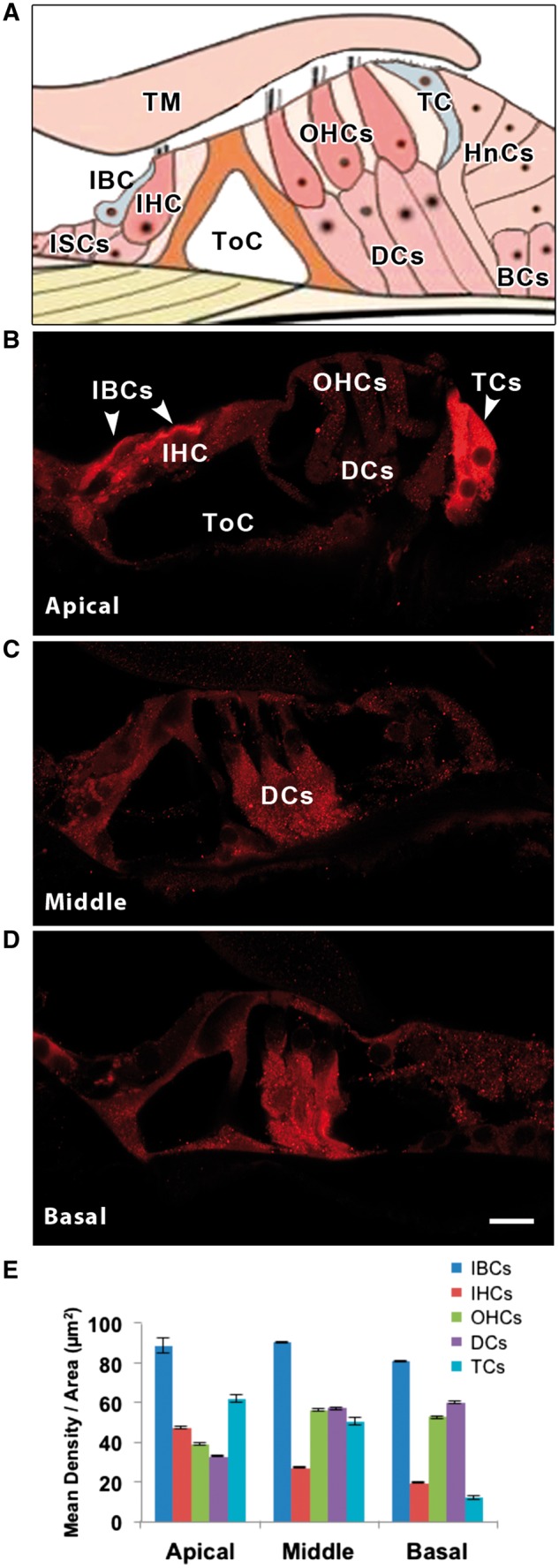
(A) Schematic diagram of the organ of Corti to show the location of tectal cells (TCs) and inner border cells (IBCs). PKCB II immuno-labelling in the three turns of the rat cochlea, apical (B), middle (C) and basal (D). Cell types are labelled in A, B and C. Note that the apical IBCs and TCs are most intensely labelled, followed by basal and middle turn Deiter’s cells (DCs). Hair cells, both inner (IHCs) and outer (OHCs), are barely above background level. Other abbreviations: tectorial membrane (TM), inner sulcus cells (ISCs), tunnel of Corti (ToC), Hensen’s cells (HnCs), Boettcher’s cells (BCs). (E) Mean density per area of PKCB II for each cochlear cell type and turn of the cochlea. Scale bar in D = 10 µm, applies to B-D.
Figure 3.
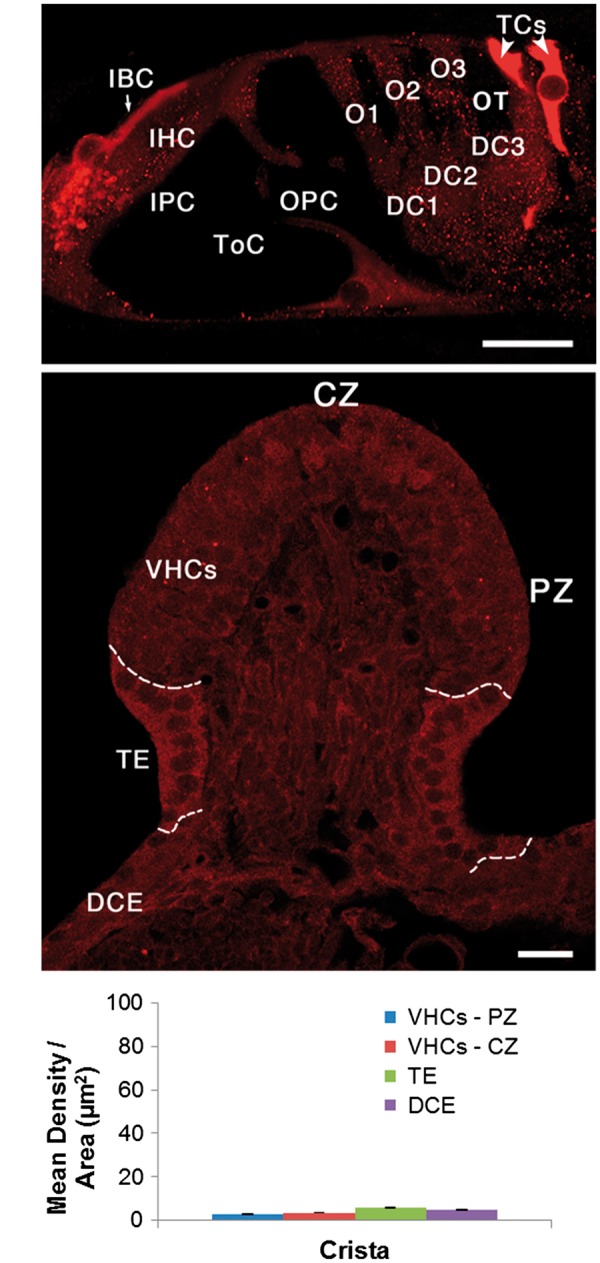
Here we directly compare cochlear PKCB II immuno-labelling in the apical turn of (A) the rat cochlea with (B) the vestibular crista ampullaris in the same animal. Note that the entire vestibular epithelium is less intensely labelled than the cochlear inner border cells (IBCs) and tectal cells (TCs). Dashed lines in B separate the vestibular sensory epithelium (VHCs) from the transitional epithelium (TE), and the TE from the dark cell epithelium (DCE). (C) Mean density per area of PKCB II in different regions of the rat crista ampullaris. Note this histogram is the same scale as that in Fig. 2E. Other abbreviations: IHC, inner hair cell; IPC, inner pillar cell; ToC, tunnel of Corti; OPC, outer pillar cell; O1, O2, O3, first, second and third row outer hair cells; DC1, DC2, DC3, first, second and third row Deiters cells; OT, outer tunnel. CZ, central zone; PZ, peripheral zone; VHCs, vestibular hair cells (sensory epithelium), TE transitional epithelium (supporting cells). Scale bars = 10 µm.
Pathway analysis of differentially expressed genes along the mouse cochlea can predict PKCB-related signalling pathways associated with tonotopy
We used gene expression datasets from Yoshimura et al. (2014), obtained from apical, middle and basal turns of the mouse cochlea, to predict signalling pathways associated with tonotopia. We extracted the significant PKCB-related signalling pathways: “Axonal Guidance Signalling”, “Alpha-Adrenergic Signalling”, “14-3-3-mediated Signalling” and “CXCR4 Signalling” (Supplementary Material, Table S4). The axonal Guidance Signalling pathway is the top candidate with 28 genes involved, since molecules that regulate spiral ganglion projections to peripheral sensory hair cells are required to establish the tonotopic connections in the auditory system.
Discussion
An essential organizing principle of cochlear structure is tonotopy, based on differences in the properties of auditory hair cells in the sensory epithelium along the apical-basal axis (2). These differences have been established in morphological and physiological studies in different species (18), including a gene expression gradient in the mammalian cochlea (3). This gene expression gradient has significant relevance for AD SNHL genes as it has also been reported for the mouse orthologues Pou4f3, Slc17a8, Tmc1, and Crym (3). Interestingly, these genes are all associated with high-frequency SNHL.
Familial MD is a rare condition and the estimated prevalence is 8-10% among sporadic cases (16,19). Most of the families have an AD inheritance with incomplete penetrance and the SNHL usually begins at low and medium frequencies (8–10). Some of the families described presented co-segregation of migraine with anticipation (14,20,21), but the family presented here does not show these features. Migraine was reported in four relatives in several generations, but as can be observed in the pedigree (Fig. 1A), it was not co-segregated with MD. Our study shows that AD FMD has an incomplete penetrance and that it has a genetic heterogeneity. Several relatives presented a partial syndrome with episodic vestibular symptoms, but no hearing loss (III-5, III-8), and they did not present the novel PRKCB variant. These findings suggest that additional factors (genetic, epigenetic or environmental) are likely to contribute to the vestibular phenotype in this family.
Familial MD has been associated with rare variants in FAM136A and DTNA genes in another Spanish pedigree, and these genes are also considered potential candidate genes for MD (22). The present study describes a second family and it confirms genetic heterogeneity in familial MD. Furthermore, we hypothesize that: a) different genes can potentially be involved in the development of the complete phenotype; or b) the interaction of environmental or epigenetic factors with candidate genes will determine differences in the phenotype.
Our recent findings (Requena et al., 2015, and the present study) are beginning to define a few of the candidate genes associated with FMD and we anticipate that different genes will be found to be involved in different families’ phenotypes.
The PRKCB gene encodes a serine and threonine-specific protein kinase that can be activated by calcium and diacylglycerol. This kinase is involved in many different cellular functions, such as neutrophil chemotaxis, melanoma cell growth and proliferation (23), or induction of apoptosis in endothelial cells (24); furthermore, studies in mice suggest that this kinase modulates neuronal functions and correlates fear-induced conflict behaviour after stress (25). The heterozygous variant (chr16: 23999898 G > T) identified in the PRKCB gene involves two of the six protein coding transcripts (ENST00000303531 and ENST 00000321728), and both are expressed in the human inner ear transcriptome (26).
It segregates a low-frequency hearing loss in this family. The likely mechanism by which heterozygous mutations are causing the loss of gene function in AD disorders is haploinsufficiency. Thus, the phenotype is developed when the gene expression of PRKCB that is essential to maintain biological function in the basal and middle turn in specific supporting cell types of the cochlea type falls below some critical level.
Our results confirm the previous immunohistochemical results of Ladrech et al. (2007), who also found PKCΒ II in Hensen cells, although their study did not differentiate specific supporting cell types. The cells in which we found the PRKCB gene product most intensely expressed in the rat cochlea, the tectal cells, were first named and described at length by the Hensons (27) in the mustached bat, Pteronotus p. parnellii, but have also been described in cats (28), mice (29), chinchillas (30) and gerbils (31), where they were also found to label with calretinin and S-100 (32). Their morphological characterization includes several features, observed in electron microscopic studies: 1) a unique position on the roof and along the lateral wall of the outer tunnel; 2) no contact with the basilar membrane: 3) isolation of adjacent cell bodies; 4) an extensive endolymphatic surface with a sparse population of short microvilli, as opposed to the rich bed of long microvilli found on the adjacent Hensen’s cells; and 5) a darkly-staining cytoplasm. Early in development (the first postnatal week in the mouse), newly formed tectal cells show several cytological characteristics suggesting increased metabolic and secretory activities (33), which include: 1) a large Golgi complex; 2) abundant amorphous material inside the cisterns of rough endoplasmic reticulum; and 3) dense granules inside the mitochondrial matrix. All these features gradually disappear, and by postnatal day 14 (P14), the tectal cells have a dark cytoplasm with few, short microvilli.
The tectal cells have been implicated in K+ transport (30,31). They show ultrastructural specializations, such as fimbriae, an abundance of small mitochondria, and tubulo-cisternal endoplasmic reticulum, characteristic of ion-transporting cells in other parts of the body (34,35), e.g., kidney proximal tubule epithelial cells or choroid plexus. Spicer et al. (2003) also noted an association of tectal cells with the third row of Deiter’s cells (DCs). Antibody labelling in tectal cells was much more intense in the apical turn than in the middle and basal turn. This pattern of protein expression was matched by a gene expression study in the mouse cochlea also showing PRKCB gene expression to be more intense in the apical and middle turns than in the basal turns (3). If protein expression were reduced in these two turns in subjects with the gene variant, then reduction of expression could correlate with the low-frequency SNHL observed at the onset of the disease in two patients.
Four signalling pathways are associated with a gradient of differentially expressed genes (DEGs, two-fold change in apex vs. basal turns) along the tonotopic axis of the cochlea, including PRKCB (Supplementary Material, Table S4). “Axonal guidance signalling” is a crucial process in the regulation of innervation of the organ of Corti and it includes several families of DEG-encoding signalling molecules, such as morphogens [bone morphogenetic proteins (BMPs), sonic hedgehog (Shh) and wingless (WNTs)], semaphorins (SEMA3A, SEMA3D), ephs and ephrins, netrins (NTNG1) and neurotrophins (BDNF and NT-3), which control the growth cone in extending neurites of the auditory projections (36). Remarkably, the Shh signalling pathway determines cochlear tonotopic organization during development in the avian basilar papilla and mammalian cochlea, although the downstream effectors of Shh signalling that establish tonotopy in the mouse organ of Corti are not known (37). “Alpha-adrenergic signalling” mediates responses to noradrenaline and adrenaline in the cochlea through α2-adrenergic receptors and they are widely expressed in inner hair cells, outer hair cells and supporting cells in the organ of Corti (38). The involvement of α2-adrenergic receptors in K+ transport and PKCB II gradient in tectal cells will require further investigations. “14-3-3 mediated signalling” in the cochlea was associated with DEG of tubulin families, suggesting a role in the cytoskeletal regulation and topographic organization of the innervation of the organ of Corti (39). “CXCR4 signalling” in the tonotopic organization of the cochlea will also need further studies.
This study provides evidences that PRKCB is a candidate gene for low frequency SNHL in familial MD and demonstrates a tonotopic gradient for PKCB II expression in tectal cells in the rat cochlea.
Materials and Methods
Patients
Blood samples were collected from patients and relatives of a Spanish family from Galicia, in the Northwest of Spain, suggesting an autosomal-dominant (AD) pattern of inheritance. The pedigree is shown in Fig. 1A. This study was approved by the Institutional Review Board for Clinical Research (PI-0496-2014), and an informed consent for donor biological samples was obtained from all subjects.
Initially, the diagnosis of Meniere’s disease (MD) was established according to the guidelines defined by the Committee on Hearing and Equilibrium of the AAO-NHS in 1995 (16). The family described included two patients with definite MD, fulfilling the criteria for familial MD (4). A complete neuro-otology assessment including otoscopy, pure-tone audiometry, oculomotor examination, saccades, smooth pursuit and caloric testing was carried out in all cases. Moreover, brain magnetic resonance imaging was performed to exclude other possible causes of neurological symptoms. Pure tone audiograms were obtained during follow-up at each visit. Hearing staging was calculated by the audiograms obtained for each patient and it was defined as the mean of a four-tone average of 0.5, 1, 2 and 3 kHz, according to the AAO- HNS criteria: stage 1, <25 dB; stage 2, 26–40 dB; stage 3, 41– 70 dB; stage 4, >70 dB.
Whole-exome sequencing and bioinformatics analyses
We performed whole-exome sequencing (WES) analyses with DNA samples from patients (II-2, II-11 and III-14) and familial controls (II-3, II-14 and III-13). Exons and flanking intron regions were selected and captured by Agilent’s All Exon 50 MB capture kit. The conditions and primer sets are available on commercial websites. Library products were sequenced with a SOLiD 5500xl platform and the sequences were analysed with LifescopeTM software 2.5 (http://www.lifetechnologies.com/lifescope). After the alignment of our data with the GRCh37 human assembly, we obtained different Variant Call Format files (VCF) to encode single nucleotide variants (SNVs) and other structural genetic variants. Each VCF file generated from LifescopeTM software 2.5 contains ∼50,000 SNVs per exome sequenced. A combination of wANNOVAR (40,41) and SeattleSeq Annotation tools (http://snp.gs.washington.edu/SeattleSeqAnnotation137/) were used to annotate the SNVs found in common between cases. Hence, to identify the candidate variants of MD phenotype and to reduce the list, we first filtered variants by exome data from our in-house control database, and minor allelic frequency (MAF) <0.0001 (42), according to the SNVs previously annotated in dbSNP 142 (http://www.ncbi.nlm.nih.gov/projects/SNP/) and in the Exome Aggregation Consortium (ExAC), a large integrated database of human exome sequencing data (http://exac.broadinstitute.org/).
To prioritize candidate variants, initially, we scored SNVs using a seven point scoring system (Pathogenic Variant or PAVAR score), according to the effect on protein structure and phylogenetic conservation: (SIFT (Sort Intolerant from Tolerant), PolyPhen2 (Polymorphism Phenotyping v2), Grantham’s Matrix, GERP+ (Genomic Evolutionary Rate Profiling), Mutation taster, PhastCons and PhyloP); and we take into account SNVs with a score > 5 (22). To improve the accuracy of the variant-prioritization, we combined the previous results with other bioinformatics tools that include phenotype information, such as Exomiser v2 (43) in which the maximum score is 1, and Variant Annotation Analysis and Search Tool (VAAST) + Phevor, bioinformatics tools that prioritize the variants and the genes affected using a ranking system (44,45).
Sanger sequencing and RT-PCR
We validated candidate variants by Sanger sequencing technology in a 3130 Genetic Analyzer (Applied Biosystems) and visualized them with Sequence Scanner Software v1.0 (Applied Biosystems), to confirm WES results (Fig. 1B). Primer pairs flanking the SNV were designed using Primer3 software (http://primer3.sourceforge.net/).
Human tissues from the basal turn of the cochlea and semicircular canals (SCs) were collected from schwannoma surgery patients and preserved immediately in RNAlater. The tissue was homogenised mechanically by adding 700 µL of Qiazol and a steel bead in a TissuLyser for 5 minutes at 50 Hz. RNA was isolated by using the QIAamp RNA miRNeasy Mini Kit (QIAGEN) and a Nanodrop spectrophotometer, and the Agilent RNA 6000 Nano Bioanalyzer chip was used to determine the integrity, quality and quantity of RNA. Complementary DNA (cDNA) was synthesized with the QuantiTect Reverse Transcription Kit (QIAGEN) by adding 60 ng of RNA to the reverse transcription master mix and following the kit instructions.
Quantitative real-time PCR (qPCR) was performed using SYBR® Green RT-PCR techniques (Life Technologies) to determine the expression of PRKCB genes in human cochlea and vestibular tissues. The primer pairs used to validate the expression of candidate genes have the following sequences: HPRT1_Fw (5’TGACACTGGCAAAACAATGCA3’) and HPRT1_Rv (5’GGTCCTT TTCACCAGCAAGCT3’) for the HPRT housekeeping gene and PRKCB_Fw (5’ GTTGTGATCCAAGATGATGACG3’) and PRKCB_Rv (5’ CTGTAAGAAGAACAGACCGATGG3’) for the PRKCB gene. The data were analysed with the ABI RQ Manager Software (Applied Biosystems). To detect genomic DNA contamination, the primer sequences for PRKCB were designed in two different exons (10 and 12) and an RT negative control of each sample was analysed in the same plate. Each sample was run in triplicate and values were expressed in ΔCt ± standard deviation. Statistical analysis was performed with IBM SPSS software (SPSS Inc, Armonk, NY).
Accession number
The PRKCB sequence in this study has been submitted to the Clinical Variant database and its accession number is SCV000264644.
Animals and tissue preparation
All animal experiments were carried out in the Department of Anatomy and Cell Biology of University of Illinois at Chicago (UIC). Rats were housed in the Biological Research Laboratory at UIC. Animal experiments were approved by the UIC Institutional Animal Care and Use Committee (IACUC) and conformed to the NIH Guide for the Care and Use of Laboratory Animals.
Wild-type rats (Rattus norvegicus) were sacrificed by an overdose of barbiturate anaesthesia and transcardially perfused. Temporal bones containing the inner ear were dissected, fixed and cryosectioned as previously described (22,46).
Immunofluorescence of rat inner ear
For immunohistochemistry, 35 μm frozen sections were stained and imaged as previously described (22,46). Briefly, sections were thawed and permeabilized with 4% Triton X-100 for 1 hr at room temperature, blocked with a solution containing 0.5% fish gelatin (Sigma, St. Louis, MO), 1% bovine serum albumin (Sigma) and 0.5% Triton X-100 in 0.1 M phosphate buffer (PB) for 1 hr, then incubated with primary antibody in the same blocking solution for 72 hrs at 4 deg C. A rabbit polyclonal antibody against PKCB II (Santa Cruz, Cat. No. sc-210, dilution 1:200) was used (47).
The primary antibody was visualized with Alexa-595-conjugated donkey anti-rabbit (Life Technologies, Cat. No. A21207, 1:200). Sections were rinsed and mounted in Mowiol 4-88 Reagent mounting medium (Calbiochem, Cat. No. 475904). For all immunoreactions, negative controls (no primary antibody incubations) were also included. A laser scanning confocal microscope (LSM 710, Carl Zeiss, Oberköchen, Germany) was used for image collection and the Zeiss browser software program ZEN was used to acquire and export data. All images were taken with the same laser intensity settings on the microscope. Final image processing and labelling were done with Adobe Photoshop CS4 (San Jose, CA).
Image analysis
Image stacks were exported from ZEN to the image analysis program ImageJ (NIH, http://imagej.nih.gov/ij/) as stacks of images. In ImageJ, individual cochlear cells or entire vestibular epithelial areas were measured on sections in which the cells showed their maximum area (see Supplementary Material, Figs S3 and S4). Measurements taken were: Area (of the cell or region measured minus cell nuclei) and Mean Density. Density values were averaged over the areas and ranged from 0 to 255; Areas were converted to μm2. In the vestibular epithelium, areas measured were per region (central vs. peripheral) since individual cell borders could not be determined. Two-tailed t-tests with unequal variances were performed to determine the significance (see mean density data and t-test results in Supplementary Material, Table S3).
Pathway analysis of DEGs along the mouse cochlea
Gene expression datasets from mouse cochlea were used to define a tonotopic gradient, according to their expression in the basal or apical turns of the cochlea (3). DEGs in the apex (adjusted for p <0.001) were used to predict the involved pathways containing the PRKCB gene. Signalling pathway analysis was performed using Ingenuity Pathways Analysis (IPA®, Qiagen, Venlo, Netherlands, http://www.ingenuity.com/products/ipa) software. The Core Analysis tool was executed using the DEG with an adjusted P value cutoff of 0.001.
Supplementary Material
Supplementary Material is available at HMG online.
Acknowledgements
We thank patients with familial Meniere’s disease for their participation in this study.
Conflict of Interest Statement. None declared.
Funding
This work was supported by grants from Fundación Progreso y Salud [PI0496-14 to T.R.], Meniere’s Society, UK [to J.A.L.E.] and the National Institutes of Health [DC013181 to A.L.]. We also acknowledge the COST Action BM1306 TINNET, which supports part of our networking activities (http://tinnet.tinnitusresearch.net/).
References
- 1.Corti A. (1851) Recherches sur l’organe de l'ouïe des mammiferes. Limaçon. Z. Wiss. Zool, 3, 106–169. [Google Scholar]
- 2.Frucht C.S., Uduman M., Kleinstein S.H., Santos-Sacchi J., Navaratnam D.S. (2011) Gene expression gradients along the tonotopic axis of the chicken auditory epithelium. J. Assoc. Res. Otolaryngol., 12, 423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshimura H., Takumi Y., Nishio S.Y., Suzuki N., Iwasa Y., Usami S. (2014) Deafness gene expression patterns in the mouse cochlea found by microarray analysis. PLoS One., 9, e92547.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez-Escamez J.A., Carey J., Chung W.H., Goebel J.A., Magnusson M., Mandala M., Newman-Toker D.E., Strupp M., Suzuki M., Trabalzini F., et al. (2015) Diagnostic criteria for Meniere's disease. J. Vestib. Res., 25, 1–7. [DOI] [PubMed] [Google Scholar]
- 5.Harcourt J., Barraclough K., Bronstein A.M. (2014) Meniere's disease. Bmj., 349, g6544.. [DOI] [PubMed] [Google Scholar]
- 6.House J.W., Doherty J.K., Fisher L.M., Derebery M.J., Berliner K.I. (2006) Meniere's disease: prevalence of contralateral ear involvement. Otol. Neurotol., 27, 355–361. [DOI] [PubMed] [Google Scholar]
- 7.Lopez-Escamez J.A., Viciana D., Garrido-Fernandez P. (2009) Impact of bilaterality and headache on health-related quality of life in Meniere's disease. Ann. Otol. Rhinol. Laryngol., 118, 409–416. [DOI] [PubMed] [Google Scholar]
- 8.Belinchon A., Perez-Garrigues H., Tenias J.M., Lopez A. (2011) Hearing assessment in Meniere's disease. Laryngoscope, 121, 622–626. [DOI] [PubMed] [Google Scholar]
- 9.Enander A., Stahle J. (1967) Hearing in Meniere's disease. A study of pure-tone audiograms in 334 patients. Acta Otolaryngol., 64, 543–556. [DOI] [PubMed] [Google Scholar]
- 10.Mancini F., Catalani M., Carru M., Monti B. (2002) History of Meniere's disease and its clinical presentation. Otolaryngol. Clin. North Am., 35, 565–580. [DOI] [PubMed] [Google Scholar]
- 11.Lopez-Escamez J.A., Dlugaiczyk J., Jacobs J., Lempert T., Teggi R., von Brevern M., Bisdorff A. (2014) Accompanying symptoms overlap during attacks in Meniere's Disease and vestibular migraine. Front. Neurol., 5, 265.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohmen J.D., White C.H., Li X., Wang J., Fisher L.M., Zhang H., Derebery M.J., Friedman R.A. (2013) Genetic evidence for an ethnic diversity in the susceptibility to Meniere's disease. Otol. Neurotol., 34, 1336–1341. [DOI] [PubMed] [Google Scholar]
- 13.Kim S.H., Marcus D.C. (2011) Regulation of sodium transport in the inner ear. Hear. Res., 280, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morrison A.W., Bailey M.E., Morrison G.A. (2009) Familial Meniere's disease: clinical and genetic aspects. J. Laryngol. Otol., 123, 29–37. [DOI] [PubMed] [Google Scholar]
- 15.Hietikko E., Kotimaki J., Sorri M., Mannikko M. (2013) High incidence of Meniere-like symptoms in relatives of Meniere patients in the areas of Oulu University Hospital and Kainuu Central Hospital in Finland. Eur. J. Med. Genet., 56, 279–285. [DOI] [PubMed] [Google Scholar]
- 16.Requena T., Espinosa-Sanchez J.M., Cabrera S., Trinidad G., Soto-Varela A., Santos-Perez S., Teggi R., Perez P., Batuecas-Caletrio A., Fraile J., et al. (2014) Familial clustering and genetic heterogeneity in Meniere's disease. Clin. Genet., 85, 245–252. [DOI] [PubMed] [Google Scholar]
- 17.Vrabec J.T. (2010) Genetic investigations of Meniere's disease. Otolaryngol. Clin. North Am., 43, 1121–1132. [DOI] [PubMed] [Google Scholar]
- 18.Mann Z.F., Kelley M.W. (2011) Development of tonotopy in the auditory periphery. Hear. Res., 276, 2–15. [DOI] [PubMed] [Google Scholar]
- 19.Lee J.M., Kim M.J., Jung J., Kim H.J., Seo Y.J., Kim S.H. (2015) Genetic aspects and clinical characteristics of familial Meniere's disease in a South Korean population. Laryngoscope, 125, 2175–2180. [DOI] [PubMed] [Google Scholar]
- 20.Arweiler-Harbeck D., Horsthemke B., Jahnke K., Hennies H.C. (2011) Genetic aspects of familial Meniere's disease. Otol. Neurotol., 32, 695–700. [DOI] [PubMed] [Google Scholar]
- 21.Frykholm C., Larsen H.C., Dahl N., Klar J., Rask-Andersen H., Friberg U. (2006) Familial Meniere's disease in five generations. Otol. Neurotol., 27, 681–686. [DOI] [PubMed] [Google Scholar]
- 22.Requena T., Cabrera S., Martin-Sierra C., Price S.D., Lysakowski A., Lopez-Escamez J.A. (2015) Identification of two novel mutations in FAM136A and DTNA genes in autosomal-dominant familial Meniere's disease. Hum. Mol. Genet., 24, 1119–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schonherr M., Bhattacharya A., Kottek T., Szymczak S., Koberle M., Wickenhauser C., Siebolts U., Saalbach A., Koczan D., Magin T.M, et al. (2014) Genomewide RNAi screen identifies protein kinase Cb and new members of mitogen-activated protein kinase pathway as regulators of melanoma cell growth and metastasis. Pigment Cell Melanoma Res, 27, 418–430. [DOI] [PubMed] [Google Scholar]
- 24.Shao B., Bayraktutan U. (2014) Hyperglycaemia promotes human brain microvascular endothelial cell apoptosis via induction of protein kinase C-ssI and prooxidant enzyme NADPH oxidase. Redox Biol., 2, 694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Birikh K.R., Sklan E.H., Shoham S., Soreq H. (2003) Interaction of “readthrough” acetylcholinesterase with RACK1 and PKCbeta II correlates with intensified fear-induced conflict behavior. Proc. Natl. Acad. Sci. USA, 100, 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schrauwen I., Hasin-Brumshtein Y., Corneveaux J.J., Ohmen J., White C., Allen A.N., Lusis A.J., Van Camp G., Huentelman M.J., Friedman R.A. (2016) A comprehensive catalogue of the coding and non-coding transcripts of the human inner ear. Hear. Res., 333, 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henson M.M., Jenkins D.B., Henson O.W., Jr. (1983) Sustentacular cells of the organ of Corti – the tectal cells of the outer tunnel. Hear. Res., 10, 153–166. [DOI] [PubMed] [Google Scholar]
- 28.Spoendlin H. (1966) The organization of the cochlear receptor. Advan. Oto-Rhino. Laryngol, 13, 1–227. [PubMed] [Google Scholar]
- 29.Kronester-Frei A. (1979) Localization of the marginal zone of the tectorial membrane in situ, unfixed, and with in vivo-like ionic milieu. Arch. Otorhinolaryngol., 224, 3–9. [DOI] [PubMed] [Google Scholar]
- 30.Spicer S.S., Smythe N., Schulte B.A. (2003) Ultrastructure indicative of ion transport in tectal, Deiters, and tunnel cells: differences between gerbil and chinchilla basal and apical cochlea. Anat. Rec. A. Discov. Mol. Cell. Evol. Biol, 271, 342–359. [DOI] [PubMed] [Google Scholar]
- 31.Spicer S.S., Schulte B.A. (1994) Ultrastructural differentiation of the first Hensen cell in the gerbil cochlea as a distinct cell type. Anat. Rec., 240, 149–156. [DOI] [PubMed] [Google Scholar]
- 32.Pack A.K., Slepecky N.B. (1995) Cytoskeletal and calcium-binding proteins in the mammalian organ of Corti: cell type-specific proteins displaying longitudinal and radial gradients. Hear. Res., 91, 119–135. [DOI] [PubMed] [Google Scholar]
- 33.Rueda J., Prieto J.J., Rubio M.E., Gutierrez A., Merchan J.A. (1993) Development of the tectal cells in the mouse cochlea. Anat. Embryol. (Berl.), 187, 425–432. [DOI] [PubMed] [Google Scholar]
- 34.Mollgard K., Rostgaard J. (1978) Morphological aspects of some sodium transporting epithelia suggesting a transcellular pathway via elements of endoplasmic reticulum. J. Membr. Biol, 40 (Special Issue), 71–89. [DOI] [PubMed] [Google Scholar]
- 35.Møllgård K., Rostgaard J. (1981), In Alfred Benzon Symposium, Copenhagen, Vol. 15. [Google Scholar]
- 36.Webber A., Raz Y. (2006) Axon guidance cues in auditory development. Anat. Rec. A. Discov. Mol. Cell. Evol. Biol., 288, 390–396. [DOI] [PubMed] [Google Scholar]
- 37.Son E.J., Ma J.H., Ankamreddy H., Shin J.O., Choi J.Y., Wu D.K., Bok J. (2015) Conserved role of Sonic Hedgehog in tonotopic organization of the avian basilar papilla and mammalian cochlea. Proc. Natl. Acad. Sci. USA, 112, 3746–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai J., Li J., Mao Y., Bai X., Xu L., Wang H. (2013) Immunohistochemical localization of alpha2-adrenergic receptors in the neonatal rat cochlea and the vestibular labyrinth. J. Mol. Neurosci., 51, 1010–1020. [DOI] [PubMed] [Google Scholar]
- 39.Jin J., Smith F.D., Stark C., Wells C.D., Fawcett J.P., Kulkarni S., Metalnikov P., O'Donnell P., Taylor P., Taylor L., et al. (2004) Proteomic, functional, and domain-based analysis of in vivo 14-3-3 binding proteins involved in cytoskeletal regulation and cellular organization. Curr. Biol., 14, 1436–1450. [DOI] [PubMed] [Google Scholar]
- 40.Yang H., Wang K. (2015) Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nature Protocols, 10, 1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang X., Wang K. (2012) wANNOVAR: annotating genetic variants for personal genomes via the web. J. Med. Genet., 49, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shearer A.E., Eppsteiner R.W., Booth K.T., Ephraim S.S., Gurrola J., 2nd., Simpson A., Black-Ziegelbein E.A., Joshi S., Ravi H., Giuffre A.C., et al. (2014) Utilizing ethnic-specific differences in minor allele frequency to recategorize reported pathogenic deafness variants. Am. J. Hum. Genet, 95, 445–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson P.N., Kohler S., Oellrich A., Sanger Mouse Genetics P., Wang K., Mungall C.J., Lewis S.E., Washington N., Bauer S., Seelow D., et al. (2014) Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res., 24, 340–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yandell M., Huff C., Hu H., Singleton M., Moore B., Xing J., Jorde L.B., Reese M.G. (2011) A probabilistic disease-gene finder for personal genomes. Genome Res., 21, 1529–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singleton M.V., Guthery S.L., Voelkerding K.V., Chen K., Kennedy B., Margraf R.L., Durtschi J., Eilbeck K., Reese M.G., Jorde L.B., et al. (2014) Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. Am. J. Hum. Genet., 94, 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lysakowski A., Gaboyard-Niay S., Calin-Jageman I., Chatlani S., Price S.D., Eatock R.A. (2011) Molecular microdomains in a sensory terminal, the vestibular calyx ending. J. Neurosci., 31, 10101–10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ladrech S., Wang J., Boukhaddaoui H., Puel J.L., Eybalin M., Lenoir M. (2007) Differential expression of PKC beta II in the rat organ of Corti. Eur. J. Neurosci., 26, 2922–2930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.