

Abstract
With the scientific community becoming increasingly aware of the need for greener products and methodologies, the optimization of synthetic design is of greater importance. Building on experimental data collected from a synthesized guaiazulene derivative, a series of analogous structures were investigated with time-dependent density functional theory (TD-DFT) methods in an effort to identify a compound with desirable photophysical properties. This in silico analysis may eliminate the need to synthesize numerous materials that, when investigated, do not possess viable characteristics. The synthesis of several computationally investigated structures revealed discrepancies in the calculation results. Further refined computational study of the molecules yielded results closer to those observed experimentally and helps set the stage for computationally guided design of organic photonic materials. Three novel derivatives were synthesized from guaiazulene, a naturally occurring chromophore, exhibiting distinct halochromic behaviour, which may have potential in a switchable optoelectronic system or combined with a photoacid generator for data storage. The protonated forms were readily excitable via two-photon absorption.
Keywords: halochromic, guaiazulene, two-photon absorption
1. Introduction
Building on conventional chemistry, effective solutions for greener chemical practices are increasingly being implemented to tackle concerns of chemical hazards, resource scarcity and climate impact on the population and the planet. In his 1998 book, Paul Anastas features 12 principles for green chemistry, the first of which states: ‘It is better to prevent waste than to treat or clean waste after it is formed’ [1]. To this end, the use of quantum chemical calculations to provide a prediction of a compound's properties prior to generating it in the laboratory could help to minimize waste generated by synthesis of impractical derivatives. The use of in silico techniques is commonly used to aid in the explanation of experimental results [2,3], though more recently studies have been directed towards identifying sustainable solvents [4], as well as designing solar cell components [5] and complex metal oxides [6].
Another of Anastas's principles, and area of keen development, is the use of chemicals from renewable sources, or otherwise coming from nature [1]. Extractable from fungi and coral [7,8], 1,4-dimethyl-7-isopropylazulene, or guaiazulene, is a natural derivative of azulene. Although azulene is an isomer of the colourless naphthalene, it exhibits a blue colour that has enchanted man since the late medieval period [9]. This is attributed to azulene's peculiar emission from the second excited state (S2), an exception to Kasha's Rule, as a result of its unusually low-lying first excited state (S1). Introducing electronic perturbing substituents on the seven-member ring and/or on the five-member ring of azulene was shown to change the electronic properties of azulene derivatives, accompanied by significant changes in their fluorescence behaviour [9,10]. Although the effect of resonantly electron withdrawing or donating groups on the HOMO, LUMO and LUMO +1 energies of azulene was reported, these theoretical calculations were initially limited to derivatives with mildly electronically perturbing (e.g. formyl or fluorine) substituents [11].
Recently, a number of studies have reported interesting optoelectronic properties of azulene derivatives having extended π-conjugated substituents that can be manipulated by protonation with strong acids. The formation of a resonance-stabilized 6-π-electron tropylium cation in protonated azulenes [12] resulted in a bathochromic shift in the S0 → S1 band of the absorption, as well as an increase in the luminescence intensity as a result of the now-dominant S1 → S0 decay pathway [13,14]. The unique electronic properties of azulene-based structures were instrumental in the development of derivatives for charge-transport, optoelectronic and sensor applications. The design of such structures is often based on theoretical calculations of the dipole moment of azulene derivatives; variation in calculated dipole moments is thought to be a result of various substituents on the azulene framework that acts as an electron bridge in a donor–acceptor–donor arrangement [12,15,16]. While these calculations predict a large dipole moment and large hyperpolarizability for 2,6-connected azulene systems, experimental investigation shows that a 4,7-connectivity often results in longer absorption and emission wavelengths, a desirable property for the development of various near-IR applications [10]. In order to avoid such discrepancies and minimize waste generated by preparing potentially impractical azulene derivatives, we exploited the naturally occurring guaiazulene to initially prepare a structurally simplified derivative tailored to guide the theoretical calculations to predict critical optical properties of more heavily conjugated systems (figure 1). Such in silico analysis could reveal any disadvantageous nature of certain derivatives, thus eliminating the wasteful necessity of producing derivatives with undesirable properties.
Figure 1.
Generic structure of a guaiazulene-terminated compound with a varied π-bridge.
In its protonated state, azulene's tropylium cation acts as an electron acceptor when conjugated through a π-bridge to an electron-rich system. With that in mind, and using our established method [17], guaiazulene 1 was condensed with 4-hexyloxybenzaldehyde 2a in the presence of potassium tert-butoxide to afford 3a (scheme 1). Thus, upon treatment with trifluoroacetic acid (TFA), the ethylene moiety acts as a π-spacer between the tropylium cation and the electron-rich benzene ring (scheme 2). This is illustrated in the absorption and emission spectra of 3a and its protonated form 3aH+ (figures 2 and 3). In addition to the weak S0 → S1 transition (depicted as a broad peak centred at 630 nm) common to the azulene family, 3a shows an absorption peak with λmax = 365 nm, hence giving the solution of 3a a typical azure blue colour. On the other hand, an intense red colour is immediately observed when the solution is treated with TFA. This is reflected in the absorption spectrum of 3aH+ where an intense peak appears at λmax = 512 nm, while that of the neutral species 3a (λabs = 630 nm) is no longer observed. Concomitantly, treatment of 3a with TFA resulted in a marked switch-on of fluorescence; in fact, a quantum yield of 0.12 was calculated for 3aH+ using the Lorentz–Lorenz equation (1.1) and (1.2), where OD is optical density, I is emission intensity, nx is the refractive index of solvent x, and φx is the proportion of solvent x in the mixture [18,19]. As seen in figure 3, a prominent emission peak is seen at λmax = 620 nm, which was chosen in order to measure the excitation anisotropy for the protonated species 3aH+ in a viscous medium (figure 3). The plateau of this trace through the main absorption band indicates that this is a single electronic transition (figure 3). Additionally, it was noted that acid treatment also results in 3a having significant two-photon absorptivity, with a two-photon absorption cross-section of approximately 170 GM at 1020 nm (figure 3). Given the unsymmetrical design of the molecule, the two-photon absorption (2PA) spectrum agrees well with that of the linear absorption.
| 1.1 |
| 1.2 |
Scheme 1.

Synthesis of guaiazulene 3a.
Scheme 2.

Structures for guaiazulene derivative 3 and its conjugate acid formed upon exposure to TFA, 3aH+.
Figure 2.
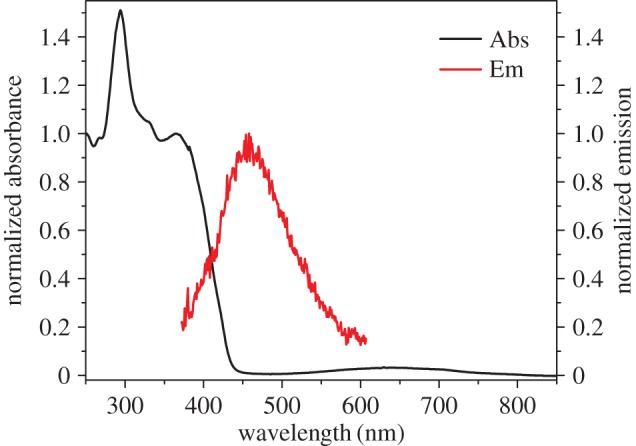
Absorption (black) and emission (red) spectra of 3a in dichloromethane (DCM).
Figure 3.
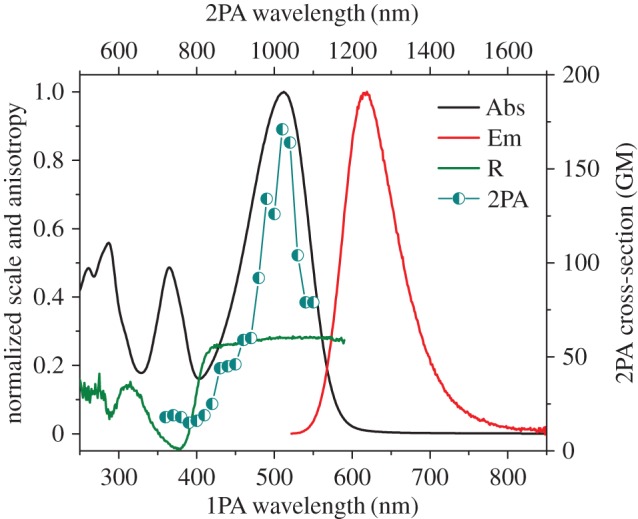
Absorption (black), emission (red), and 2PA spectra (blue circles) for 3aH+ in 10% TFA/DCM, and excitation anisotropy (green) in acidified silicone oil.
The quantum yields of photodecomposition measured for 3a and 3aH+ show significant disparity, separated by three orders of magnitude (table 1). Though a value is not explicitly stated, the photostability of guaiazulene has been investigated previously; the sample was seen to persist for a longer period of time than 1 at an equivalent or greater irradiance but no quantitative value was reported [20]. It is possible that change of the solvent from methanol to dichloromethane (DCM) is responsible for the discrepancy: DCM can promote photooxidation and C–Cl bonds can be broken using UV light, leading to reactive species and additional damage to the compound in solution [21]. In the case of 3aH+, longer wavelength light was used, thus avoiding the degradation of the solvent, and obtaining a value similar to those of other fluorescent dyes that have been previously reported [22].
Table 1.
Photophysical parameters measured for 3a and 3aH+ in DCM and 10% TFA/DCM, respectively.
| a (nm) | a (nm) | Δλb (nm) | c (M−1 cm−1) | Φdf | τe (ns) | ΦPh (10−6) | |
|---|---|---|---|---|---|---|---|
| 3a | 365, 630 | 458 | 93 | 29 000, 690 | <0.01 | —f | 1000 |
| 3aH+ | 512 | 619 | 93 | 25 000 | 0.12 | 0.87 | 1.5 |
aAbsorption and emission maxima ±1 nm.
bStokes shift ±2 nm.
cExtinction coefficients ±5%.
dFluorescence quantum yields ±10%.
eFluorescence lifetimes ±10%.
fNot determined.
Examination of the optimized structures for 3a and 3aH+ reveals that the neutral form does not exhibit a planar conformation (figure 4a), but, instead, a distinct dihedral angle, ϕ, between the guaiazulene moiety and the vinyl bond (ϕ ≈ 38°) is noted. When guaiazulene is protonated, the optimized structure indicates that planarity is restored (ϕ ≈ 1°, figure 4b), hence amplifying electron donation from the adjacent anethole group.
Figure 4.
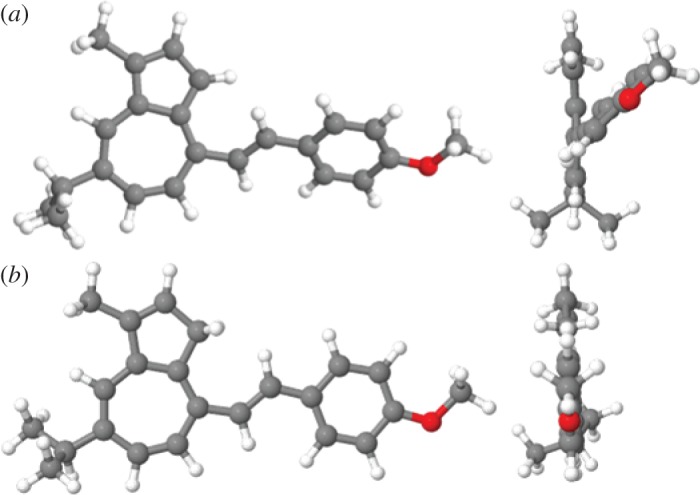
Optimized structures for 3a (a) and 3aH+ (b).
The ground state stationary dipole for the two forms is consistent in magnitude (1.90 D and 2.20 D for 3a and 3aH+, respectively) despite the presence of an electron deficient tropylium cation in 3aH+. However, the dipole of 3aH+ is polarized toward the cyclopentene ring rather than the anethole moiety.
The absorption spectra generated through time-dependent density functional theory (TD-DFT) calculations show that the weak intensity band of the S0 → S1 transition is present, though the bands at shorter wavelength are resolved better than seen experimentally (figure 5a; electronic supplementary material, table S1). In the case of 3aH+ (figure 5b; electronic supplementary material, table S2), the absorption band centred at approximately 600 nm is anticipated by the calculation; however, the short wavelength bands are less well aligned.
Figure 5.
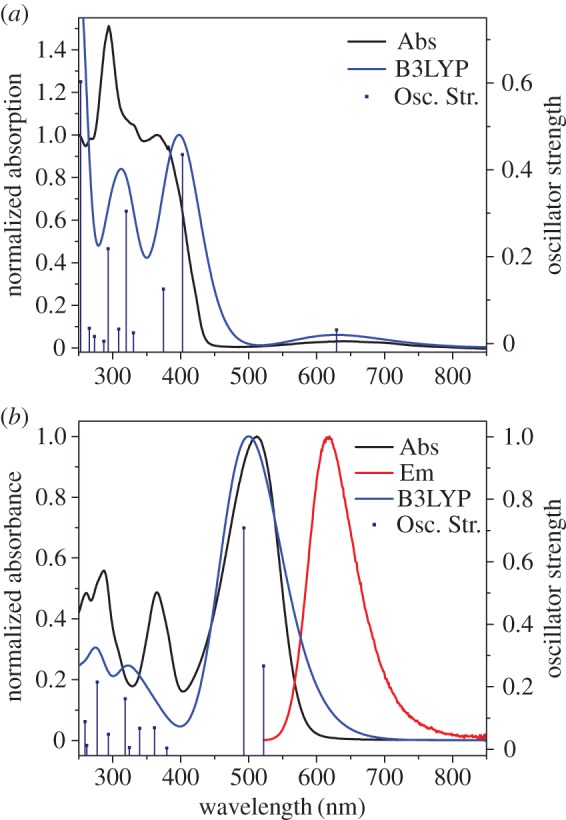
Calculated absorption spectra (blue line) and oscillator strengths (blue bars) overlaid with experimental absorption (black) and emission (red) spectra for 3a (a) and 3aH+ (b).
The consistency found between the experimental and calculated photophysical properties of 3a could, therefore, allow for prediction of the electronic properties of a series of structures similar to 3a and having various π-spacers (figure 6). Among potential spacers, phenyl rings offer a greater degree of rigidity over polyene groups; compared with these spacers, the fluorene moiety can be a better alternative as it confers higher rigidity given the bridging carbon in the 9-position. Alternatively, the use of thiophene, an isostere of benzene, is shown to result in a bathochromic shift of the linear absorption spectrum [21,22]. The length of conjugation is known to shift the absorption spectrum, but it can also lead to increased two-photon absorption cross-sections [23,24]. Eleven structures were studied in their neutral and protonated forms. Of this π-extended series, two structures were synthesized and their experimentally determined properties were compared with calculated properties.
Figure 6.

Compounds designed using 3 as a basis.
Analysis of the results from TD-DFT calculations (table 2) reveals that the varying length of the chromophore system has little effect on the ground state dipole of the neutral form for this series. However, the protonated forms of the longer systems (vi–xiv) show a marked increase relative to their neutral counterparts, ranging from approximately threefold to eightfold increases. There is also a notable trend in the dihedral angles observed for the optimized structures: the structures with a thiophene ring adjacent to the guaiazulene system (i, iv, v, x and xiii) do not exhibit a change in dihedral angle between these systems upon protonation, instead both forms are planar. Conversely, structures with either a phenyl ring or fluorene moiety adjacent to the guaiazulene undergo approximately 25° rotation to relax the dihedral angle upon protonation. A possible explanation is that thiophene withdraws electron density from the vinyl hydrogen closest to the guaiazulene, thereby facilitating a planar conformation of the neutral form. Structure xiv, however, appears to be an exception to this trend, behaving similarly to the phenyl-substituted guaiazulenes.
Table 2.
TD-DFT calculation data for the series of compounds shown in figure 6.
| Cmpd | dipole (D) | dihedral angle | first transition (nm) | (nm) |
|---|---|---|---|---|
| neutral forms | ||||
| i | 1.62 | 0.639 | 639 | 418 |
| ii | 3.13 | 23.0 | 650 | 459 |
| iii | 3.11 | 19.5 | 653 | 485 |
| iv | 3.14 | 0.074 | 677 | 493 |
| v | 4.32 | 0.001 | 679 | 510 |
| vi | 2.45 | 22.7 | 637 | 437 |
| vii | 3.68 | 24.1 | 644 | 476 |
| viii | 3.02 | 18.8 | 657 | 487 |
| ix | 2.59 | 37.0 | 647 | 405 |
| x | 3.59 | 0.553 | 682 | 519 |
| xi | 3.11 | 17.2 | 661 | 508 |
| xii | 2.00 | 23.1 | 656 | 448 |
| xiii | 3.15 | 0.050 | 686 | 540 |
| xiv | 1.39 | 17.8 | 685 | 557 |
| protonated forms | ||||
| iH+ | 1.39 | 0.024 | 527 | 483 |
| iiH+ | 8.34 | 5.05 | 753 | 738 |
| iiiH+ | 6.56 | 3.56 | 758 | 732 |
| ivH+ | 6.13 | 0.040 | 683 | 668 |
| vH+ | 4.24 | 0.017 | 688 | 668 |
| viH+ | 8.48 | 0.004 | 676 | 657 |
| viiH+ | 15.6 | 0.000 | 1009 | 999 |
| viiiH+ | 16.7 | 0.016 | 966 | 949 |
| ixH+ | 13.0 | 0.011 | 1048 | 1041 |
| xH+ | 13.9 | 0.011 | 847 | 842 |
| xiH+ | 26.1 | 0.012 | 1473 | 730 |
| xiiH+ | 23.9 | 0.001 | 1563 | 736 |
| xiiiH+ | 23.0 | 0.004 | 1273 | 688 |
| xivH+ | 19.1 | 0.006 | 1354 | 709 |
These results also include the first transition wavelength and the peak of the calculated absorption spectrum. Although there is some fine variation, the first transition of the neutral forms, corresponding to the S0 → S1 transition, remains in the region of 650–680 nm, consistent with experimental results for 3a and other studies of azulene derivatives [9,12,14]. Results generally indicate that the absolute λmax steadily increased as the length of the chromophore increased, as expected (figure 7a). This implies that the energy gap between S0 and S2 is decreasing while that between S0 and S1 remains constant.
Figure 7.
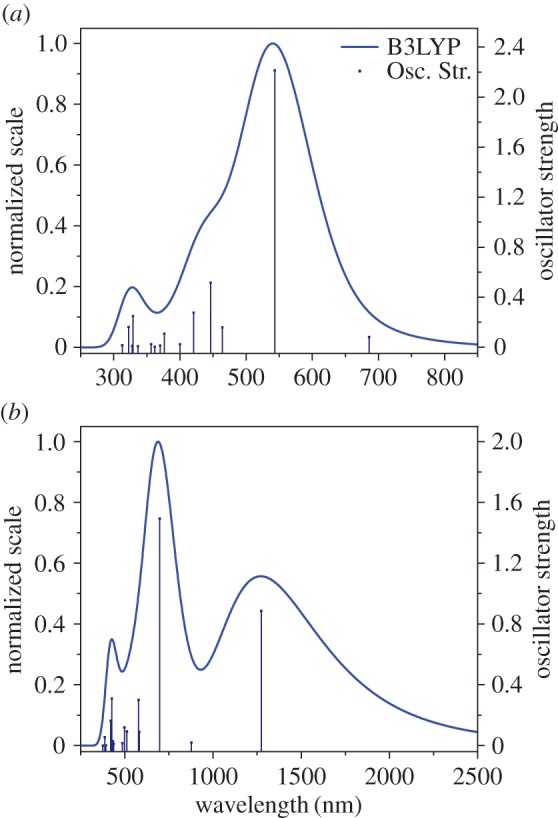
Calculated absorption spectra for xiii (a) and xiiiH+ (b) using the B3LYP method.
Protonation induces the same bathochromic shift seen for compound 3a in the calculated absorption data. Disappearance of the weak S0 → S1 band of the azulene at approximately 650 nm and emergence of a new band from the azulenium cation extending into the NIR are observed [23]. In the case of the longer species, such as xiiiH, the first transition no longer represents the peak absorption as it has been superseded by a higher transition (figure 7b).
To confirm the validity of these results, as well as probe further into the photophysical properties of these extended guaiazulene derivatives, compounds similar to structures vii and xiii were synthesized and characterized. These compounds were selected as they represent the midpoint and the extreme of the series, as well as having two and three moieties, respectively, in addition to guaiazulene. Given the increased length of the conjugated systems, and the inclusion of a thiophene moiety (in the case of xiii), these structures should have greater 2PA properties over 3a.
The approach for the synthesis of 3b is depicted in scheme 3. In this synthetic route, a Wittig reaction was employed to prepare styrene 4 by condensing benzaldehyde 2 with methyltriphenylphosphonium bromide under basic conditions. Alternatively 6, which was prepared by a lithium-halogen exchange reaction employing intermediate 5 in the presence of n-BuLi, was coupled with 4 to furnish the aldehyde 7. Finally, a condensation of 7 with guaiazulene 1 afforded the target compound 3b. It is noteworthy that the formation of 3b can be preliminarily observed as change in colour of the spot corresponding to the desired product from green to dark blue when exposing the compound spotted on a thin layer chromatography (TLC) plate to TFA vapour.
Scheme 3.

Synthetic scheme for compound 3b.
Similarly, the synthesis of guaiazulene 3c involved a convergent approach (scheme 4). Namely, intermediate 7 was transformed into the corresponding vinyl derivative 8; commercially available 5-bromo-thiophene-2-carboxyaldehyde 9 was condensed with guaiazulene 1 to afford 10 bearing a bromo substituent. The two components 8 and 10 were then subjected to Heck cross-coupling conditions to furnish π-extended guaiazulene 3c.
Scheme 4.

Synthetic scheme for compound 3c.
The experimental absorption spectra of 3b and 3c appear hypsochromically shifted in relation to their computationally determined counterparts (electronic supplementary material, tables S3–S6), though they share a somewhat similar structure (figures 8a and 9a). With the intense S0 → S2 bands pushed to lower wavelength, the S0 → S1 bands are more visible, appearing at approximately 650–660 nm in both instances. Weak emission was observed through excitation of the S0 → S2 band, though no emission was apparent for the S0 → S1 transition. Upon protonation, the absorption spectrum was altered in a manner similar to 3a, though there was no evidence of the absorption bands in the NIR (figures 8b and 9b). These protonated forms also fluoresce upon excitation of the azulenium cation bands at 586 nm and 664 nm, respectively (table 3).
Figure 8.
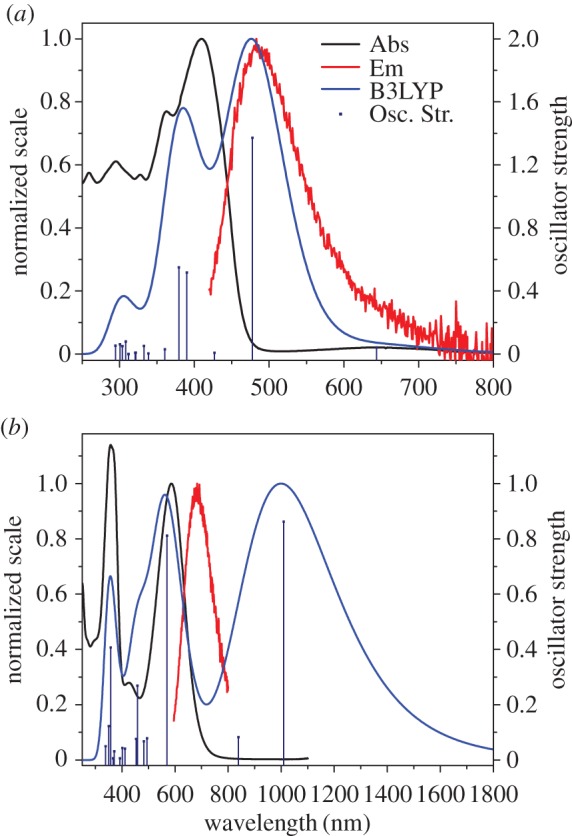
Absorption (black) and emission (red) spectra recorded for 3b in DCM (a) and 3bH+ in 10% TFA/DCM (b) overlaid with their respective calculated absorption spectra (blue line) and oscillator strengths (blue bars).
Figure 9.
Absorption (black) and emission (red) spectra recorded for 3c in DCM (a) and 3cH+ in 10% TFA/DCM (b) overlaid with their respective calculated absorption spectra (blue).
Table 3.
Photophysical parameters measured for 3b and 3c and 3bH+ and 3bH+ in DCM and 10% TFA/DCM, respectively.
| a (nm) | a (nm) | Δλb (nm) | c (M−1 cm−1) | Φfd | τe (ns) | ΦPh (10−6) | |
|---|---|---|---|---|---|---|---|
| 3b | 408, 644 | 500 | 92 | 31 800, 820 | 0.01 | —f | 85 |
| 3bH+ | 355, 586 | 700 | 114 | 38 000, 25 400 | 0.03 | 2.06 | —f |
| 3c | 448, 656 | 486 | 38 | 16 100, 470 | 0.10 | —f | 780 |
| 3cH+ | 385, 664 | 764 | 100 | 18 200, 16 900 | 0.01 | 1.52 | 13 |
aAbsorption and emission maxima ±1 nm.
bStokes shift ±2 nm.
cExtinction coefficients ±5%.
dFluorescence quantum yields ±10%.
eFluorescence lifetimes ±10%.
fNot determined.
It was shown that lengthening the conjugated system of the chromophore increased the molar absorptivity of 3b and 3bH+ and the inclusion of a thiophene ring imposed a detrimental effect on 3c and 3cH+ molar absorptivity (tables 1 and 3). These effects are unexpected as the addition of a thiophene ring has been noted to generally increase molar absorptivity [24,25]; while increasing the length of the conjugated system has been shown to have the opposite effect [26]. Across the series of compounds, trends in fluorescence quantum yield can be seen (tables 1 and 3). The decreased fluorescence quantum yield with increased conjugation length can be correlated to the higher degree of flexibility in the structure, allowing for more rotation and vibration, though the inversion of this trend in the neutral forms is equally counterintuitive.
With the increased conjugation length, an increase in photodecomposition quantum yield also occurs (table 3). Although a longer wavelength was utilized for the excitation (405 nm), the neutral forms exhibit lower photostability than their conjugate acids. Despite this, 10% TFA/DCM solutions of the compounds were noted to degrade between experiments, though this is more likely to be a case of chemical stability in the present of an acid.
The excitation anisotropy trace calculated for 3bH+ presents a gradient within the region of the long wavelength absorption band, inferring that this is an overlap of more than one electronic transition (figure 10a). The two-photon absorption (2PA) spectrum obtained in 10% TFA/DCM of 3bH+ seems erratic, which may be explained by a variety of possible transitions. Unlike 3bH+, the excitation anisotropy of 3cH+ plateaus within the long wavelength absorption band, in accordance with a sole transition. This plateau also suggests that the absorption and emission are collinear, given its anisotropy value of 0.4 [18]. The 2PA spectrum of 3cH+ initially shows reasonable agreement with the linear absorption spectrum, though appears erratic beyond 1300 nm (figure 10b).
Figure 10.
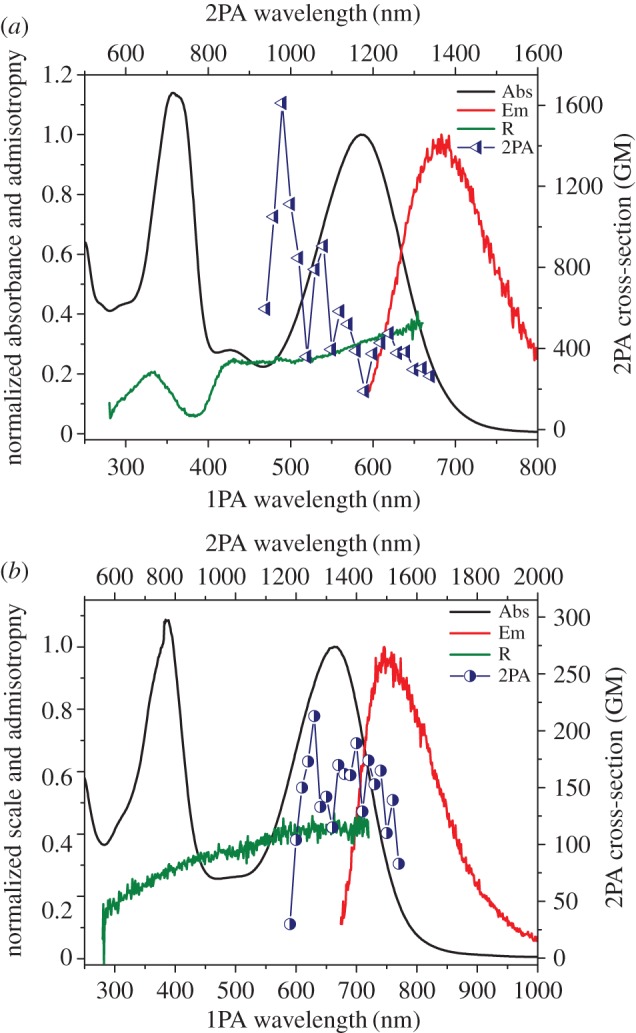
Absorption (black), emission (red), and 2PA spectra (dark blue points) for 3bH+ (a) and 3cH+ (b) in 10% TFA/DCM, and excitation anisotropy trace (dark green) in acidified silicone oil.
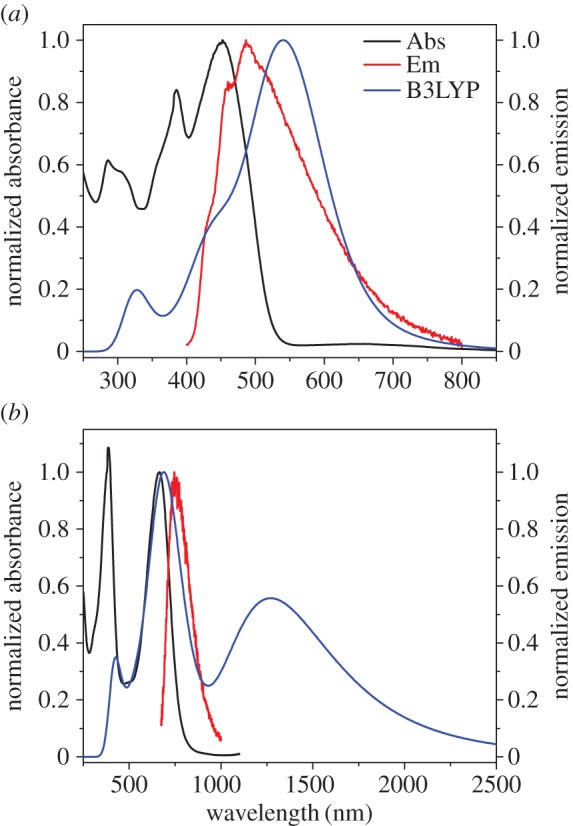
In an effort to resolve the difference observed between the experimental results and those of the B3LYP TD-DFT calculation (figures 7 and 8), a series of additional methods were implemented by varying the proportion of Hartree–Fock (HF) calculations. Ab initio methods, like HF, yield results that are less erratic than semiempirical methods, though at the cost of time [27]. Hybrid functions, including B3LYP (20% HF), have been developed to bridge this gap, where the ratios can be adjusted. With respect to spectroscopic data, increasing the ab initio portion decreases, or blue shifts, the wavelengths of the transitions.
When the TD-DFT calculation was repeated for 3bH+ using the M06-HF method (100% HF), the calculated spectrum was in closer semblance to the experimental spectrum than previous results (figure 11a). The first transition appeared hypsochromically shifted from the experimental main absorption band, as is the shorter wavelength transition. There is greater difference in the experimental and calculated 2PA spectrum; in the region of the main linear absorption band, accounting for the wavelength shift, they were similar, though at higher energy the data diverges.
Figure 11.
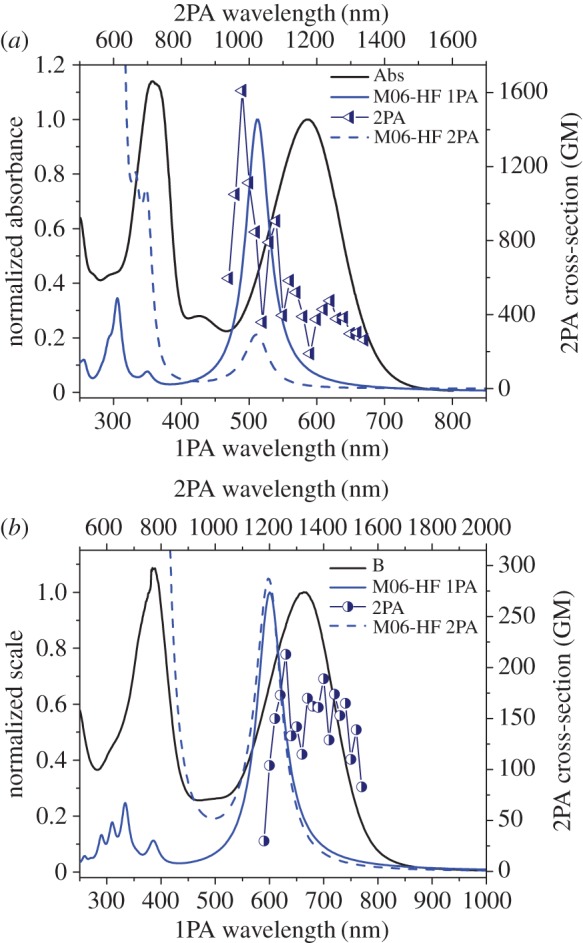
Experimental (black) and M06-HF calculated one-photon absorption (solid blue), and experimental (dark blue points) and M06-HF calculated two-photon absorption (dashed blue) for 3bH+ (a) and 3cH+ (b).
Likewise, the results obtained for 3cH+ using the same M06-HF method show a hypsochromic shift compared with the experimental data, for both the one- and two-photon absorption spectra (figure 11b). Here, the 2PA data are more closely matched, with results becoming erratic towards the limit of the germanium detectors (1600 nm).
The use of a method with a higher proportion of HF provided greater similarity between the calculated and experimental results for these compounds because of the presence of the large dipole moments that are more akin to charge transfer, a phenomenon that traditional DFT methods struggle to depict [28,29].
2. Experimental
2.1. General information
Reagents and solvents were purchased from commercial sources and used without further purification unless otherwise specified. TLC was performed on SiO2-60 F254 aluminium plates with visualization by UV light or TFA staining. Flash column chromatography was performed using a Teledyne CombiFlash Rf 200 unit. Melting points (mp.) were determined on a Fisher Scientific melting point apparatus. In total, 400 (100) MHz 1H (13C) NMR spectra were recorded on Bruker Avance III 400 spectrometer. In total, 500 (125) MHz 1H (13C) NMR were recorded on Varian VNMRS 500 spectrometer. Chemical shifts (δ) are given in parts per million (ppm) relative to TMS and referenced to residual protonated solvent (CDCl3: δH 7.26 ppm, δC 77.23 ppm). Abbreviations used are s (singlet), d (doublet), t (triplet), q (quartet), quin (quintet), hp (heptet), b (broad) and m (multiplet). ESI-TOF-MS spectra were recorded on Agilent 6210 TOF spectrometer. 4-(Hexyloxy)benzaldehyde 2 [30], 1-(hexyloxy)-4-vinylbenzene 4 [28], 7-bromo-9,9-dihexyl-9H-fluorene-2-carbaldehyde 6 [29] were prepared according to literature procedures.
2.2. Synthesis
2.2.1. (E)-9,9-Dihexyl-7-(4-(hexyloxy)styryl)-9H-fluorene-2-carbaldehyde (7)
Pd(OAc)2 (0.05 mmol) was added to a solution of 1-(hexyloxy)-4-vinylbenzene 4 (1.50 mmol), 7-bromo-9,9-dihexyl-9H-fluorene-2-carbaldehyde 6 (1.00 mmol), K2CO3 (2.0 mmol) and Bu4NBr (2.00 mmol) in DMF (10 ml) under Ar. The reaction mixture was heated at 140°C for 8 h, upon which the solvent was evaporated, and the resulting crude was suspended in DCM (25 ml) and passed through a silica plug (25 g). The solvent was evaporated and the crude product was purified using flash column chromatography (hexanes : DCM, 9 : 1) to afford the title product as colourless oil that crystallized upon standing (60%). 1H NMR (400 MHz, CDCl3) δ 10.05 (s, 1H), 7.87 (dd, J = 1.4, 0.7 Hz, 1H), 7.84 (dd, J = 7.8, 1.4 Hz, 1H), 7.79 (dd, J = 7.7, 0.6 Hz, 1H), 7.74–7.71 (m, 1H), 7.20–7.04 (m, 2H), 6.94–6.88 (m, 2H), 3.98 (t, J = 6.6 Hz, 2H), 2.03 (ddd, J = 10.5, 6.4, 3.9 Hz, 4H), 1.79 (ddt, J = 9.0, 7.9, 6.4 Hz, 2H), 1.53–1.42 (m, 2H), 1.40–1.29 (m, 4H), 1.21–0.98 (m, 13H), 0.97–0.87 (m, 2H), 0.74 (t, J = 7.0 Hz, 6H), 0.61 (tq, J = 11.1, 7.0 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 192.3, 159.1, 152.7, 147.4, 138.8, 135.1, 130.6, 129.8, 128.8, 127.8, 126.6, 125.6, 123.0, 121.2, 120.6, 119.8, 114.8, 68.1, 55.2, 40.3, 31.6, 29.6, 29.3, 25.7, 23.8, 22.6, 14.1.
2.2.2. (E)-9,9-Dihexyl-2-(4-(hexyloxy)styryl)-7-vinyl-9H-fluorene (8)
The title compound was prepared starting from intermediate 7 according to literature procedure [31]. Colourless oil (70%). 1H NMR (400 MHz, CDCl3) δ 7.66 (dd, J = 7.8, 6.1 Hz, 2H), 7.55–7.37 (m, 6H), 7.20–7.04 (m, 2H), 6.94 (d, J = 8.7 Hz, 2H), 6.84 (dd, J = 17.6, 10.9 Hz, 1H), 5.83 (d, J = 17.6 Hz, 1H), 5.29 (d, J = 10.9 Hz, 1H), 4.02 (t, J = 6.6 Hz, 2H), 2.10–1.93 (m, 4H), 1.83 (dt, J = 14.7, 6.7 Hz, 2H), 1.51 (ddt, J = 13.0, 9.7, 5.8 Hz, 2H), 1.39 (dq, J = 7.2, 3.6 Hz, 4H), 1.21–1.03 (m, 12H), 1.00–0.93 (m, 3H), 0.79 (t, J = 7.0 Hz, 6H), 0.74–0.64 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 159.0, 151.7, 151.5, 141.0, 140.4, 137.6, 136.8, 136.5, 130.3, 127.8, 127.7, 127.3, 125.5, 125.4, 120.6, 120.6, 120.0, 119.8, 114.9, 113.1, 68.3, 55.1, 40.7, 31.8, 31.7, 29.9, 29.4, 25.9, 23.9, 22.8, 22.8, 14.2, 14.2.
2.3. General procedure for the preparation of guaiazulenes 3a-c and 10
A solution of t-BuOK (3.0 mmol) in t-amyl alcohol (10 ml) was heated at 105°C for 30 min. To this solution was added the appropriate aldehyde (3.0 mmol) followed by the dropwise addition of a solution of guaiazulene 1 (1.0 mmol) in t-amyl alchol (5 ml). The solution was heated for 3 h at 105°C, then cooled to room temperature and poured into dilute HCl (1 N). The mixture was extracted with DCM (2 × 25 ml) and the solvent was removed under reduced pressure. The resulting crude mixture was purified using Flash column chromatography using hexanes and EtOAc (0–3%).
2.3.1. (E)-2-Bromo-5-(2-(7-isopropyl-1-methylazulen-4-yl)vinyl)thiophene (10)
Green needles (mp. 72–74°C, 25%). 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 1.9 Hz, 1H), 7.80–7.68 (m, 2H), 7.51 (dd, J = 11.1, 2.0 Hz, 1H), 7.47–7.37 (m, 3H), 7.04 (d, J = 3.8 Hz, 1H), 6.94 (d, J = 4.0 Hz,1H), 3.13 (p, J = 6.9 Hz, 1H), 2.71 (s, 3H), 1.42 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 144.6, 140.9, 140.4, 137.1, 136.7, 136.5, 134.9, 133.3, 130.8, 129.5, 127.6, 126.2, 126.1, 119.8, 112.6, 112.0, 38.5, 29.9, 13.2. HRMS (ESI) (m/z): [M + H]+ calcd for C20H19BrS C28H35O 370.0391; found, 370.0408.
2.3.2. (E)-4-(4-(Hexyloxy)styryl)-7-isopropyl-1-methylazulene (3a)
Green needles (mp. 55.0–58.0°C, 43%). 1H NMR (CDCl3, 400 MHz) δ 8.21 (s, 1H), 7.92 (d, J = 16.1 Hz, 1H), 7.69 (d, J = 3.9 Hz, 1H), 7.60 (d, J = 8.6 Hz, 2H), 7.57–7.49 (m, 3H), 7.38 (d, J = 16.1 Hz, 1H), 6.97 (d, J = 8.6 Hz, 2H), 4.04 (t, J = 6.6 Hz, 2H), 3.14 (sept, J = 6.9 Hz, 1H), 2.72 (s, 3H), 1.84 (dd, J = 8.5, 6.4 Hz, 2H), 1.56–1.33 (m, 13H), 1.00–0.93 (m, 2H). 13C NMR (CDCl3, 100 MHz) δ 159.7, 142.4, 139.9, 136.8, 136.6, 136.3, 135.0, 133.7, 133.1, 130.0, 128.5, 127.3, 125.9, 120.3, 115.0, 112.0, 68.3, 38.4, 31.7, 29.9, 29.4, 25.9, 24.9, 22.8, 14.2, 13.2. HRMS (ESI) (m/z): [M + H]+ calcd for C28H35O 387.2682; found, 387.2688.
2.3.3. 9,9-Dihexyl-2-((E)-4-(hexyloxy)styryl)-7-((E)-2-(7-isopropyl-1-methylazulen-4-yl)vinyl)-9H-fluorene (3b)
Green solid (25%, 72.0–75.0°C). 1H NMR (CDCl3, 400 MHz): δ 8.20 (d, J = 1.4 Hz, 1H), 8.06 (d, J = 16.1 Hz, 1H), 7.73–7.61 (m, 4H), 7.59–7.44 (m, 9H), 7.19–7.04 (m, 2H), 6.95–6.87 (m, 2H), 3.99 (t, J = 6.6 Hz, 2H), 3.12 (p, J = 6.9 Hz, 1H), 2.70 (s, 3H), 2.04 (dd, J = 11.3, 5.7 Hz, 4H), 1.87–1.73 (m, 2H), 1.48 (dd, J = 10.5, 4.8 Hz, 2H), 1.40 (d, J = 6.9 Hz, 6H), 1.38–1.31 (m, 4H), 1.16–1.03 (m, 11H), 0.95–0.90 (m, 4H), 0.81–0.65 (m, 10H). 13C NMR (CDCl3, 100 MHz): δ 159.2, 152.1, 152.0, 140.3, 137.0, 136.4, 135.2, 135.0, 130.5, 128.9, 128.1, 128.0, 127.4, 126.2, 125.8, 121.7, 120.9, 120.6, 120.4, 120.3, 115.1, 77.6, 68.5, 55.4, 40.9, 38.7, 32.0, 31.9, 30.1, 29.6, 26.1, 25.1, 24.2, 23.0, 14.4, 14.4, 13.4. Calcd for C55H68O [M + H]+ = 745.5343; found [M + H]+ = 745.5189.
2.3.4. 2-((E)-2-(9,9-Dihexyl-7-((E)-4-(hexyloxy)styryl)-9H-fluoren-2-yl)vinyl)-5-((E)-2-(7-isopropyl-1-methylazulen-4-yl)vinyl)thiophene (3c)
Green solid (39%, 154.0–156.0°C). 1H NMR (CDCl3, 400 MHz): δ.18 (d, J = 1.8 Hz, 1H), 7.82 (d, J = 15.8 Hz, 1H), 7.73–7.60 (m, 3H), 7.53–7.41 (m, 10H), 7.27 (dd, J = 16.0, 0.7 Hz, 1H), 7.19–7.00 (m, 5H), 6.90 (d, J = 8.7 Hz, 2H), 3.98 (t, J = 6.6 Hz, 2H), 3.10 (p, J = 6.9 Hz, 1H), 2.72–2.64 (s, 3H), 2.07–1.93 (m, 4H), 1.85–1.75 (m, 2H), 1.52–1.43 (m, 2H), 1.37 (m, 10H), 1.19–0.99 (m, 11H), 0.95–0.88 (m, 4H), 0.80–0.73 (t, J = 8.0 Hz, 6H), 0.73–0.63 (m, 4H). Calcd for C55H68O [M + H]+ = 852.5304; found [M + H]+ = 852.5373.
2.4. Photophysical characterization
Linear photophysical properties were measured in solution (approx. 10−6 M) in 10 mm quartz cuvettes using spectroscopic grade solvent. An Agilent 8453 was used to collect absorption spectra, while an Edinburgh Instruments FLS 980 for steady-state luminescence emission, excitation anisotropy and fluorescence lifetimes. These measurements used a red-sensitive photomultiplier tube (PMT), and a liquid-nitrogen-cooled Hamamatsu R5509–72; all measurements were corrected for detector response.
Fluorescence quantum yields were calculated using a relative method, with 9,10-DPA (Φf = 0.95) as a reference [18]. Anisotropy measurements were performed in a viscous solvent, namely silicone oil, to hamper the rotational relaxation of the molecules. Fluorescence lifetimes were measured using a 470 nm laser for 3aH, a 510 nm laser for 3bH and a 670 nm laser for 3cH.
Photodecomposition quantum yields, ΦPh, were measured by irradiating into the main absorption band of solutions with a LOCTITE 97034 UV-lamp (λex = 366 nm, I0(λ) ≈ 13 mW cm−2), a green diode laser (λex = 532 nm, I0(λ) ≈ 98 mW cm−2), or a red diode laser (λex = 650 nm, I0(λ) ≈ 62 mW cm−2). Spectra were recorded at incremental time intervals, and the data were utilized in equation (2.1), where D(λ, 0) and D(λ,tir) are the initial and final optical density of the solution, (λ) is the extinction coefficient (dm3 mol−1 cm−1), t is irradiation time (s) and λ is excitation wavelength (cm), NA is Avogadro's number, tir is total irradiation time, I0(λ) is the spectral distribution of the excitation irradiance [32].
| 2.1 |
Two-photon absorption spectra were collected through an open aperture z-scan set-up (previously detailed [33]) using solutions (10−2 M) in a 1 mm cuvette translated through the focal point of the output beam of an optical parametric amplifier (OPA) pumped by a 1 kHz, approximately 100 fs Coherent, Inc. Legend Elite, that was seeded by a Coherent, Inc. Mira Ti:sapphire laser [34].
2.5. Quantum chemical calculations
Taking the output from the GaussView5 program, structures were optimized to the B3LYP/D95* level with the Gaussian09 software package [35]. TD-DFT calculations looking at the first 15 transitions utilized these optimized conformations, and, initially, the B3LYP method. Further calculations used M05, M06, M06-2X and M06-HF methods. The alkoxy and alkyl chains in the para-position of phenyl and 9-position of fluorene were shortened to methoxy and methyl groups, respectively, due to the minimal impact these groups have on the results of the calculations [36]. The calculated 2PA spectrum was determined from the permanent and state-to-state transition dipoles, which were obtained using a posteriori Tamm–Dancoff approximation (ATDA) in a locally modified version of Gaussian09 [37].
3. Conclusion
Through the use of quantum chemical calculations, a library of guaiazulene derivatives based on an initial synthesized compound led to the discovery of molecules with high linear absorption dipoles. Measurements on two synthesized structures from the series illustrated issues in the calculations performed. As a result of the numerous terms that factor into nonlinear absorption, the larger dipole did not correspond to the great 2PA.
Despite the discrepancies between experimental and computational results, the use of quantum chemical calculations to consider the properties of designed, yet unsynthesized, compounds is promising. Conducting in silico analysis and prediction, combined with deriving materials from renewable resources, is a noteworthy course of action for chemists in this increasingly ecologically and sustainability-conscious world.
Supplementary Material
Acknowledgements
We thank Prof. Artëm E. Masunov (University of Central Florida) for helpful discussions.
Data accessibility
Computational data and molecular orbital illustrations along with 1H and 13C NMR spectra are provided in the electronic supplementary material available from the publisher.
Authors' contributions
E.H.G.Z. carried out the molecular laboratory work, including synthesis and characterization, participated in data analysis, participated in the design of the study and helped draft the manuscript; A.W.W. and M.V.B. conducted computational and photophysical experimental work, participated in data analysis, participated in the design of the study and helped draft the manuscript; K.D.B. conceived and designed the study, coordinated the study and drafted the manuscript. All authors gave final approval for publication.
Competing interests
The authors declare no competing interests.
Funding
K.D.B. acknowledges support from the National Science Foundation (CBET-1517273 and CHE-0832622). M.V.B. acknowledges support from the National Academy of Sciences of Ukraine (grants 1.4.1.В/153 and VC/157), and FP7–Marie Curie Actions: ITN ‘Nano2Fun’ GA #607721.
References
- 1.Anastas PT, Warner JC. 1998. Green chemistry: theory and practice. Oxford, UK: Oxford University Press. [Google Scholar]
- 2.Das S, Nag A, Sadhu KK, Goswami D, Bharadwaj PK. 2007. Metal induced enhancement of fluorescence and modulation of two-photon absorption cross-section with a donor–acceptor–acceptor–donor receptor. J. Organomet. Chem. 692, 4969–4977. (doi:10.1016/j.jorganchem.2007.07.013) [Google Scholar]
- 3.Moreshead WV, Przhonska OV, Bondar MV, Kachkovski AD, Nayyar IH, Masunov AE, Woodward AW, Belfield KD. 2013. Design of a new optical material with broad spectrum linear and two-photon absorption and solvatochromism. J. Phys. Chem. C 117, 23 133–23 147. (doi:10.1021/jp406500t) [Google Scholar]
- 4.Moity L, Molinier V, Benazzouz A, Barone R, Marion P, Aubry J-M. 2014. In silico design of bio-based commodity chemicals: application to itaconic acid based solvents. Green Chem. 16, 146–160. (doi:10.1039/C3GC41442F) [Google Scholar]
- 5.Monti A, de Groot HJM, Buda F. 2014. In-silico design of a donor–antenna–acceptor supramolecular complex for photoinduced charge separation. J. Phys. Chem. C 118, 15 600–15 609. (doi:10.1021/jp505105a) [Google Scholar]
- 6.Collins C, Dyer MS, Demont A, Chater PA, Thomas MF, Darling GR, Claridge JB, Rosseinsky MJ. 2014. Computational prediction and experimental confirmation of B-site doping in YBa2Fe3O8. Chem. Sci. 5, 1493–1505. (doi:10.1039/c3sc52734d) [Google Scholar]
- 7.Harmon AD, Weisgraber KH, Weiss U. 1980. Preformed azulene pigments of Lactarius indigo (Schw.) Fries (Russulaceae, Basidiomycetes). Experientia 36, 54–56. (doi:10.1007/BF02003967) [Google Scholar]
- 8.Fusetani N, Matsunaga S, Konosu S. 1981. Bioactive marine metabolites I. Isolation of guaiazulene from the gorgonianEuplexaura erecta. Experientia 37, 680–681. (doi:10.1007/BF01967917) [Google Scholar]
- 9.Amir E, Amir RJ, Campos LM, Hawker CJ. 2011. Stimuli-responsive azulene-based conjugated oligomers with polyaniline-like properties. J. Am. Chem. Soc. 133, 10 046–10 049. (doi:10.1021/ja203267g) [DOI] [PubMed] [Google Scholar]
- 10.Wang F, Lai Y-H. 2003. Conducting azulene−thiophene copolymers with intact azulene units in the polymer backbone. Macromolecules 36, 536–538. (doi:10.1021/ma025662i) [Google Scholar]
- 11.Shevyakov SV, Li H, Muthyala R, Asato AE, Croney JC, Jameson DM, Liu RSH. 2003. Orbital control of the color and excited state properties of formylated and fluorinated derivatives of azulene. J. Phys. Chem. A 107, 3295–3299. (doi:10.1021/jp021605f) [Google Scholar]
- 12.Koch M, Blacque O, Venkatesan K. 2013. Impact of 2,6-connectivity in azulene: optical properties and stimuli responsive behavior. J. Mater. Chem. C 1, 7400–7408. (doi:10.1039/c3tc31610f) [Google Scholar]
- 13.Amir E, Murai M, Amir RJ, Cowart JS, Chabinyc ML, Hawker CJ. 2014. Conjugated oligomers incorporating azulene building blocks—seven- vs. five-membered ring connectivity. Chem. Sci. 5, 4483–4489. (doi:10.1039/C4SC02210F) [Google Scholar]
- 14.Wang X, Ng JK-P, Jia P, Lin T, Cho CM, Xu J, Lu X, He C. 2009. Synthesis, electronic, and emission spectroscopy, and electrochromic characterization of azulene−fluorene conjugated oligomers and polymers. Macromolecules 42, 5534–5544. (doi:10.1021/ma900847r) [Google Scholar]
- 15.Dias JR. 2007. Electronic and structural properties of biazulene, terazulene, and polyazulene isomers. J. Phys. Org. Chem. 20, 395–409. (doi:10.1002/poc.1159) [Google Scholar]
- 16.Dutta S, Lakshmi S, Pati SK. 2008. Comparative study of electron conduction in azulene and naphthalene. Bull. Mater. Sci. 31, 353–358. (doi:10.1007/s12034-008-0055-8) [Google Scholar]
- 17.Ghazvini Zadeh EH, Tang S, Woodward AW, Liu T, Bondar MV, Belfield KD. 2015. Chromophoric materials derived from a natural azulene: syntheses, halochromism and one-photon and two-photon microlithography. J. Mater. Chem. C 3, 8495–8503. (doi:10.1039/C5TC01459J) [Google Scholar]
- 18.Lakowicz JR. 2007. Principles of fluorescence spectroscopy. Berlin, Germany: Springer. [Google Scholar]
- 19.Lorentz HA, Teubner BG. 1909. The theory of electrons and its applications to the phenomena of light and radiant heat: a course of lectures delivered in Columbia University, New York, in March and April 1906. London, UK: B. G. Teubner; David Nutt; G. E. Stechert, Leipzig. [Google Scholar]
- 20.Fiori J, Gotti R, Albini A, Cavrini V. 2008. Study on the photostability of guaiazulene by high-performance liquid chromatography/mass spectrometry and gas chromatography/mass spectrometry. Rapid Comm. Mass Spectrom. 22, 2698–2706. (doi:10.1002/rcm.3661) [DOI] [PubMed] [Google Scholar]
- 21.Dean JA, Lange NA. 1999. Handbook of chemistry, 15th edn New York, NY: McGraw-Hill. [Google Scholar]
- 22.Belfield KD, Bondar MV, Morales AR, Padilha LA, Przhonska OV, Wang X. 2011. Two-photon STED spectral determination for a new V-shaped organic fluorescent probe with efficient two-photon absorption. ChemPhysChem 12, 2755–2762. (doi:10.1002/cphc.201100456) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang F, Lin TT, He C, Chi H, Tang T, Lai Y-H. 2012. Azulene-containing organic chromophores with tunable near-IR absorption in the range of 0.6 to 1.7 μm. J. Mater. Chem. 22, 10 448–10 451. (doi:10.1039/c2jm31098h) [Google Scholar]
- 24.Wang S, Guo J, He L, Wang H, Zhao J, Lu C. 2013. Influence of thiophene and benzene unit in triphenylamine dyes on the performance of dye-sensitized solar cells. Synth. Met. 168, 1–8. (doi:10.1016/j.synthmet.2013.02.010) [Google Scholar]
- 25.Zhao L, Wang W, Yuan M-S. 2015. Asymmetric multibranched conjugated molecules: synthesis, structure and photophysical properties. Spectrochim. Acta Mol. Biomol. Spectrosc. 135, 63–68. (doi:10.1016/j.saa.2014.06.153) [DOI] [PubMed] [Google Scholar]
- 26.Padilha LA, et al. 2010. Efficient two-photon absorbing acceptor-π-acceptor polymethine dyes. J. Phys. Chem. A 114, 6493–6501. (doi:10.1021/jp100963e) [DOI] [PubMed] [Google Scholar]
- 27.Young DC. 2001. Computational chemistry: a practical guide for applying techniques to real world problems (e-book). John Wiley & Sons, Inc (doi:10.1002/0471220655.fmatter_indsub) [Google Scholar]
- 28.Ferla S, Aboraia AS, Brancale A, Pepper CJ, Zhu J, Ochalek JT, DeLuca HF, Simons C. 2014. Small-molecule inhibitors of 25-hydroxyvitamin D-24-hydroxylase (CYP24A1): synthesis and biological evaluation. J. Med. Chem. 57, 7702–7715. (doi:10.1021/jm5009314) [DOI] [PubMed] [Google Scholar]
- 29.Yue X, Armijo Z, King K, Bondar MV, Morales AR, Frazer A, Mikhailov IA, Przhonska OV, Belfield KD. 2015. Steady-state and femtosecond transient absorption spectroscopy of new two-photon absorbing fluorene-containing quinolizinium cation membrane probes. ACS Appl. Mater. Interfaces 7, 2833–2846. (doi:10.1021/am508093p) [DOI] [PubMed] [Google Scholar]
- 30.Mangalum A, Gilliard RJ Jr, Hanley JM, Parker AM, Smith RC. 2010. Metal ion detection by luminescent 1,3-bis(dimethylaminomethyl) phenyl receptor-modified chromophores and cruciforms. Org. Biomol. Chem. 8, 5620–5627. (doi:10.1039/c0ob00156b) [DOI] [PubMed] [Google Scholar]
- 31.Tian Y, Chen C-Y, Yang C-C, Young AC, Jang S-H, Chen W-C, Jen AKY. 2008. 2-(2′-Hydroxyphenyl)benzoxazole-containing two-photon-absorbing chromophores as sensors for zinc and hydroxide ions. Chem. Mater. 20, 1977–1987. (doi:10.1021/cm702527m) [Google Scholar]
- 32.Corredor CC, Belfield KD, Bondar MV, Przhonska OV, Yao S. 2006. One- and two-photon photochemical stability of linear and branched fluorene derivatives. J. Photochem. Photobiol. A 184, 105–112. (doi:10.1016/j.jphotochem.2006.03.036) [Google Scholar]
- 33.Belfield KD, Bondar MV, Morales AR, Yue X, Luchita G, Przhonska OV, Kachkovsky OD. 2012. Two-photon absorption and time-resolved stimulated emission depletion spectroscopy of a new fluorenyl derivative. ChemPhysChem 13, 3481–3491. (doi:10.1002/cphc.201200405) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belfield KD, Bondar MV, Hernandez FE, Przhonska OV, Yao S. 2007. Two-photon absorption cross section determination for fluorene derivatives: analysis of the methodology and elucidation of the origin of the absorption processes. J. Phys. Chem. B 111, 12 723–12 729. (doi:10.1021/jp074456f) [DOI] [PubMed] [Google Scholar]
- 35.Frisch MJ, et al. 2009. Gaussian 09, revision A.02. Wallingford, CT: Gaussian, Inc.
- 36.Belfield KD, Bondar MV, Hernandez FE, Masunov AE, Mikhailov IA, Morales AR, Przhonska OV, Yao S. 2009. Two-photon absorption properties of new fluorene-based singlet oxygen photosensitizers. J. Phys. Chem. C 113, 4706–4711. (doi:10.1021/jp8102832) [Google Scholar]
- 37.Mikhailov IA, Bondar MV, Belfield KD, Masunov AE. 2009. Electronic properties of a new two-photon absorbing fluorene derivative: the role of Hartree–Fock exchange in the density functional theory design of improved nonlinear chromophores. J. Phys. Chem. C 113, 20 719–20 724. (doi:10.1021/jp906875b) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Computational data and molecular orbital illustrations along with 1H and 13C NMR spectra are provided in the electronic supplementary material available from the publisher.