

Abstract
The Epstein-Barr Nuclear Antigen 1 (EBNA1) is a critical protein encoded by the Epstein-Barr Virus (EBV). During latent infection, EBNA1 is essential for DNA replication and transcription initiation of viral and cellular genes and is necessary to immortalize primary B-lymphocytes. Nonetheless, the concept of EBNA1 as drug target is novel. Two EBNA1 crystal structures are publicly available and the first small-molecule EBNA1 inhibitors were recently discovered. However, no systematic studies have been reported on the structural details of EBNA1 “druggable” binding sites. We conducted computational identification and structural characterization of EBNA1 binding pockets, likely to accommodate ligand molecules (i.e. “druggable” binding sites). Then, we validated our predictions by docking against a set of compounds previously tested in vitro for EBNA1 inhibition (PubChem AID-2381). Finally, we supported assessments of pocket druggability by performing induced fit docking and molecular dynamics simulations paired with binding affinity predictions by Molecular Mechanics Generalized Born Surface Area calculations for a number of hits belonging to druggable binding sites. Our results establish EBNA1 as a target for drug discovery, and provide the computational evidence that active AID-2381 hits disrupt EBNA1:DNA binding upon interacting at individual sites. Lastly, structural properties of top scoring hits are proposed to support the rational design of the next generation of EBNA1 inhibitors.
Keywords: EBNA1 “druggability” assessment, Molecular docking, Computational approaches to pocket finding, Structure-based drug design
Introduction
Epstein-Barr Nuclear Antigen 1 (EBNA1) is a viral onco-protein expressed by the lymphotropic gamma-herpes virus, Epstein Barr Virus (EBV) or human herpes virus 4 (HHV-4) [1], whose infections are associated with many serious diseases and are very common worldwide. Indeed, the Centers for Disease Control estimated an infection rate of 95 % in the adult population of the United States. When one is infected during childhood, usually only mild symptoms are presented. However, if one is infected as a teenager or young adult, EBV infection may cause severe conditions, like infectious mononucleosis (IM). Once infected, EBV establishes a lifelong latency in infected cells. In the majority of cases, however, EBV infections remain asymptomatic for many years, the EBV genome persisting as a circular, multi-copy episome. In rare instances, however, EBV is associated with cancer, including endemic Burkitt’s lymphoma, Hodgkin’s disease, nasopharyngeal carcinoma and several lymphomas associated to immunosuppressed patients.
EBNA1 is the only viral protein that is consistently expressed in all forms of latency and acts to promote survival and proliferation in infected cells. In particular, EBNA1 binds to DNA in a sequence specific manner to a 18 base-pair (bp) palindromic recognition site present at the EBV origin of plasmid replication (OriP) [1, 2], the site of initiation for DNA replication. EBNA1 is essential for the immortalization of primary B-lymphocytes [3, 4].
EBNA1 is a 641-amino acid protein and is reported to be a stable dimer both in solution or bound to DNA [5–7]. The DNA-binding and dimerization domains of EBNA1 are located at the C-terminal region of EBNA1, comprising residues 459 through 607 [8]. Two crystal structures of EBNA1 are currently available in the Protein Data Bank (PDB) [9]: VHI is the apo or unbound structure of EBNA1 [5] and 1B3T corresponds to EBNA1 co-crystallized with the 18 bp palindromic DNA recognition sequence (GGGAAGCAT|ATGCTTCCC) [6].
Analysis of these structures indicates the presence of two interacting domains (Fig. 1, panel a): the first one, termed the Core Domain (CD), is located at the dimerization interface (amino acids 504–607) and is structurally composed of eight-strands (four in each monomer) in an antiparallel β-barrel and two α-helices per monomer. One of these helices carries the primary residues involved in the DNA contacts (Lys514, Tyr518 and Arg522) and therefore is termed the Recognition Helix (RH) [5–7].
Fig. 1.

Structural Organization of the EBNA1 Protein. a Functional EBNA1 dimer bound to DNA: the tri-nucleotide (tri-nt) bp sequence (3′-TGC-5′)113 located in the center of the DNA Binding Site is shown in stick rendering. The Core Domain (CD, residues 504–607), comprising the Recognition Helix (RH), as well as the Flanking Domain (FD, residues 461–503), constituted by the helix perpendicular to DNA forming the Extended Chain (EC) loop that embraces the DNA molecule, are shown. b Superimposition of apo (PDB ID 1VHI) and DNA-bound (PDB ID 1B3T) crystal structures of EBNA1. In the apo conformation, the EC loop is unstructured and (partially) unsolved. In the bound-conformation, the EC loop adopts a conformation embracing the DNA helix (the DNA molecule was removed from the visualization). The tri-nucleotide (tri-nt) bp sequence (3′-TGC-5′)113 located in the center of the DNA Binding Site is shown
The second region, the Flanking Domain (FD), spans amino acids 461–503 and consists of an α-helix, perpendicular to the main DNA axis (Fig. 1, panel a). The flanking domain terminates with a flexible amino-acid segment, called the Extended Chain (EC; Fig. 1, panel b), which adopts a conformation that extends into the minor groove of DNA. Several amino acids belonging within the extended chain (Lys477, Lys461, Gly463 and Arg469) provide additional favorable base contacts to the DNA [5–7].
Although its pivotal role in maintaining the EBV genome during latency has been widely recognized, the concept of EBNA1 as a therapeutic target for EBV infections and related malignancies is relatively novel. To the best of our knowledge, only a few examples are available in the literature of drug design strategies that led to the discovery of novel chemical agents to therapeutically inhibit EBNA1.
In previous studies [10, 11], EBNA1 was established as an attractive candidate for targeting inhibition of EBV latent infections, and a virtual High Throughput Screening (vHTS) approach was reported that led to the identification of the first known small molecule inhibitors of the DNA binding activity of EBNA1 protein. Tested in vitro by a homogeneous Fluorescence Polarization (FP) assay, these chemical entities showed an inhibitory concentration range of 20–100 μM, and their ability to disrupt the EBNA1:DNA complex was further confirmed by Electrophoresis Mobility Shift Assay (EMSA). Furthermore, top Virtual Screening (VS) hits were found successful in inhibiting EBNA1 transcription activation in a cell-based assay and, when incubated with a Burkitt lymphoma cell line, they showed the ability to significantly reduce EBV genome copy number [10]. More recently, Kang et al. [12] used a cellular High Throughput Screening (HTS) to show that Roscovitine, a potent inhibitor of Cyclin-Dependent Kinase (CDK) types 1, 2, 5 and 7, inhibits Ori-P dependent episome maintenance by suppressing CDK-mediated phosphorylation of EBNA1 on Serine 393. Phosphorylation, as well as other post-translational modifications, influences the activities of EBNA1 including transcription and regulation functions, although their exact mechanisms remain poorly understood [13].
Lastly, Yasuda et al. [14] proposed a series of pyrrole-imidazole polyamides containing N-methylpyrrole (Py) and N-methylimidazole (Im) that serve as DNA minor groove binders to prevent DNA-EBNA1 binding, thus targeting DNA sequence as opposed to a direct interaction with EBNA1 protein.
Regardless of the mechanism by which EBNA1 is prevented from binding to sequence-specific DNA (or by which its transcriptional function is regulated), these studies contributed to a growing body of evidence indicating that inhibition of EBNA1 is a very attractive strategy in drug discovery. Indeed, as we previously mentioned [10, 11], inhibition of EBNA1 function can be selectively achieved by using small molecules that directly interact with EBNA1, thus preventing this carcinogenic protein from binding to DNA, and this data further confirmed the high “druggability potential” of EBNA1.
The concept of “druggable genome” proposed by Hopkins and Groom [15] refers to a subset of proteins encoded by the human genome that constitute biological targets suitable for drug design. Thus, according to this definition, a “druggable target” is a protein with a high propensity to bind small molecules with high affinity.
“Protein druggability” resides in the ability of surface binding sites to be complementary, in terms of their physicochemical properties (i.e. size, shape, electrostatics and hydrophobicity), to “drug-like” or even “fragment-like” compounds in order to successfully bind them with high affinity thus exerting desired biological effects [15]. However, despite the fact that Bochkarev et al. [5, 6] published crystal structures of EBNA1 back in the mid 90’s, no structural studies are nowadays available that report systematic analysis of EBNA1 binding sites to be exploited by small molecule or protein–protein interaction inhibitors.
As a keystone for Structure-Based Drug Design (SBDD) we have applied various computational methods to identify an initial set of putative binding sites of EBNA1. Visual inspection and structural characterization of potential binding pockets allowed us to narrow the focus to a smaller number of candidates, which meet all the requirements for being considered “druggable binding sites”.
Furthermore, we assessed the “druggability” of our binding sites by performing molecular docking and Molecular Dynamics (MD) simulations of compounds that were tested by fluorescence polarization-based biochemical high-throughput dose response assay for inhibitors of EBNA1 (PubChem BioAssay Identification AID 2381) [16]. Compounds of the AID 2381 set were docked against the top “druggable” EBNA1 binding-pockets, and then evaluated by MD simulations along with Molecular Mechanics Generalized Born Surface Area (MM-GBSA) calculations. For the two most “druggable” active sites, best scoring hits were identified, and their structural features enabled a better understanding of ligand functionalities critical for EBNA1 inhibition while providing a rationale for future design of novel, potent inhibitors of EBNA1/DNA binding.
Methods
Protein structures preparation and alignment
Two high-resolution crystal structures of EBNA1 were downloaded from the PDB web site: 1VHI [5] (2.5 Å resolution) and 1B3T [6] (2.2 Å resolution). In addition, we included in in our study a second crystallographic structure of apo-EBNA1 (1.9 Å resolution) solved in house (Messick, T, The Wistar Institute, unpublished structure).
All these structures were prepared according to the Protein Preparation Wizard [17] available in Schrödinger (Linux-x86 platform). The preparation included: addition of hydrogen atoms, removal of water molecules and adjustments of ligand bond-orders and formal-charges. The program Prime was used to run side chain conformation predictions for missing side chains. Last, the impref utility by Impact [17] was used to run OPLS2005 [18–20] restrained minimizations (0.30 Å RMSD atom displacement for convergence). During preparation, 1B3T structure was used as reference template for protein structures alignment on Cα atoms.
Pocket finding by MOE site finder
Site Finder [21] predictions were computed for all the available crystal structures of EBNA1 by applying default settings. The Probe Radius 1, which indicates the radius of a hypothetical hydrophilic hydrogen-bonding atom, was set to 1.4 Å. The Probe Radius 2, which indicates the radius of a hypothetical hydrophobic atom, was set to 1.8 Å. The Isolated Donor/Acceptor parameter, which indicated the maximum distance set to discard hydrophilic alpha spheres that have no hydrophobic alpha spheres within the specified distance, was set to 3.0 Å. The Connection Distance, the distance limit to merge individual clusters containing initial alpha spheres, was set to 3.0 Å. The “Minimum Site Size and Radius” indicate the maximum number of individual alpha spheres and the maximum radius allowed to bounding spheres per cluster, respectively. Clusters not matching these criteria were discarded.
Pocket finding by Schrödinger SiteMap
SiteMap [22] calculations were performed on all the crystal structures available for EBNA1. The Protein Preparation Wizard was used to prepare input proteins. Searches were conducted on the entire protein structures by choosing “Identify top-ranked potential receptor binding sites” from the Task menu of the SiteMap tool. By default, at least 15 points were used to define each reported site. A more restrictive definition of hydrophobicity was chosen, defining a fine grid. The OPLS 2005 force field was used during calculations.
Ligands preparation
All chemical structures of compounds in the AID 2381 set were downloaded from the PubChem Bioassay web site: http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=2381. LigPrep [23] was used to prepare all the structures submitted to molecular docking simulations (molecular probes, DNA sequence (3′-TGC-5′)113 and AID 2381). Stereoisomers and alternative ring conformations were generated. The tool Epik [24] was used to produce different ionization and tautomeric states at physiological pH. Finally, energy minimizations of all structures were performed with the OPLS 2005 force field.
Molecular docking
Molecular docking simulations were run using the tool Glide [25], at Extra Precision (XP) level [26–28]. While docking molecular probes and compounds in the AID 2381 sets, ligands were treated flexibly and Epik state penalties were included for re-scoring. While re-docking the DNA sequence (3′-TGC-5′)113, the ligand was rigidly docked. Docking grid in the Primary Site was set by choosing the default size of the enclosing box around the DNA sequence (3′-TGC-5′)113 and by excluding the co-crystallized ligand pocket. A number of grids for docking against the Secondary Site were defined by selecting from the amino acid residues: Tyr590, Lys586, Asn480, Ile481, Lys477, Gly484, Ser516, Leu520, and Asn519. Default sizes of the enclosing boxes were accepted. In all docking simulations performed, the OPLS 2005 force field was used to perform post docking energy minimizations.
Ligand sets
Binding modes and free energy values of several compounds were evaluated against both the Primary and Secondary binding pockets. In particular two sets were defined that contained top scoring hits retrieved against Primary and Secondary Sites, respectively. For each compound set, Molecular Mechanics Generalized Born Surface Area (MM-GBSA) calculations and molecular dynamics (MD) simulations were performed against one or both binding pockets. Secondary Site hits were fragmented to obtain substructures that were further used to assess druggable structural features of the pocket upon running MM-GBSA calculations and MD simulations.
MM-GBSA binding free energy calculations
Similarly to what reported by others, the MM-GBSA approach, as implemented in the program Prime [29–31], was used to estimate relative binding free energies of ligands in docking complexes of Primary and Secondary Sites (Glide pose viewer files, XP level). Calculations were run in parallel using two different set-ups. In one set of jobs, the protein was treated rigidly. In the other, protein residues comprised within 5 Å of ligand atoms were treated flexibly. Then, the “Minimize” sampling method was used.
MD simulations and scoring by ensemble docking and MM-GBSA
The program Desmond [32], as distributed by Schrödinger, and the OPLS-2005 all-atom force field were used to run 10 ns molecular dynamics (MD) simulations of hits against Primary and/or Secondary Sites. In addition, docking complexes of fragment substructures against the Secondary Site were simulated. For each protein–ligand complex, the best binding mode (Glide, XP level) was used as the starting configuration for MD simulations. All systems were embedded in orthorhombic boxes of TIP3P solvent [33]. After equilibration, simulations were performed in the NPT ensemble, at 300.0 K temperature and 1.01325 bar pressure.
Simulation quality analyses (SI-7, SI-8 and SI-9) were performed in Desmond, from the Maestro interface. For each simulated complex, the final snapshot was saved and minimized before evaluating ligand binding free energy by molecular docking with Glide XP and/or by Prime MM-GBSA calculations. Hydrogen bonds and contacts analyses were performed using the hydrogen bond plugin (http://www.ks.uiuc.edu/Research/vmd/plugins/hbonds/) available in VMD [34] as previously described [30].
Secondary Site hits were fragmented and substructure fragments were selected to probe specific interactions with complementary residues in the pocket. MD simulations (10 ns each) were run for all EBNA1/fragment complexes generated. Initial geometries were derived form docking complexes of the non-fragmented compounds. After 10 ns MD simulations, the last frame of each complex was saved before re-docking the compounds with Glide XP to derive docking scores and ligand efficiency values [35].
Secondary site hits: induced fit docking
The induced fit docking (IFD) methodology available in Schrödinger [36, 37] was used to perform docking simulations of Secondary Site hits. Many authors, including us [36, 38], used IFD to incorporate side-chain flexibility into the docking procedure. In our IFD case, side chains of residues 519, 586 and 590 were treated flexibly during the initial Glide docking step, by temporary substitution of these residues to alanine amino acids. For each residue, the top 20 docking poses were submitted to the steps of Prime protein-refinement and Glide re-docking. First, residues temporarily mutated to alanine were returned to their original amino acid type. All residues within 5 Å of any ligand poses were chosen for conformational refinement, consisting of side-chains re-orientation and minimization. All complexes within a 30 kcal/mol window of the minimum energy structure were ranked by Prime energy, before re-docking (Glide, XP level). The IFD composite scoring function [36] was used to rank the final protein/ligand complexes.
Results and discussion
Comparison of apo versus DNA-bound EBNA1 structures
Two crystallographic structures of EBNA1 are publicly available: the apo structure (PDB ID 1VHI [5], 2.50 Å resolution) and the crystal complex of EBNA1 bound to DNA (PDB code 1B3T [6], 2.20 Å resolution). In addition, we included in the study a second crystallographic structure of apo-EBNA1 (1.90 Å resolution) solved in a different space-group (Messick, T; Lieberman, P.M. The Wistar Institute, unpublished structure).
Analysis of apo structures revealed high similarity (0.44 Å RMSD of the C-alpha atoms of structurally aligned residues). Comparison of apo versus DNA-bound EBNA1 structures revealed two major sources of structural variation (details in Supporting Information 1, SI-1): the EC Loop and the Proline Reach (PR) Loop [39, 40] (545PGPGPQPGP553), located between helix 2 and the β-barrel bundle of the EBNA1 core structure (Figure S1). Putatively, they both relate to conformational changes occurring upon DNA binding, and therefore they deserve special consideration since their impairment would possibly compromise the ability of EBNA1 to bind DNA. We envision that further understanding of the precise mechanisms of regulation for these events would allow their exploitation from a drug design perspective.
Identification of “druggable” binding sites
Putative binding sites definition by pocket finding
We investigated the available crystal structures of EBNA1 with the aim of identifying “druggable” binding sites that might serve as binding pockets and that therefore might be targeted by small molecule inhibitors of EBNA1 activation and DNA binding. To achieve this, we used two independent computational methods for “pocket finding”, which allowed the identification of a number of potential sites on either the apo or the DNA-bound EBNA1 structures, after they were superimposed based on sequence alignment. Then, we computationally assessed the potential “druggability” of multiple pockets by docking, against each one of them, a set of molecular probes, i.e. small molecules experimentally known to inhibit EBNA1. Lastly, this information was used to rank the identified potential binding sites, and to select the top four EBNA1 pockets showing highest “druggability” potential.
We selected two distinct computational approaches to “pocket finding”, based on different and complementary methodologies. First, we used Site Finder [21], as distributed with the Molecular Operating Environment (MOE; https://www.chemcomp.com/) of Chemical Computing Group. The MOE Site Finder tool is classified as a purely geometric method [41] and indeed does not rely on energy models. Instead, it uses the geometric concept of alpha spheres, first applied to computational chemistry by Edelsbrunner [42] in 1995, and Liang [43] 1998, combined with a Voronoi decomposition of the 3D space according to Clarkson’s extension [44] of the well-known Gram-Schmidt orthogonalization algorithm. This procedure is used to generate an initial collection of spheres that are successively classified based on the hydrophobic/hydrophilic properties of the areas in the protein structure that they belong to. Lastly, a clustering method is applied to group together collections of sites, each containing at least one hydrophobic spot (Table S1, SI-2) [41].
As a second approach, we chose SiteMap [22], distributed by Schrödinger (http://schrodinger.com/). Unlike Site Finder, SiteMap implements a GRID [45] based algorithm, and therefore is aimed at calculating interaction energies between a predefined set of probe atoms and the receptor protein to locate energetically favorable receptor sites. SiteMap operates in three steps [46, 47]: Site Finding, Site Mapping and Site Assessment. In the Site Finding stage, the protein is embedded in a grid of points, which are used to identify a set of initial sites. The grid points are classified as inside or outside the protein, and only the inside points are retained and merged to constitute a set of potential receptor pockets. Then, during Site Mapping, the sites are characterized by calculating hydrophobic and hydrophilic potentials that are used to define three types of regions: hydrophobic, hydrophilic (likely to participate in hydrogen bond donor or acceptor interactions), and regions showing no preference for hydrophobic or hydrophilic interactions. Finally, in the Site Assessment stage, the sites are evaluated for a number of features that contribute to define two important properties, the SiteScore and the DScore (Druggability Score), which collectively provide indications of the overall “druggability” propensity of the target sites. A full description of the SiteMap methodology, along with the mathematical derivation and the physical–chemical meaning of the scores, can be found elsewhere [46, 47].
The MOE Site Finder identified a total of 24 putative binding sites either on the apo or the DNA bound structures of the EBNA1 dimer, and the Schrödinger SiteMap tool proposed five distinct pockets, two of which were found adjacent one to another, and they were classified as belonging to the same predicted cavity. While visual inspection of individual pockets, initially proposed by MOE Site Finder (Table S1), allowed us to prioritize a subset of 14 of them for further analyses, all Schrödinger SiteMap pockets were retained at this stage.
Putative binding sites assessment by docking molecular probes and structural analyses
A number of successful Blind Docking (BD) [48, 49] applications are currently known, where locations of binding hot spots on protein receptors are not based on their solved crystal structures in complexes with natural ligands or small molecule binders, but instead they are assumed based upon computational predictions. To further assess these putative binding sites (14 proposed by Site Finder and 5 by SiteMap) for their potential to constitute “druggable” pockets for use in SBDD, and inspired by the success of BD applications (especially an effective study performed by other authors [50] on protein binding-site assessment by fragment docking), we performed molecular docking of a chemically diverse [51, 52] set of compounds selected to serve as molecular probes (Table S2, SI-3). Then, we took the distribution of binding modes obtained against individual EBNA1 pockets as a measure of their propensity to serve as high-affinity binding sites for either “drug-like” or “fragment-like” compounds (SI-3).
The overall localization of docking poses, obtained for the molecular probes compounds against putative EBNA1 pockets, highlighted the presence of a highly populated primary site matching the location of the DNA minor groove in the protein/DNA complex (Fig. 2). Structural analysis of this region indicated a quite large protein region centered on a tri-nucleotide (tri-nt) bp sequence (3′-TGC-5′)113. Interestingly, this surface almost extended up to reach a secondary site, located at the bottom of the FD where it intersects the longitudinal axis of the RH. Additional hot spots, i.e. regions of the protein surface populated by a significantly high number of docking poses, were also identified. While tiny, hydrophobic regions distributed over the entire protein surface constituted most of these hot spots, one of them (which we term the Tertiary Site) was of a particular interest since it represented a large, quite flat area that extended up to the protein–protein dimerization interface of the EBNA1 dimer.
Fig. 2.

Four Putative Binding Sites identified on EBNA1. Maps were generated using SiteMap (www.schrodinger.com). Blue ovals indicate the Primary Binding Site. White ovals indicate the Secondary Binding Site. Yellow ovals indicate the Tertiary Binding Site. Pink ovals indicate the Quaternary Binding Site. a, b show the crystal structure of EBNA1 co-crystallized in complex with DNA (PDB ID 1B3T). In b, the DNA molecule was removed to show the maps generated by SiteMap (blue ovals). c, d show the crystal structure of EBNA1 unbound to DNA (PDB ID 1VHI and The Wistar Institute proprietary structure, respectively)
Consensus of features to select EBNA1 binding sites
Using a combination of the above-mentioned criteria (namely, pocket finding, docking of molecular probes and structural analyses), we significantly narrowed our scope by requiring maximal feature consensus within the sites initially identified, finally selecting four putative binding pockets likely to serve as “druggable” targets for high affinity binding of EBNA1 inhibitors (Fig. 2). First, Schrödinger SiteMap predicted all these sites, using either the crystal structure of the apo or the DNA-bound EBNA1. In agreement with these results, MOE SiteFinder predicted most of the sites, with partial volume overlap (SI-2). In addition, all the selected sites showed at least one binding mode with a good or excellent docking score from the molecular probes docking. Finally, structural analyses of all these sites allowed us to exploit the functional group composition of these binding sites, and speculate on possible mechanisms for their involvement in achieving EBNA1 inhibition. We summarize here the main features that provide the structural and functional characterization of each “druggable” binding site identified on EBNA1 (Fig. 2). Based on their ranking by our consensus of features, they were named Primary to Quaternary Site.
Primary site
Identified by both computational tools for pocket finding [21, 22] a very wide and open valley characterizes the Primary Site (Fig. 2, panels a and b; Figure S2, SI-4). Symmetrically stretched between the two units of the EBNA1 dimer, this pocket accommodates the DNA double helix. Therefore, it is also called the DNA Binding Site and, not surprisingly, it was previously described elsewhere [7]. Centered on the tri-nucleotide bp sequence (3′-TGC-5′)113, the Primary Site presents a number of amino acid residues that contribute to engage in binding interactions with the tri-nucleotide sequence located across the DNA minor groove (Figure S2, SI-4). In particular, the RH residues Lys514 and Tyr518 [7], but also Arg522, being spaced apart by four residues, i.e. the helical turn, can orient their side chains in a parallel arrangement and offer the opportunity to engage in multiple interactions with a ligand counterpart.
Far away from the main surface defining the Primary Site on the RH, additional residues, namely like Lys477, Lys461, Gly463 and Arg469, are located on the EC of the FD and are known to contribute to establishing important DNA binding interactions (Figure S2, SI-4). Re-docking the tri-nucleotide bp sequence (3′-TGC-5′)113 against this site is shown in Figure S3 (SI-5), and provided a score of −10.4 kcal/mol.
Although an obvious choice as putative binding pocket for rational drug design, the EBNA1 Primary Site does not provide the conventional features that generally define easy-to address-targets. Indeed, very wide and open, it lacks a well-defined hydrophobic surface common to “druggable” pockets. Also, to engage in DNA binding, a proper arrangement of the RH embracing the DNA minor grove is needed to contribute fundamental determinants responsible for molecular recognition (like Lys477, Lys461, Gly463 and Arg469). In the absence of DNA however, no structural information is available to predict the conformation adopted by this loop, which is very flexible. The lack of such an important structural element represents a considerable challenge in designing small molecule inhibitors that might target this pocket. One way to overcome this limitation could be the use of Fragment-Based Drug Design strategies (FBDD), which typically place “fragment-like” molecules within multiple sub-pockets of a receptor site, and aims to successively link them together to build a final “drug-like” molecule. Indeed, FBDD has been demonstrated to be a very powerful tool in targeting largely exposed and poorly defined binding pockets, like those located at protein–protein interaction surfaces [53].
Secondary site
Initially identified by Schrödinger Site-Map, then confirmed by partial overlap with MOE SiteFinder results and visual analysis of EBNA1 structures, as well as by docking against a number of molecular probes, the Secondary Site is defined by a tiny, mostly hydrophobic hole where the terminal base of one DNA strand interfaces with the protein (Fig. 2 and Figure S4, SI-6). It is located right across the volume enclosed by the RH and one edge of the FD Helix and, considering its location we named it the RH Site.
Despite its modest size, the “druggability” score calculated by SiteMap [22, 46, 47] for this site was remarkable (SiteScore 0.7, Dscore 0.6), and its propensity to serve as a binding pocket might be explained by the amino acid composition of a few residues facing the site.
The first two residues, Asn519 and Leu520, belong to the RH. Of these, the side chain of Leu520 offers its hydrophobic contribution by appearing right in the center of the site. At the same time, the polar side chain of Asn519 represents the opportunity to engage in strong interactions with hydrogen bond acceptor moieties of ligand molecules. The side chains of residues Thr590 and Lys586, both localized on the CD, represent additional sources of hydrogen bond donors. Last, the bifurcated side chain of Asn480 facing from the FD Helix provides both hydrogen bond donor and acceptor moieties (Figure S4, SI-6). Given its key location at the interface between protein and DNA, we envision for this pocket a possible role in regulating protein/DNA interactions, and therefore small molecules that bind to this site might severely compromise EBNA1 ability to bind DNA. Clearly, the EBNA1 Secondary Site represents an invaluable opportunity for drug design, and in consideration of its size, we suggest that it might constitute an ideal target for FBDD approaches.
Tertiary site
Located at the interface between the monomer units of EBNA1, the Tertiary Site is also named the Dimerization Site, and is constituted by a wide surface that extents over part of the 8 stranded antiparallel β-barrel structure in the CD (Fig. 2). While presenting a limited hydrophobic surface right in the middle, it primarily shows a mixed hydrophobic and hydrophilic character.
Interestingly, although the Tertiary Site was identified by computational mappings on both the crystal structures of EBNA1 unbound to DNA that were analyzed (1VHI and unpublished structure), no “druggable” cavity was detected at the protein–protein interface of EBNA1 in complex with DNA, suggesting that conformational changes might occur upon DNA binding or release.
Given the pivotal role of protein dimerization in EBNA1 activation and DNA binding, small molecule inhibitors of protein–protein interactions might be a winning strategy to EBNA1 inhibition. Thus, we conceive that also the Dimerization Site holds great potential as a target for drug design.
Quaternary site
Last, we highlight presence of a Quaternary Site, which was identified by Schrödinger SiteMap, and further confirmed by providing quite a number of molecular probes docking modes (Fig. 2). Very small and almost entirely solvent exposed, it is located opposite to the DNA molecule, in a protein region that is delimited by the top of the FD Helix, the second helix of the CD and the loop sequence connecting the FD to the CD elements.
No structural information is currently available to explain a possible role of this pocket in EBNA1 function or DNA binding, and therefore it is not clear whether it could represent a potentially “druggable” site. We wonder whether it may constitute an allosteric site, in which case it could be possible to inactivate EBNA1 by targeting against small molecule inhibitors.
Evaluation of AID 2381 compounds by docking against multiple binding sites
Once we had identified a set of multiple binding sites on the EBNA1 protein that might be targeted to prevent its activation by interfering with DNA binding, we tested our findings using SBDD. We focused on the two most “druggable” of the ligand-binding pockets identified, the Primary and Secondary Sites, and we used a computational protocol combining docking, MD simulations and MM-GBSA calculations to evaluate a set of compounds experimentally tested for their ability to serve as EBNA1 inhibitors by selectively preventing DNA binding [16].
The compound set under investigation was available for download at the NCBI PubChem web site (http://pubchem.ncbi.nlm.gov/assay) under the BioAssay ID (AID) 2381. It comprised a total of 127 organic small molecules that were tested in a “Fluorescence Polarization-Based Biochemical High-Throughput Confirmation Assay to Identify Inhibitors of EBNA1” [16]. Of these, 61 compounds were found to have half maximal inhibitory concentration (IC50) equal or lower to 10 μM, while the remaining 66 had IC50 greater than 10 μM.
The purpose of the screening was to determine dose/response curves to confirm compounds identified as active in a previous assay, in particular as part of the “Fluorescence Polarization-Based Biochemical High-Throughput Confirmation Assay to Identify Inhibitors of the Epstein-Barr Virus Nuclear Antigen 1 (EBNA1)” campaign (AID 2292) [54]. In addition, these compounds were earlier identified as inactive when tested against ZTA Protein, a biological target involved in an EBNA1-related pathway, namely the “Counter-screen for Inhibitors of EBNA1: Fluorescence Polarization-Based Biochemical High Throughput Assay for Inhibitors of the Epstein-Barr Virus-encoded Protein ZTA, in Triplicate” (AID 2338) [55] suggesting a potential for these compounds to constitute selective inhibitors of the EBNA1 function.
Although the ability of these compounds to inhibit EBNA1 was clearly established by in vitro testing, no structural information was known to explain, at a molecular level, the mechanisms by which these ligand molecules may interact with the EBNA1 protein receptor, as well as to shed light on how these interactions might affect DNA binding.
Molecular docking has been proven a powerful tool in generating binding mode hypothesis that highlight molecular principles responsible for binding and recognition, even in response to challenging situations, as in the case of Ensemble Docking (EnsD) [56] of multiple protein conformations, which was introduced to account for protein flexibility in drug design.
As a variation of classical EnsD methods, here we applied a docking approach against an ensemble of multiple receptor sites, which we computationally identified on EBNA1. Clearly, the difference resides in the fact that we docked a set of compounds against multiple receptor sites of the same protein, as opposed to multiple receptor conformations that a flexible protein might adopt. Indeed, our aim was to derive, for the Primary and the Secondary Binding Sites identified on EBNA1, binding mode hypotheses of the most active compounds likely to bind to each site. We envision that understanding the molecular determinants characterizing individual binding pockets will provide a strong foundation to rationally design the next generation of potent and selective EBNA1 inhibitors.
The distribution of IC50 values (μM) experimentally determined for the 127 compounds in the AID 2381 set is reported as a histogram in Fig. 3, panel a. The threshold to define active compounds against EBNA1 was set to equal or lower than 10 μM [16], reducing active compounds to a limited space in the activity range. Also, only 9.4 % of the entire set (or less than 20 % of the active compounds) fell into the category of tight binders (with IC50 \ 5 μM), and just one of which with an IC50 lower than 1 μM.
Fig. 3.

Histogram Distributions of Activity and Score Values for AID 2381 Compounds. a Experimental Activities (μM IC50); b Docking Scores (kcal/mol)
Generating correlations models that link experimentally determined activities to predicted ligand binding affinities could be quite a challenging task in SBDD. In our case, this was particularly difficult, not only because the active compounds accumulated in a very narrow range of values, but also in regard to the fact that it was not possible to assume a priori that all these compounds were binding to a unique binding pocket.
As a consequence, our primary goals have shifted towards the identification, within the active sub-set of the AID 2381 compounds, of the best candidate ligands for binding to individual EBNA1 pockets, in particular the Primary (or DNA Binding) and the Secondary (or Recognition Helix) Sites.
Binding modes against the primary site: DNA binding site
The Primary Site or DNA Binding Site of EBNA1 is shown in Figs. 2 and S2. To assess our docking system for its use in SBDD, we extracted the tri-nucleotide single strand DNA sequence (3′-TGC-5′)113 from the crystal structure of EBNA1 in complex with DNA (PDB ID 1B3T) and we re-docked this molecule against the EBNA1 Primary Site.
Molecular docking simulations were performed using Glide [25], at extra precision mode XP [26–28] (details in “Methods” section). Since the purpose of this simulation was not to explore possible ligand conformations matching the binding pocket, but instead to test the method in reproducing crystallographic interactions for this molecule, the ligand was treated rigidly. Docking complex (Figure S3) reproduced almost perfectly both protein/ligand orientations and binding interactions of the crystallographic complex, with an XP score of −10.4 kcal/mol (scores of about −8.0 kcal/mol or lower generally indicate tight-binding), indicating a strong accuracy and reliable predictive power of the docking system.
Frequency distributions of activities and docking scores for the compounds in the AID 2381 are shown in Fig. 3, panel b. While docking scores of most of the compounds were predicted in the range of −7.0 to −4.0 kcal/mol, only a few compounds fell below a value of −7.0 kcal/mol. Interesting to note, this distribution might include compounds that actually bind to EBNA1 pockets other than the Primary Site, and these results should be interpreted with that in mind.
In order to set a threshold for the docking score to separate binder from non-binder molecules, we generated our own decoys set constituted by a number of compounds with physicochemical properties similar to the AID 2381 molecules that, when tested in house by the FP assay against EBNA1 (data not shown), were all found to be completely inactive (IC50 > 166 μM). Not surprisingly, the majority of these molecules showed a docking score of about −2.0 kcal/mole, and none of them obtained a value lower than −5.0 kcal/mol. In addition to that, binding modes proposed for these compounds never overlaid the DNA backbone of the crystallographic molecule. Given these considerations, we regarded a score of −6.5 kcal/mol as an appropriate threshold to accept compounds as potential binders of the EBNA1 Primary Site, provided that their preferred binding orientations overlaid the DNA backbone conformation of the X-ray ligand (Figure S3).
Binding modes of the top scoring compounds targeting the EBNA1 Primary Site are shown in Fig. 4, and their structures are reported in Table 1 along with docking scores (with or without Epik penalties applied) and IC50 values. Despite their structural diversity, all these compounds shared surprisingly similar binding orientations that closely superposed onto the crystallographic DNA ligand. In addition to that, all these compounds engage in favorable interactions by hydrogen bonding to fundamental residues of the RH (Lys514, Tyr518 or Arg522), and some of them also gain additional interactions with residues located in the EC of the FD.
Fig. 4.

Docking Complexes of Top Scoring Compounds against EBNA1 Primary Binding Site. Chemical structures, docking scores and IC50 values are reported in Table 1. a 15944862; b 3167530; c 5973599; d 3186499; e 5826369; f 3185712; g 1432744; h 1919699; i 7256141; j 3185717; k 1452563; l 1107365
Table 1.
Docking results for top AID 2381 compounds classified as binders of the EBNA1 primary site
| CMPD IDa | Chemical structure | IC50 (μM) | Scoreb kcal/mol | Glide Score c kcal/mol |
|---|---|---|---|---|
| 15944862 |
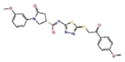
|
5.9 | −9.4 | −9.6 |
| 3167530 |
|
2.0 | −7.8 | −7.8 |
| 5973599 |
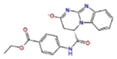
|
5.5 | −7.2 | −7.5 |
| 3186499 |
|
5.8 | −7.1 | −7.3 |
| 5826369 |
|
7.2 | −6.9 | −6.9 |
| 3185712 |
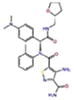
|
13.7 | −6.8 | −6.8 |
| 1432744 |
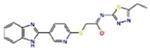
|
17.1 | −6.8 | −6.9 |
| 1919699 |
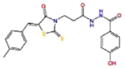
|
11.7 | −6.7 | −6.7 |
| 7256141 |
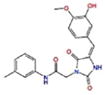
|
9.1 | −6.7 | −6.7 |
| 3185717 |
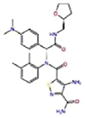
|
2.7 | −6.6 | −6.6 |
| 1452563 |
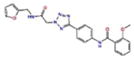
|
0.6 | −6.6 | −6.6 |
| 1107365 |
|
15.1 | −6.5 | −6.5 |
PubChem Compound ID (CID)
XP Docking Score (Epik penalties applied)
Glide XP Docking Score
Binding modes against the secondary site: recognition helix site
With respect to the small size of the Secondary Site and making it suitable for binding to molecules with fragment-like properties (fragment molecules typically evince poor docking scores), the threshold of acceptable docking score that we set to −6.5 kcal/mol was regarded as particularly significant. Binding modes of top candidate compounds binding the Secondary Site, or Recognition Helix Site, are shown in Fig. 5, and their structures are reported in Table 2 along with IC50 (μM) values and docking scores (kcal/mol).
Fig. 5.

Docking complexes of top scoring compounds against EBNA1 secondary binding site. Chemical structures, docking scores and IC50 values are reported in Table 2. a 2122620; b 647423; c 1335528; d 1272394; e 3186499; f 4879718; g 2122620 shown within the surface binding site; h 647423 shown within the surface binding site
Table 2.
Docking results for top AID 2381 compounds classified as binders of the EBNA1 secondary site
| CMPD IDa | Chemical Structure | IC50 (μM) | Scoreb kcal/mol | Glide Scorec kcal/mol | IFD (kcal/mol) |
|---|---|---|---|---|---|
| 2122620 |
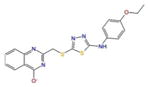
|
27.0 | −7.5 | −8.9 | −611.6 |
| 647423 |
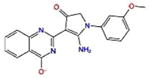
|
10.2 | −6.0 | −8.2 | −605.9 |
| 1335528 |
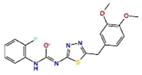
|
6.7 | −6.6 | −7.2 | −607.5 |
| 1272394 |

|
41.5 | −6.7 | −6.9 | −607.4 |
| 3186499 |
|
5.8 | −6.8 | −6.9 | −612.2 |
| 4879718 |
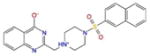
|
5.6 | −5.3 | −6.9 | −610.6 |
PubChem Compound ID (CID)
XP Docking Score (Epik penalties applied)
Glide XP Docking Score
A common feature of all these docking complexes is the presence of a heterocyclic system (often aromatic) that locates right in the center of the ligand pocket to engage in hydrophobic interactions offered by the side chain of Leu520. Also, a high number of these compounds (70 %) showed additional hydrogen bonding interactions, in particular with Asn519. The majority of these hits (1335528, 1272394, 3186499 and 4879718) reported very similar docking scores, in the range of −6.9 to −7.2 kcal/mol (XP score), with IC50 activity values between 5.6 and 6.7 μM (Table 2). However, one compound, predicted to have a very strong docking score (−8.2 kcal/mol), was found to only barely fall across the activity threshold of 10 μM IC50 (647423 10.2 μM IC50), while two additional compounds (1272394 and 2122620), reported to have inhibitory concentrations of 27.0 and 41.5 μM in experimental assay, did show strong docking scores (in particular, 2122620, with XP docking score of −8.9 kcal/mol and IC50 27.0 μM).
A possible explanation to this behavior derives from the fact that the Secondary Site is very tiny and mostly suitable for accommodating low MW compounds (typically MW \ 300 kDa). Indeed, all the docking complexes generated for the AID 2381 set, showed that ligands were never entirely accommodated within the receptor pocket, but instead only their heterocyclic portions were able to fill it, leaving the rest of the compounds completely solvent exposed. Thus, in spite of their IC50s, we found these compounds particularly worthy of consideration for drug design, and therefore we submitted them to further computational work.
Searching the minimum ligand scaffold for binding to the secondary site
In consideration of the fact that the Secondary Site is very tiny and that only a substructure of each of the compounds selected from the AID 2381 set was able to engage in significant protein/ligand interactions, one question naturally arises: for each top-scoring compound investigated, what are the structural features necessary and sufficient to gain strong binding affinity?
To address this question, we developed the Minimum Ligand Scaffold (MLS) approach, shown in Figs. 6 and 7. We took the structure hits from selected docking complexes and we submitted them to a fragmentation procedure (typically based on retrosynthetic [57] and/or topological [58] approaches) that resulted in “fragment-like” compounds.
Fig. 6.

Minimum Ligand Scaffold (MLS) Approach used to Target the Secondary Binding Site (I). Compounds from initial docking (Hits) were fragmented into smaller “fragment-like” molecules, which were successively docked against the Secondary Site. If they obeyed the Minimum Scaffold Requirements, then a new molecular framework was designed directly within cavity by properly linking them together
Fig. 7.

Minimal Ligand Scaffold (MLS) Approach used to Target the Secondary Binding Site (II). Two active compounds, classified by multiple binding-sites docking as putative Secondary Site binders, were fragmented to generate MLSs 1 and 2. Ideally, by joining them together upon exploring their synthetic feasibility (or eventually looking for similar synthetically accessible building blocks), the expectation is to obtain compounds demonstrating new binding interactions (Linked Fragments column). Overlapping substructures shown in green
Then, in search for the MLS, we docked the generated “fragment-like” molecules against the Secondary Site of EBNA1. To become a MLS, a fragment structure must (1) reproduce the same binding mode that was originally proposed for the entire molecule, (2) maintain approximately the same docking score, and (3) preserve all key interactions that are held responsible for binding in their parent molecules (Fig. 6). Examples of MSLs derived from AID 2381 compounds are reported in Fig. 7.
The next level of our fragment-based approach was to explore the opportunity of gaining additional interactions by joining together two (or even more) of the fragments generated by MLS design. Clearly, this approach prompts concerns as to the synthetic feasibility of compounds obtained by fragment-linking strategies. To partially address the challenge, we used retro-synthetic approaches, expected to generate synthetically accessible building blocks. Alternatively, virtual structures resulting from joining non-building blocks could still be used as query reference molecules in search for real compounds, like using Shape Signatures [59, 60] screening of the ZINC database [61, 62] or any other compound collections, including corporate databases. By sharing similar shape as the parent structures, the newly identified compounds are likely to evince same binding behaviors towards the EBNA1 Secondary Site.
As an example, Fig. 7a shows the docking complex generated for one MLS (named MLS-1) derived by fragmenting compound 2122620. As expected, the Glide XP docking score for MLS-1 was nearly the same as the parent compound (−8.9 vs −8.2 kcal/mol; Fig. 6), and its binding mode was guided by hydrogen bonding to Asn519, Thr590 and Lys586. Similarly, Fig. 7b shows a second MLS (named MLS-2) arising from the fragmentation of compound 15944862, with Glide XP docking score of −5.9 kcal/mol (−6.4 kcal/mol for the parent compound) and binding mode guided by hydrogen bonding to Asn519 and Ser513. Interestingly, Fig. 7c–e show the chemical moieties putatively active in MLS-1 and MLS-2, and their superposition within the Secondary Site of EBNA1. Clearly, the best scenario would be to suitably join the two fragments together (Fig. 7f) in such a way to maintain all the interactions gained by the putatively active chemical moieties (Asn519, Thr590 and Lys586, derived from MLS-1 and Ser513 derived from MLS-2), while getting rid of those substructures supposedly not contributing to stabilizing binding.
Besides suggesting new potential-scaffolds, MLS design contributed important details for the characterization of molecular determinants responsible for binding at the secondary site, and revealed new insights into the rational design of EBNA1 inhibitors. Indeed, chemical and structural features common to our MLS scaffolds include: (1) a heterocyclic ring (or a fused ring) that accommodates into the inner region of the pocket (Fig. 7) by complementing the entirely hydrophobic character of the side chains facing the lumen (Fig. 2); (2) substituents that can capture chemical interactions with charged/polar resides, like Lys586 and Asn519 that regulate access to the hydrophobic site. Additional groups (3) can be envisioned to gain further interactions with solvent exposed residues, such as Ser513 or Thr590.
Accounting for protein flexibility in druggability assessment
Incorporating protein flexibility of binding sites and accounting for conformational changes upon ligand binding are crucial aspects to assess pockets druggability [63]. A number of approaches have been recently developed that address these challenges, and include the use of sets of compounds (either small organic molecules or fragment like compounds) to identify ‘hot spots’ targeted by multiple probes [48, 49]. Here, we used the top scoring hits of each site as molecular probes to identify amino-acid residues that are crucial to pocket druggability. Thus, we evaluated the ability of these residues to provide main stabilizing interactions, and we assessed whether protein motion could translate into local conformational changes increasing pockets druggable features.
Our approach incorporated protein flexibility at multiple levels. We performed 10-ns long MD simulations of each complex under investigation (SI-7, SI-8 and SI-9), and we derived hydrogen bond patterns established over the course of the simulations (SI-10 and SI-11). To support unambiguous attribution of hits to a specific site, we used MM-GBSA calculations that estimated ligand binding-affinities of protein/ligand configurations either derived from the initial docking poses or selected upon hydrogen-bond analysis of MD trajectories.
Given the size of the Secondary Site being suitable for accommodating fragment-like compounds, we performed additional work (SI-9, SI-12 and SI-13) to compare hits versus substructure fragments, and to probe the Secondary Site druggability in conditions where the protein was explicitly treated flexibly. Thus, we used IFD docking of hits against the Secondary binding site (Table 2 and SI-12) to evaluate alternative binding modes upon introducing side chain flexibility. In addition, we performed MD simulations (SI-11 and SI-13) of EBNA1 as in complexes with the molecular fragments (10 ns each) obtained by MLS approach. Last, we used ligand efficiency values, calculated by Glide (SI-14), as a metric to compare initial hits versus derived fragments.
Our approach allowed identifying best candidate residues of each protein site to be targeted for rational design of EBNA1 inhibitors
a. Structural features of the primary site that determine pocket druggability
Visual inspection of MD trajectories indicated different possible behaviors adopted by compounds bound to the Primary Site during 10 ns of trajectories. For the majority of the compounds, binding conformations adopted during the simulated times (10 ns each) slightly deviated from original docking modes. Molecules affiliated to this class were: 3167530, 5826369, 5973599, 7256141, 15944862, and 1107365. Hydrogen bond analysis (SI-10) indicated that, overall, the most frequently interacting residues were Glu556, Leu536, Arg522, Arg521, Tyr518, Gly472, Gly470, Lys467, Trp464 and Phe465. Less frequently, also Lys514, an important residue responsible for DNA binding and recognition [7], was listed as critical for ligand binding. With the exception of residues located on the extended chain, namely Gly472, Gly470, Lys467, Phe465 and Trp464, all the remaining amino acids faced the Primary site in a location that is known to host DNA binding. Noteworthy, we recall that Arg522, Tyr518 and Lys514, located on the RH, are known to offer essential contact to DNA binding [5–7]. In addition, EBNA1 mutants of these residues are known that show severe reductions in EBNA1 DNA binding activities [7], suggesting that binding of these compounds against Primary Site most likely interfere with DNA binding. One additional compound, i.e. 1919699, adopted a similar binding behavior. Indeed, analysis of the simulation trajectory showed that 1919699 interacted primarily with Leu536, Arg521 and Tyr518 located on the core domain or on the recognition helix and Arg469 on the extended chain loop (SI-10).
Of the remaining hits, two derivatives of the same chemical class, namely 3185712 and 3185717, showed binding preferences for residues of the core domain at the interface between two EBNA1 subdomains (Fig. 1), thus loosing interactions with the extended chain. Primary residues involved in hydrogen bonds were Arg538, Glu556, Leu536 and Pro535 (SI-10).
In contrast, compound 1432744 showed binding preferences for residues located across the recognition helix, in particular Arg521, Tyr518, or the extended chain loop, such as His468, Arg469 and Gly470 (SI-10).
Finally, two hits required additional attention for unambiguous attribution to a binding site. First, we found that rankings of compound 1452563 by Glide docking score (Table 1) and by Prime MM-GBSA ΔG Bind (SI-15) were in disagreement. In addition, analysis of simulation trajectory for compound 1452563 showed that the molecule did not stably bind to the Primary Site, and indeed it was observed to leave the pocket during the time between 2 and 3 ns of simulation. Based on the observed binding behavior we concluded that 1452563 likely constitutes as a false positive hit of the Primary Site. Given the high potency of 1452563 in experimental assay (0.61 μM, Table 1), we assumed the existence of alternative binding preferences of 1452563. Thus, we simulated the compound as in a complex with the EBNA1 Secondary Site (10 ns). Hydrogen bond analysis of MD trajectory indicated Asn480, Ser516, Asn519, Lys586, and Thr590 as the Secondary Site residues most frequently contributing to binding. Being this hydrogen-bonding pattern consistent with what we found to characterize the Secondary Site (see dedicated paragraph for details), we speculated that compound 1452563 likely binds to this pocket.
Second, compound 3186499 was selected in the top scoring hits of both the Primary and the Secondary Sites. We used an MM-GBSA scoring approach based on hydrogen bond frame selection [64] to suggest 3186499 as a Secondary Site hit (SI-15 & SI-16, and SI-17 & SI-18). To do so, we selected representative MD conformations of 3186499 as in complex with either the Primary or the Secondary Sites that maximized hydrogen bond networks and we scored these binding modes using Prime MM-GBSA. This data (SI-16 and SI-17) along with MM-GBSA binding affinities estimated for the initial docking complexes (SI-15 and SI-18), as well as visual inspection of respective MD trajectories and hydrogen bond networks, supported our attribution.
b. Structural features of the secondary site that determine pocket druggability
To account for protein motion involving side chain flexibility, we performed IFD (SI-12) as well as MM-GBSA calculations (SI-18). Binding modes and protein–ligand interaction diagrams are shown in the SI-12, and relative scores are listed in Table 2.
Given the size of the Secondary Site being suitable to accommodate fragment-ligands, we applied the MLS approach to generate substructure fragments, dock and simulate by MD all the Secondary Site hits (Table 2). It is worth to mention that the primary goal here was not to generate synthetically accessible fragments, but instead to “probe” structural features of the Secondary Site by using substructures of compounds previously tested in vitro.
Analyses of MD simulation trajectories for both classes of compounds, namely hits (Table 2; SI-8) and fragments (SI-9), along with MM-GBSA and IFD calculations all agreed to indicate Lys586, Asn480, Lys77, Asn519 as the most critical residues in establishing interactions with ligands and the Secondary Site. In addition, a few more resides, such as Thr590, Thr590, Ser516 and Ala588, were highlighted as important structural features to increase the druggability of this pocket.
In addition, we used the Glide ligand efficiency [35] as a metric to compare the original hits and the relative substructure fragments bound to the Secondary Site (SI-14). When comparing LE values, fragments with a higher LE are generally preferred since LE is expected to decrease during optimization [35]. However, being Glide LE calculated over the Glide score, lower values are preferred for fragments. Analysis of the Glide LE values for the Secondary Site hits and derived fragments (SI-14) showed invariantly better (lower) values for the fragments than for the original ligand hits, indicating that a fragment based approach can be successfully employed for rationally designing novel EBNA1 inhibitors of the Secondary Site. In particular, fragments with top LEs values were reported to be FRAGM_1335528, FRAGM_4879718 and FRAGM_2122620 (SI-14). Of the EBNA1 residues involved, Asn519 and Lys477 most frequently served as counterparts in hydrogen bonding.
Conclusions
In this work, we compared two crystal forms of the EBNA1 dimer: the apo-EBNA1 conformation and the EBNA1/DNA complex. We found that these structures differ primarily due to conformations adopted by two structurally distinct regions, and we speculated on their possible roles in relation to DNA binding.
The first one is the flexible loop that constitutes the EC of the FD, known to be involved in DNA binding, while the second is the PR Loop adopting different conformations in the two monomers of both the apo-structures of EBNA1 analyzed. Interestingly, the EBNA1 PR Loop was shown to contribute to DNA binding as well as in mediating interactions with accessory proteins, like cellular transcription factors [5, 34]. Since both protein regions are involved in conformational changes occurring upon DNA binding, we conclude that their impairment would negatively affect the ability of EBNA1 to bind DNA.
As a second step, we applied computational methods for pocket finding, followed by blind docking of molecular probe compounds that allowed the identification of four putative ligand binding pockets of the EBNA1 protein. Their structural analysis allowed us to confirm their “druggability” potentials, and to propose rationales for their involvement in various functions regulating EBNA1 activation and DNA binding. In particular, we focused out attention on two of these sites, which we named the DNA Binding Site (or Primary Site) and the Recognition Helix Site (or Secondary Site). Indeed, structural analyses of these pockets indicated that they were both directly involved in binding to DNA, and therefore it follows that small-molecule binding to these pockets would result in disrupting EBNA1/DNA interactions.
We tested our binding site hypotheses by docking the AID 2381 compounds, previously tested in vitro for their ability to inhibit EBNA1/DNA binding, against both Primary and Secondary Sites of EBNA1 and we identified the best compounds putatively binding to each one of them.
As for the Primary Site (DNA Binding Site), we confirmed the primary role of amino acid residues located on the RH, in particular Lys514, Tyr518 and Arg522, as well as additional residues located on the flexible EC Loop that constitutes an essential part of the binding pocket. In absence of DNA, this loop is completely unstructured and its conformation in EBNA1 when in complex with small molecules is not currently known.
We also identified candidate compounds binding to the Secondary Site (Recognition Helix Site) of EBNA1, and we developed a unique fragment-based drug design approach that enabled us to identify a set of MLSs to be used as starting points to design novel potential inhibitors of EBNA1 directly within the Secondary Site by fragment linking and growing methods. Further work can be addressed to shed lights on characterizing the Tertiary and Quaternary Sites as additional target pockets for SBDD.
Once identified the best candidate hits of the Primary and Secondary Sites by molecular docking, we supported compounds attribution as well as we explored alternative binding modes by running MD simulations of the generated complexes. In only one case, a compound (1452563) was found to leave the binding pocket (Primary Site) during the simulation time. Indeed, further calculations supported the hypothesis that this compound preferentially binds to a different pocket (Secondary Site). Additional work was performed in the case of the Secondary Site, namely by IFD and MLS approach paired with prediction of LE. These calculations confirmed the propensity of the pocket to accommodate fragment-like compounds, therefore suggesting the application of drug design approaches based on fragment screening.
To the best of our knowledge, this is a first attempt to validate fully EBNA1 as a biological target for lead design and discovery. We envision that our results will provide a strong foundation for future design of potent and selective EBNA1 inhibitors.
Lastly, it is worth to mention that a confirmation of our binding sites predictions was provided later by experimental evidence (proprietary work by Messick, T. and Lieberman, P.M. at The Wistar Institute). Interestingly, the resolution of crystal structures of EBNA1 co-crystallized with fragment-like molecules (structures and manuscript in preparation) revealed highly significant matches with our computational results, and confirmed our predicted EBNA1 binding pockets. Although describing the fragment-based approach implemented to target EBNA1 [65] exceeds the scope of the present work, the experimental confirmation to our predictions constitutes an important piece of information towards validating EBNA1 druggability, and shows that combining computational and experimental approaches can be a key to success in modern drug discovery.
Supplementary Material
Acknowledgments
EG and RZ gratefully acknowledge The Wistar Institute (www.wistar.org) for providing funding in partial support of this work. Also, EG and RZ thank Dr. Elia Eschenazi, Dr. Preston B. Moore and Dr. Vojislava Pophristic at the University of the Sciences, Philadelphia for useful discussions. TEM and PML acknowledge support from the Wellcome Trust Seeding Drug Discovery program (096496/Z/11/Z). TEM acknowledges supports from American Cancer Society (ACS-IRG-96-153-10).
Abbreviations
- CD
Core domain
- EBNA1
Epstein-Barr nuclear antigen 1
- EBV
Epstein Barr virus
- EC
Extended chain FBDD Fragment-based drug design
- FD
Flanking domain
- FP
Fluorescence polarization
- HTS
High-throughput screening
- MLS
Minimum ligand scaffold
- PR
Proline rich
- SBDD
Structure-based drug design
- vHTS
Virtual high-throughput screening
- VS
Virtual screening
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10822-016-9899-y) contains supplementary material, which is available to authorized users.
References
- 1.Young LS, Rickinson AB. Nat Rev Cancer. 2004;4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 2.Linder SE, Sugden B. Plasmid. 2007;58:1–12. doi: 10.1016/j.plasmid.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kempkes B, Pich D, Zeidler R, Hammerschmidt W. Proc Natl Acad Sci USA. 1995;92:5875–5879. doi: 10.1073/pnas.92.13.5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Humme S, Reisbach G, Feederle R, Delecluse HJ, Bousset K, Hammerschmidt W, Schepers A. Proc Natl Acad Sci USA. 2003;100:10989–10994. doi: 10.1073/pnas.1832776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bochkarev A, Barwell JA, Pfuetzner RA, Furey W, Jr, Edwards AM, Frappier L. Cell. 1995;83:39–46. doi: 10.1016/0092-8674(95)90232-5. [DOI] [PubMed] [Google Scholar]
- 6.Bochkarev A, Bochareva E, Frappier L, Edwards AM. J Mol Biol. 1998;284:1273–1278. doi: 10.1006/jmbi.1998.2247. [DOI] [PubMed] [Google Scholar]
- 7.Cruickshank J, Shire K, Davidson AR, Edwards AM, Frappier L. J Biol Chem. 2000;275:22273–22277. doi: 10.1074/jbc.M001414200. [DOI] [PubMed] [Google Scholar]
- 8.Ambinder RF, Shah WA, Rawlins DR, Hayward GS, Hayward SD. J Virol. 1990;64:2369–2379. doi: 10.1128/jvi.64.5.2369-2379.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. [Accessed on 22 May 2013];The Research Collaboratory for Structural Bioinformatics Protein Data Bank, RCSB PDB. http://www.rcsb.org/pdb.
- 10.Li N, Thompson S, Schults DC, Zhu W, Jiang H, Luo C, Lieberman PM. PLoSONE. 2010;(5):4, e10126. doi: 10.1371/journal.pone.0010126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson S, Messick T, Schultz DC, Reichman M, Lieberman PM. J Biomol Screen. 2011;15:1107–1115. doi: 10.1177/1087057110379154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang MS, Lee EWK, Soni V, Lewis TA, Koehler AN, Srinivasan V, Kieff E. J Virol. 2011;85:2859–2868. doi: 10.1128/JVI.01628-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duellman S, Thompson KL, Coon JJ, Burgess RR. J Gen Virol. 2009;90:2251–2259. doi: 10.1099/vir.0.012260-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yasuda A, Noguchi K, Minoshima M, Kashivazaki G, Kanda T, Katayama K, Mitsuhashi J, Bando T, Sugiyama H, Sugimoto Y. Cancer Sci. 2011;102:2221–2230. doi: 10.1111/j.1349-7006.2011.02098.x. [DOI] [PubMed] [Google Scholar]
- 15.Hopkins AL, Groom CR. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 16.National Center for Biotechnology Information. [Accessed 27 May 2013];PubChem BioAssay Database; AID = 2381, Source = Scripps Research Institute Molecular Screening Center, Assay Provider: Paul Lieberman, Wistar Institute. http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=2381.
- 17.Schrödinger Suite 2010 Protein Preparation Wizard; Epik version 2.1. Schrödinger, LLC; New York, NY: 2010. [Google Scholar]; Impact, version 5.6. Schrödinger, LLC; New York, NY: 2005. [Google Scholar]; Prime, version 2.2. Schrödinger, LLC; New York, NY: [Google Scholar]
- 18.Jorgensen WJ, Maxwell DS, Tirado-Rives J. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 19.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 20.Shivakumar D, Williams J, Wu J, Damn W, Shelly J, Sherman W. J Chem Theory Comput. 2010;6:1509–1519. doi: 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]
- 21.Site Finder: Molecular Operating Environment (MOE), 2012.10. Chemical Computing Group Inc; 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7: 2012. [Google Scholar]
- 22.SiteMap, version 2.5. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- 23.LigPrep, version 2.5. Schrödinger, LLC; New York: 2012. [Google Scholar]
- 24.Epik, version 2.2. Schrödinger, LLC; New York: 2012. [Google Scholar]
- 25.Glide, version 5.7. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- 26.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelly M, Perry JK, Shaw DE, Francis P, Shenkin PS. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 27.Halgren TA, Murphy RB, Friesner RA, Behard HS, Frye LL, Pollard WT, Banks JL. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 28.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 29.Lyne PD, Lamb ML, Saeh JC. J Med Chem. 2006;49:4805–4808. doi: 10.1021/jm060522a. [DOI] [PubMed] [Google Scholar]
- 30.Gianti E, Zauhar RJ. J Comput Aided Mol Des. 2015;29:451–470. doi: 10.1007/s10822-015-9835-6. [DOI] [PubMed] [Google Scholar]
- 31.Sirin S, Kumar R, Martinez C, Karmilowicz MJ, Ghosh P, Abramov YA, Martin V, Sherman W. J Chem Inf Model. 2014;54:2334–2346. doi: 10.1021/ci5002185. [DOI] [PubMed] [Google Scholar]
- 32.Desmond Molecular Dynamics System, version 3.8. D. E. Shaw Research; New York, NY: 2014. [Google Scholar]; Maestro-Desmond Interoperability Tools, version 3.8. Schrödinger; New York, NY: 2014. [Google Scholar]
- 33.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 34.Humphrey W, Dalke A, Schulten K. J Molec Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 35.Schultes S, de Graaf C, Haaksma EEJ, de Esch IJP, Leurs R, Krämer O. Drug Discov Today Technol. 2010;7:e157–e162. [Google Scholar]
- 36.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 37.Schrödinger Suite 2014-2 Induced Fit Docking protocol; Glide version 6.3. Schrödinger, LLC; New York, NY: 2014. [Google Scholar]; Prime version 3.6. Schrödinger, LLC; New York, NY: 2014. [Google Scholar]
- 38.Gianti E, Zauhar RJ. J Chem Inf Model. 2012;52:2670–2683. doi: 10.1021/ci3002342. [DOI] [PubMed] [Google Scholar]
- 39.Han SJ, Hu J, Pierce B, Weng Z, Renne R. J Gen Virol. 2010;91:2203–2215. doi: 10.1099/vir.0.020958-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ceccarelli DF, Frappier L. J Virol. 2000;74:4939–4948. doi: 10.1128/jvi.74.11.4939-4948.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Labute P, Santavy M. Chemical Computing Group Inc; http://www.chemcomp.com/journal/sitefind.htm. [Google Scholar]
- 42.Edelsbrunner H, Facello M, Fu R, Liang J. Proceedings of the 28th annual Hawaii international conference on systems science; 1995. pp. 256–264. [Google Scholar]
- 43.Liang J, Edelsbrunner H, Woodward C. Protein Sci. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clarkson KL. Proceedings of 31st annual IEEE symposium on foundations of computer science; 1992. pp. 387–395. [Google Scholar]
- 45.Goodford PJ. J Med Chem. 1985;28:849–857. doi: 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- 46.Halgren T. J Chem Inf Model. 2009;49:377–389. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- 47.Halgren T. Chem Biol Drug Des. 2007;69:146–148. doi: 10.1111/j.1747-0285.2007.00483.x. [DOI] [PubMed] [Google Scholar]
- 48.Hetény C, Van Der Spoel C. FEBS Lett. 2006;580:1447–1450. doi: 10.1016/j.febslet.2006.01.074. [DOI] [PubMed] [Google Scholar]
- 49.Hetény C, Van Der Spoel C. Protein Sci. 2002;11:1729–1737. doi: 10.1110/ps.0202302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang N, Jacobson MP. PLoS ONE. 2010;5(4):e10109. doi: 10.1371/journal.pone.0010109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Del Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 52.Congreve M, Carr R, Murray C, Jhoti H. Drug Discov Today. 2003;8:876–877. doi: 10.1016/s1359-6446(03)02831-9. [DOI] [PubMed] [Google Scholar]
- 53.Valkov E, Sharpe T, Marsh M, Greive S, Hyvönen M. Top Curr Chem. 2012;317:145–179. doi: 10.1007/128_2011_265. [DOI] [PubMed] [Google Scholar]
- 54.National Center for Biotechnology Information. Assay Provider: Paul Lieberman. Wistar Institute; [Accessed on 27 May 2013]. PubChem BioAssay Database; AID = 2292, Source = Scripps Research Institute Molecular Screening Center. http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=2292. [Google Scholar]
- 55.National Center for Biotechnology Information. Assay Provider: Paul Lieberman. Wistar Institute; [Accessed on 27 May 2013]. PubChem BioAssay Database; AID = 2338, Source = Scripps Research Institute Molecular Screening Center. http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=2338. [Google Scholar]
- 56.Huang SY, Zou X. Proteins. 2007;66:399–421. doi: 10.1002/prot.21214. [DOI] [PubMed] [Google Scholar]
- 57.Lewell XQ, Judd DB, Watson SP, Hann MM. J Chem Inf Comput Sci. 1998;38:511–522. doi: 10.1021/ci970429i. [DOI] [PubMed] [Google Scholar]
- 58.Gianti E, Sartori L. J Chem Inf Model. 2008;48:2129–2139. doi: 10.1021/ci800219h. [DOI] [PubMed] [Google Scholar]
- 59.Zauhar RZ, Moyna G, Tian L-F, Li Z-W, Welsh WJ. J Med Chem. 2003;46:5674–5690. doi: 10.1021/jm030242k. [DOI] [PubMed] [Google Scholar]
- 60.Zauhar RJ, Gianti E, Welsh WJ. J Comput Aided Mol Des. 2013;12:1009–1036. doi: 10.1007/s10822-013-9698-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Irwin JJ, Shoichet BK. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. J Chem Inf Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grove LE, Hall DR, Beglov D, Vajda S, Kozakov D. Bioinformatics. 2013;29:1218–1219. doi: 10.1093/bioinformatics/btt102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adasme-Carreño F, Muñoz-Gutierrez C, Caballero J, Alzate-Morales JH. Phys Chem Chem Phys. 2014;16:14047–14058. doi: 10.1039/c4cp01378f. [DOI] [PubMed] [Google Scholar]
- 65.Messick TE. Cambridge Healthtech Institutes’s tenth annual drug discovery chemistry, protein–protein interactions conference.2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.