

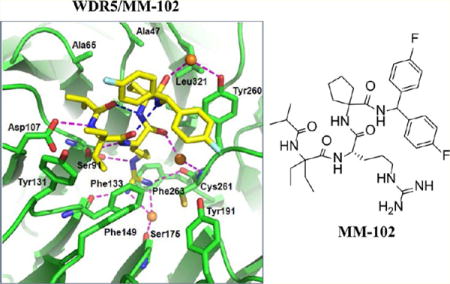
Abstract
Mixed lineage leukemia 1 (MLL1) is a histone H3 lysine 4 (H3K4) methyltransferase, and targeting the MLL1 enzymatic activity has been proposed as a novel therapeutic strategy for the treatment of acute leukemia harboring MLL1 fusion proteins. The MLL1/WDR5 protein–protein interaction is essential for MLL1 enzymatic activity. In the present study, we designed a large number of peptidomimetics to target the MLL1/WDR5 interaction based upon –CO-ARA-NH–, the minimum binding motif derived from MLL1. Our study led to the design of high-affinity peptidomimetics, which bind to WDR5 with Ki < 1 nM and function as potent antagonists of MLL1 activity in a fully reconstituted in vitro H3K4 methyltransferase assay. Determination of co-crystal structures of two potent peptidomimetics in complex with WDR5 establishes their structural basis for high-affinity binding to WDR5. Evaluation of one such peptidomimetic, MM-102, in bone marrow cells transduced with MLL1-AF9 fusion construct shows that the compound effectively decreases the expression of HoxA9 and Meis-1, two critical MLL1 target genes in MLL1 fusion protein mediated leukemogenesis. MM-102 also specifically inhibits cell growth and induces apoptosis in leukemia cells harboring MLL1 fusion proteins. Our study provides the first proof-of-concept for the design of small-molecule inhibitors of the WDR5/MLL1 protein–protein interaction as a novel therapeutic approach for acute leukemia harboring MLL1 fusion proteins.
Graphical abstract
INTRODUCTION
Histones are the most basic units for packing DNA into nucleosomes, which are further organized into several higher order chromatin structures. It is now well recognized that covalent modifications of histones, such as methylation, acetylation, and phosphorylation, are essential for many important cellular processes, including gene regulation.1,2 Mounting evidence also indicates that misregulation or mutation of histone-modifying enzymes plays a key role in the development of a wide range of human diseases, including cancer.3,4 Chemical modulation of histone-modifying enzymes is thus a promising approach for the development of new therapeutics.
Mixed lineage leukemia 1 (MLL1) is a histone H3 lysine 4 (H3K4) methyltransferase (HMT) that catalyzes mono-, di-, and trimethylation (Figure 1a) through its evolutionarily conserved SET domain.5 MLL1 is essential for definitive hematopoiesis by regulating transcription activation of Hox genes and associated cofactors (e.g., HoxA9 and Meis-1), which encode transcription/regulatory factors promoting hematopoietic stem cell expansion. Deregulation of MLL1 is associated with acute lymphoid leukemia (ALL) and acute myeloid leukemia (AML). MLL1 abnormality accounts for 5–10% of AML in adults and 70% of ALL in infants.6 In most cases, chromosome translocations occur on one MLL1 allele and result in leukemogenic MLL1 fusion proteins (e.g., MLL1-AF9, MLL1-AF4, and MLL1-ENL).7 MLL1 fusion proteins, which lack the C-terminal SET domain and thus the H3K4 HMT enzymatic activity, cooperate with the remaining copy of wild-type MLL1 in leukemogenesis (Figure 1b).8,9 It was recently shown that, in leukemia cells transformed by MLL1-AF9, both wild-type MLL1 and oncogenic MLL1-AF9 fusion proteins were recruited to Hox gene loci.8 Furthermore, wild-type MLL1 is required for Hox gene over-expression through persistent H3K4 methylation and for viability of MLL1-AF9-transformed leukemia cells.8 Thus, targeting H3K4 HMT activity of MLL1 can be a promising new strategy for the treatment of leukemia carrying MLL1 fusion protein.
Figure 1.
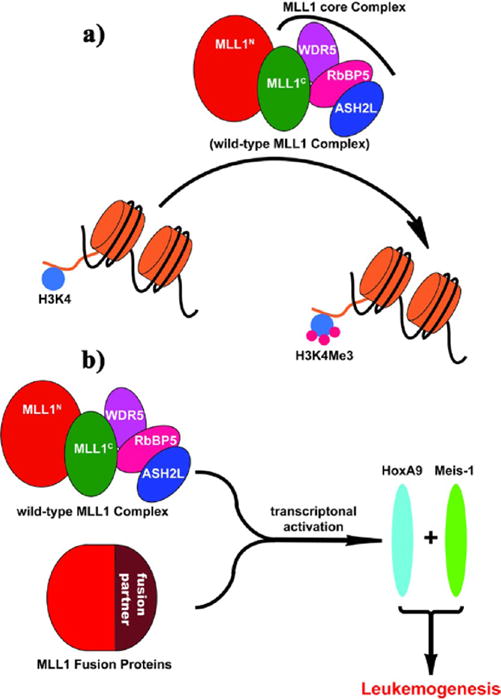
Schematic diagram of MLL1 in histone 3 lysine 4 (H3K4) methylation and in leukemogenesis. (a) Wild-type MLL1 complex methylates H3K4, and the core complex is required for robust catalytic activity. (b) MLL1 fusion proteins cooperate with wild-type MLL1 complex to activate MLL1 target genes, leading to leukemogenesis. MLL1N, N-terminus of MLL1; MLL1C, C-terminus of MLL1.
The H3K4 HMT activity of MLL1 is tightly controlled by a core complex consisting of MLL1, WDR5 (WD (Trp-Asp) repeat domain 5), RbBP5 (retinoblastoma binding protein 5), and ASH2L (absent small or homeotic-2-like) (Figure 1a).7,10 While MLL1 protein alone has weak enzymatic activity, its H3K4 HMT activity can be greatly enhanced with formation of the core complex.10 The structural integrity of the MLL1 core complex depends on a well-defined interaction between WDR5 and MLL1 proteins.7,10 Indeed, disruption of the protein– protein interaction between WDR5 and MLL1 by mutating key residues on WDR5 effectively dissociates the MLL1 core complex and results in dramatic inhibition of the MLL1 H3K4 HMT activity.11
The co-crystal structures of an MLL1 peptide complexed with WDR512,13 show that the interaction between WDR5 and MLL1 involves a well-defined pocket in WDR5 and a WDR5 interacting (WIN) motif, comprised of approximately 12 amino acid residues in MLL1. In our previous study,14 we explored the binding of MLL1 peptides to WDR5 and determined that the –CO-ARA-NH– motif within MLL1 (residues 3764–3766) is both necessary and sufficient for MLL1 binding with WDR5. Our previous study14 led to the identification of a tripeptide, Ac-ARA-NH2, which binds to WDR5 with Ki = 120 nM and represents a promising lead structure for further optimization.
Currently, there is no high-affinity and cell-permeable chemical probe for the MLL1/WDR5 interaction, and such a compound could be invaluable to further investigate the role of MLL1 in leukemogenesis, as well as in normal cellular processes. In the present study, we have performed extensive modifications of Ac-ARA-NH2 to probe the WDR5 binding site. Our study has led to the identification of a number of peptidomimetics, which bind to WDR5 with Ki ≤ 1 nM and are >100 times more potent than Ac-ARA-NH2. We further demonstrated that high-affinity peptidomimetics efficiently inhibit H3K4 methyltransferase activity of the MLL1 complex in a fully reconstituted in vitro HMT assay. Co-crystal structures of two peptidomimetics complexed with WDR5 provide the structural basis for their high-affinity binding to WDR5. Using one of these peptidomimetics, MM-102, we show that the compound effectively inhibits the expression of HoxA9 and Meis-1, two critical MLL1 target genes, in bone marrow cells transduced with an MLL1-AF9 fusion gene. Significantly, MM-102 selectively inhibits cell growth in leukemia cells carrying MLL1 fusion proteins. Taken together, our study provides a critical proof-of-concept that small-molecule inhibitors of the WDR5/MLL1 interaction can effectively inhibit MLL1-mediated gene transcription in leukemia cells harboring MLL1 fusion proteins and represent a novel therapeutic strategy for acute leukemia.
MATERIALS AND METHODS
A. Chemistry
All the synthesized compounds were characterized with 1H NMR, 13C NMR (300 MHz, Bruker), and HRMS (ESI+) (Agilent Q-TOF Electrospray). These data are provided in the Supporting Information (Table S2). Chemical shifts were reported in ppm relative to TMS. D2O (4.79 ppm) and CD3OD (3.31 ppm) were used as internal standards for 1H NMR, and D2O (1,4-dioxane, 66.7 ppm) and CD3OD (49.2 ppm) for 13C NMR spectra.
1. Solid-Phase Peptide Synthesis of Compounds in Tables 1–5
Table 1.
Binding Affinities of Ac-ARA-NH2 Analogues Designed to Investigate the P1, P2, and P4 Sites in WDR5a
| A1 modifications
|
R2 modifications
|
A3 modifications
|
||||||
|---|---|---|---|---|---|---|---|---|
| peptide | IC50 ± SD (μM) | Ki ± SD (μM) | peptide | IC50 ± SD (μM) | Ki ± SD (μM) | peptide | IC50 ± SD (μM) | Ki ± SD (μM) |
| 1a, GRA | 77.4 ± 13.2 | 16.7 ± 2.8 | 2a, AN1eA | >300 | >65 | 3a, ARG | 10.3 ± 1.2 | 2.2 ± 0.3 |
| 1b, AbuRA | 0.28 ± 0.02 | 0.06 ± 0.001 | 2b, AOrnA | >300 | >65 | 3b, ARAbu | 0.03 ± 0.008 | 0.006 ± 0.002 |
| 1c, VRA | 0.24 ± 0.03 | 0.05 ± 0.005 | 2c, AKA | >300 | >65 | 3c, ARV | 0.11 ± 0.02 | 0.02 ± 0.003 |
| 1d, CRA | 1.83 ± 0.30 | 0.4 ± 0.6 | 2d, ACitA | >300 | >65 | 3d, ARC | 0.08 ± 0.01 | 0.02 ± 0.003 |
| 1e, LRA | 10.8 ± 0.40 | 2.3 ± 0.3 | 3e, ARL | 34 ± 6 | 7.3 ± 1.2 | |||
| 1f, ChgRA | 8.7 ± 0.60 | 1.9 ± 0.1 | 3f, ARChg | 280 ± 70 | 61 ± 16 | |||
| 1g, HRA | 1.7 ± 0.3 | 0.4 ± 0.03 | 3g, ARF | 280 ± 40 | 60 ± 8 | |||
| 1h, FRA | 3.1 ± 0.3 | 0.7 ± 0.07 | 3h, ARS | 1.8 ± 0.03 | 0.4 ± 0.07 | |||
| 1i, SRA | 2.9 ± 0.61 | 0.6 ± 0.1 | 3i, ART | 0.08 ± 0.003 | 0.02 ± 0.001 | |||
| 1j, TRA | 2.9 ± 0.4 | 0.6 ± 0.09 | 3j, ARE | >100 | >20 | |||
For ARA, IC50 = 0.54 ± 0.03 μM, Ki = 0.12 ± 0.01 μM. Abbreviations: 2-Abu, 2-aminobutyric acid; Chg, cyclohexylglycine; Nle, nor-leucine; Orn, ornithine; Cit, citrulline.
Table 5.
Structures and Binding Affinities of ARA Peptide Analogues with N-Terminal Modifications
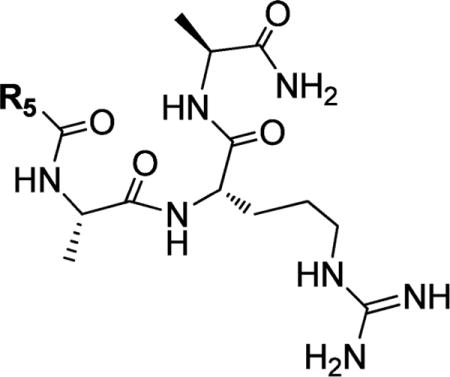
| |||
|---|---|---|---|
| Compound | R5 | IC50 ± SD (μM) | Ki ± SD (μM) |
| ARA |
|
0.54 ± 0.03 | 0.12 ±0.01 |
| 7a |
|
0.17 ±0.06 | 0.04 ± 0.01 |
| 7b |
|
0.5 ± 0.03 | 0.11 ±0.01 |
| 7c |
|
5.1 ±0.5 | 1.1 ± 0.1 |
| 7d |
|
0.7 ± 0.02 | 0.16 ±0.01 |
| 7e |
|
23.4 ± 1.7 | 5.1 ±0.4 |
| 7f |

|
22±2 | 4.7 ± 0.5 |
| 7g |
|
>300 | >65 |
| 7h |
|
>100 | >5 |
| 7i |
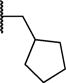
|
>100 | >20 |
| 7j |
|
2.5 ± 0.4 | 0.54 ± 0.09 |
| 7k |
|
17.4 ±0.3 | 3.74 ± 0.06 |
All the peptides were synthesized manually using Fmoc chemistry. Rink amide resin was used as the solid support. DIC/HOAt was used as the coupling reagent. All the peptides were cleaved from the resin by treatment with a TFA:DTT:TIS:H2O (17.5 mL:0.5 g:0.5 mL:1 mL) cleavage cocktail, which also resulted in removal of the protecting group(s). The cleavage solution was evaporated, and the crude product was precipitated with diethyl ether followed by RP-HPLC purification (Waters, Sunfire Prep C18, 19 × 150 mm, 5 μm).
2. Synthesis of Compounds in Tables 6 and 7
Table 6.
Structures and Binding Affinities of ARA Peptide Analogues with C-Terminal Amide Modifications
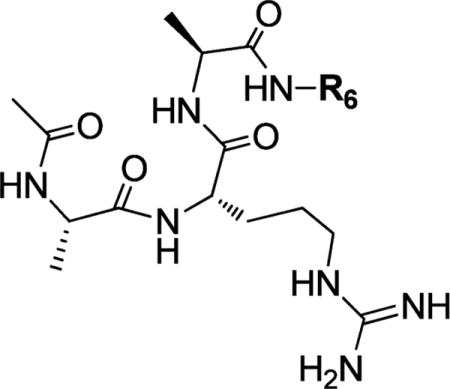
| |||
|---|---|---|---|
| Compound | R6 | IC50 ± SD (μM) | Ki ± SD (μM) |
| ARA | −H | 0.54 ± 0.03 | 0.12 ±0.01 |
| 8a |
|
0.70 ±0.14 | 0.15 ±0.03 |
| 8b |
|
0.16 ±0.1 | 0.05 ± 0.01 |
| 8c |
|
0.10 ±0.06 | 0.02 ±0.01 |
| 8d |
|
0.28 ± 0.06 | 0.06 ±0.01 |
| 8e |
|
0.34 ± 0.08 | 0.06 ±0.02 |
| 8f |
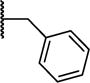
|
0.12 ±0.02 | 0.03 ± 0.004 |
| 8g |
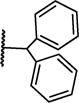
|
0.04 ± 0.01 | 0.007 ± 0.002 |
Table 7.
Structures and Binding Affinities of Peptidomimetics
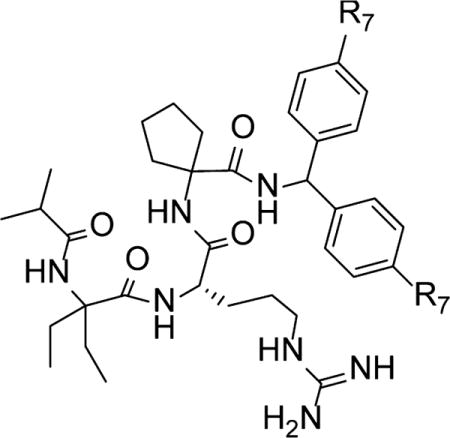
| |||
|---|---|---|---|
| compound | R7 | IC50 ± SD (nM) | Ki(nM) |
| MM-101 | −H | 2.9 + 1.4 | <1 |
| MM-102 | −F | 2.4 + 1.7 | <1 |
| MM-103 | −Cl | 4.5 + 0.6 | <1 |
Ac-Ala-Arg(Pbf)-Ala-COOH (IV) peptide was synthesized using Fmoc solid-phase chemistry and 2-chlorotrityl chloride (I) resin as the solid support (Scheme 1). Fmoc-Ala-OH (3 equiv) was loaded to the resin in a reaction vessel and shaken overnight in CH2Cl2 (DCM) and in the presence of 3 equiv of DIPEA. MeOH was then added to this mixture, and the reaction vessel was shaken for 30 min to endcap unreacted 2-chlorotrityl group on the resin. Next, classical chain elongation was carried out with Fmoc chemistry. The carboxylic acid intermediate IV was cleaved from the resin by treatment of III with 4 mL of 1% TFA in DCM (3 × 10 min). The filtrate was evaporated and purified by HPLC using a C18 reverse-phase column. The Ac-AR(Pbf)A-COOH (IV, 0.2 mmol) was dissolved in 10 mL of THF, and 3 equiv of HATU, 3 equiv of HOAt, 5 equiv of DIEA, and 3 equiv of the corresponding amines were added. The reaction mixture was stirred at room temperature for 24 h, the solvent was evaporated, and the crude product was purified by RP-HPLC. The Pbf protecting group was removed from the arginine by treatment with the cleavage cocktail TFA:DTT:TIS:H2O (17.5 mL:0.5g:0.5 mL:1 mL), followed by HPLC purification (Waters, Sunfire Prep C18, 19 × 150 mm, 5 μm), yielding 8a–8g.
Scheme 1.
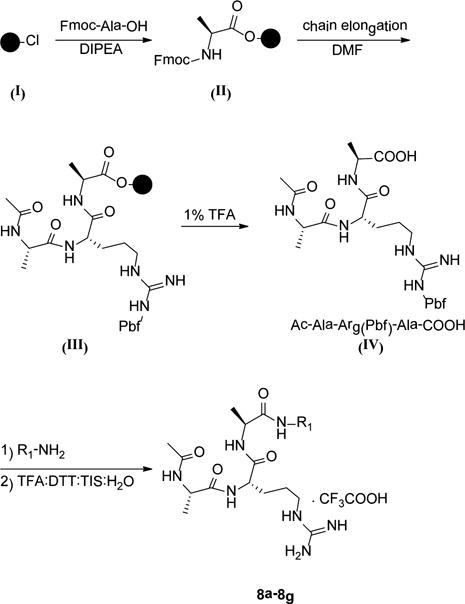
Synthesis of Compounds in Table 6
A similar procedure was used for the synthesis of the peptidomimetics in Table 7, as shown in Scheme 2. For the synthesis of C-MM-102, a control compound for MM-102, the same procedure was used with the exception that the D-arginine was used.
Scheme 2.
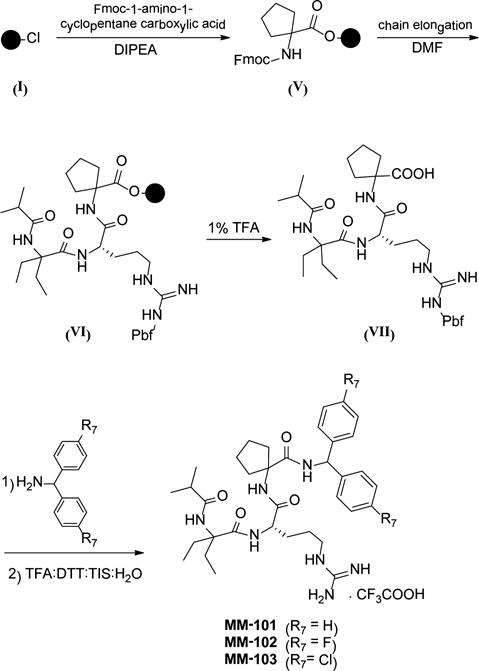
Synthesis of Peptidomimetics (Table 7)
B. Competitive Binding Assay
Binding affinities of all the synthesized compounds were determined using a fluorescence-polarization (FP)-based competitive binding assay; the details of this assay have been described earlier.14
C. In Vitro H3K4 Methyl Transferase Assay with the Reconstituted MLL1 Core Complex
1. Protein Expression
Full-length constructs of both RbBP5 (residues 1–538) and Ash2L (residues 1–635) were used for their expression. Truncated WDR5 (residues 23–334) and MLL1 (residues 3762–3969) constructs, which are sufficient for the formation of the MLL1 core complex and for robust HMT activity of the core complex, were used. MLL1, WDR5, RbBP5, and ASH2L were expressed as His-SUMO fusions from the pET28A-SUMO vector. Proteins were expressed from BL21 DE3 pLyss codon (+) at 16 °C overnight after induction with 0.1 mM IPTG in the mid-log phase of bacterial growth. For each protein, cells were harvested and the protein was purified by the His tag on Ni-NTA resin (Qiagen). The SUMO tag was removed from RbBP5, ASH2L, and MLL1 proteins by incubation with the ULP1 protease at 4 °C overnight. The protease and cleaved SUMO-His tag were collected by batch binding with the Ni-NTA resin for 1 h.
2. In Vitro Histone Methyltransferase (HMT) Assay
The HMT assay was performed in 50 mM HEPES pH 7.8, 100 mM NaCl, 1.0 mM EDTA, and 5% glycerol at 22 °C. Each reaction contained 1.5 μCi of the co-factor,3H-S-adenosylmethionine (Perkin-Elmer). H3 10-residue peptide was used as the substrate at 50 μM. Compounds were added at concentrations ranging from 0.125 to 128 μM and incubated with the pre-assembled WDR5/RbBP5/ASH2L complex at a final concentration of 0.5 μM for each protein for 2–5 min. Reactions were initiated by addition of the MLL1 protein at a final concentration of 0.5 μM and allowed to proceed for 30 min before preparing scintillation counting. To count samples, reactions were spotted on separate squares of P81 filter paper (Whatman) and precipitated by submerging in freshly prepared 50 mM sodium bicarbonate buffer with pH 9.0. After washing and drying, samples were vortexed in Ultima Gold scintillation fluid and counted. As a negative control, assays were performed using 0.5 μM MLL1/WDR5/RbBP5/ASH2L complex assembled with the non-interacting mutant, WDR5D107A.15,16
D. Determination of Crystal Structures of MM-101 and MM-102 in Complex with WDR5
1. Protein Preparation
WDR5 (Δ23aa, NCBI Reference Sequence: NP_060058.1) was cloned into a pET28b-based vector with a 6xHis-SUMO tag fused at the N-terminus. E. coli BL21(DE3) cells bearing expression plasmids were induced for 16 h with 0.1 mM IPTG at 25 °C. The protein was purified by Ni-NTA affinity resin and on-bead digestion using Ulp1 protease, followed by gel filtration chromatography on Hiload Superdex 75 equilibrated with 25 mM Tris-HCl pH 8.0 and 150 mM NaCl. The purified protein was concentrated to 30 mg/mL and stored at −80 °C for crystallization.
2. Crystallization and Structural Determination
WDR5/MM-101 binary complex was obtained by mixing WDR5 and MM-101 at a molar ratio of 1:3 immediately before crystallization. The complex was crystallized at 25% PEG4000, 0.1 M sodium acetate pH 4.6, and 0.2 M ammonium sulfate. The crystals were harvested in the same buffer with 20% glycerol. The 3.4 Å data set was collected at Advanced Photon Source beamline 21ID-F and was processed by HKL2000.17 The crystal belongs to the P1 space group. The structure was solved by molecular replacement by Phaser18 using the previously published WDR5 structure (PDB ID: 2H14). Iterative cycles of refinement and modeling were carried out using Phenix and Coot.19,20
WDR5/MM-102 binary complex was obtained by mixing WDR5 and MM-102 at a molar ratio of 1:3 before crystallization. The complex was crystallized at 30% PEG8000, 0.2 M ammonium sulfate. The crystals were harvested in the same buffer with 20% glycerol. The 2.6 Å data set was collected at Advanced Photon Source beamline 21ID-D and was processed by HKL2000.17 The crystal belongs to the P21 space group. The structure was solved by molecular replacement by Phaser18 using the previously published WDR5 structure (PDB ID: 2H14). There are four WDR5 molecules in one asymmetric unit. Iterative cycles of refinement and modeling were carried out using Phenix and Coot.19,20
The coordinates for the co-crystal structures for MM-101 and MM-102 in complex with WDR5 have been deposited into the Protein Data Bank as PDB IDs 4GM3 and 4GM8.
E. Computational Methods
The crystal structure of WDR5 in complex with the WIN peptide (PDB ID: 3EG6) was used to construct the WDR5/Ac-ARA-NH2 model using Sybyl (Tripos, Inc.). Solvent accessibility calculations were performed by Naccess21 using this model and the crystal structure of WDR5/MM-102 complex.
F. qRT-PCR Analysis of HOXA9 and MEIS-1 Genes
Murine MLL1-AF9 transformed bone marrow cells were obtained by transducing normal murine bone marrow cells with MLL1-AF9 oncogene according to the procedures described by Tan et al.22 MM-102 and C-MM-102 were dissolved in DMSO. The transformed cells were treated with MM-102 (25 μM, 50 μM), C-MM-102 (50 μM), and Mock (0.2% DMSO), giving a final concentration of 0.2% DMSO in all the samples. Total RNA was isolated from MLL1-AF9 transduced mouse bone marrow cells after 96 h treatment using Trizol (Invitrogen) and the RNEASY kit (QIAGEN) according to the protocol described earlier.23 The cDNA was generated using random priming with the SuperScript III kit (Invitrogen). Real-time PCR amplifications of HoxA9, Meis1, and GAPDH genes were carried out with primers specific for each gene in the presence of SYBR dye. Relative quantification of each gene transcript was carried out as described in our previous work.10 The results were presented as relative expression to Mock treatment after normalizing to an internal loading control (e.g., GAPDH or total input RNA).
G. Cell Growth and Apoptosis Studies of Leukemia Cell Lines
MV4;11, KOPN8, and K562 cells were a generous gift from Dr. Jolanta Grembecka (University of Michigan). MV4;11, KOPN8, and K562 cells were cultured in RPMI 1640 medium (ATCC) supplemented with 10% fetal bovine serum and 100 U/L penicillin-streptomycin and incubated at 37 °C under 5% CO2. Cells were seeded into 12-well plates for suspension at a density of 5 × 105 per well (1 mL) and treated with either vehicle control (DMSO, 0.2%) or MM-102 for 7 days. The medium was changed every 2 days, and compounds were resupplied.
The CellTiter-Glo Luminescent Cell Viability Assay kit (Promega) was used following the manufacturer’s instruction. First, 100 μL of the assay reagent was added into each well, and the content was mixed for 2 min on an orbital shaker to induce cell lysis. After 10 min incubation at room temperature, the luminescence was read on a microplate reader (SpectraMax M5, Molecular Devices).
Annexin V–propidium iodide assay was used to determine the effect of compounds on apoptosis induction. Cells were plated in 12-well plates with 5 × 105 cells per well (1 mL per well), treated with MM-102 or Mock control (0.2% final concentration of DMSO for all samples), and incubated for 96 h at 37 °C in a 5% CO2 incubator. After incubation, 1.5 × 105 cells were harvested and resuspended in 100 μL of 1× annexin V binding buffer from the Annexin V–FITC Apoptosis kit (BD Biosciences Pharmingen), incubated with 4 μL of Annexin V–FITC and 6 μL of propidium iodide (Sigma-Aldrich) at 25 °C in the dark for 10 min, and analyzed by flow cytometry on an LSR II instrument. Data analysis was performed using WinList software (Verity Software House).
RESULTS AND DISCUSSION
Probing the Binding of Ac-ARA-NH2 to WDR5 at Ala1, Arg2, and Ala3 Positions
Based upon the crystal structure of MLL1 peptide in complex with WDR5,12 we predicted the binding model of Ac-ARA-NH2 in complex with WDR5 (Figure 2a). Analysis of this binding model suggests that the key interactions between Ac-ARA-NH2 and WDR5 may be roughly divided into five sub-pockets (P1–P5 in Figure 2).
Figure 2.

(a) Predicted binding model of WDR5 complexed with Ac-ARA-NH2 peptide (sticks) and five binding pockets (P1–P5) in WDR5 (magenta for polar surface, green for hydrophobic surface and red for exposed surface). (b) Detailed interaction of Ac-ARA-NH2 peptide with residues of WDR5.
The methyl group of Ala1 in Ac-ARA-NH2 interacts with a small binding pocket (P1) formed by Y131 and F149 (Figure 2b). To probe this pocket, we replaced Ala1 with residues of different sizes, shapes and hydrophobicities (Table 1). Replacement of Ala1 with Gly (1a) decreases the binding affinity by a factor of >100. In contrast, substitution of Ala1 with either 2-aminobutyric acid (2-Abu) (1b) or Val (1c) improves the binding affinity by a factor of 2. However, residues with a side chain larger than the isopropyl group as in Val decreases the binding affinity significantly, as evident from the much reduced binding affinity of 1e and 1f. Changing Ala1 to Cys (1d) decreases the binding affinity by 3 times. Residues with a polar side chain such as Ser (1i) and Thr (1j) in this position also weaken the binding by 5 times as compared to Ala, and by approximately 10 times as compared to Val and Abu. A hydrophobic aromatic residue Phe (1h) or a polar aromatic residue His (1g) decreases the binding by 3–6 times as compared to Ala. Hence, the most favorable residues for binding in the P1 pocket are either 2-Abu or Val.
We next modified Arg2. In the crystal structure of WDR5 bound to MLL112 and in our predicted binding model of Ac-ARA-NH2 with WDR5,14 the Arg side chain inserts into a deep cavity in WDR5 and has extensive interactions with the P2 channel of WDR5 (Figure 2). The guanido group in Arg forms cation-π interactions with F133 and F263 (Figure S1). Two ω-nitrogen atoms in Arg also form hydrogen bonds with the backbone carbonyl groups in F133 and C261 and, via two water molecules, with the backbone carbonyl groups of S175 and S218 (Figure 2b). The δ-nitrogen in Arg forms a hydrogen bond with the backbone carbonyl group of S91. There are also hydrophobic contacts between the hydrophobic portion in the side chain of Arg2 and the I305 side chain. Since the P2 channel is narrow, we have selected a number of residues with linear side chains to probe this site (Table 1). Replacement of the guanido of Arg2 with a methyl group (2a) or removal of the amidine portion of the guanido group (2b) leads to complete loss of binding. Replacement of Arg2 with Lys yields another inactive compound (2c). Finally, replacement of the Arg guanido with urea group (2d), which would remove one of the hydrogen bonds and cation–π interactions, also fails to bind to WDR5. These results clearly show that the side chain of Arg2 is critical for binding to WDR5, consistent with the extensive interactions between Arg2 and WDR5 observed in our binding model.
Next, we modified Ala3, whose side chain is projected to a hemispherical hydrophobic P4 pocket formed mainly by the side chains of A47 and L321 in WDR5 (Figure 2). Replacement of the methyl group in Ala3 by hydrogen (3a) decreases the binding by >10-fold. Changing this methyl to an ethyl group (3b), however, improves the binding affinity by a factor of 20. Further increase in the size of the ethyl group to an isopropyl group (3c) or to a hydroxylethyl group (3i) reduces the binding affinity 3-fold as compared to 3b, although both 3c and 3i are still 5 times more potent than the ARA peptide. These results suggest the importance of the ethyl group in the side chain. A similar result was observed when Ala3 was replaced with Cys (3d). Peptides with a side chain larger than an isopropyl group, for example, ARL (3e), ARChg (3f), and ARF (3g), all have much weaker affinities than the ARA peptide. Finally, replacement of Ala3 with Ser (3h) reduces the binding by 3 times as compared to ARA, while Glu at this position (3j) decreases the binding by >100 times. Therefore, we conclude that an ethyl side chain as in Abu is the most favorable group for binding to WDR5 at this site.
Modifications Using Constrained Hydrophobic Side Chains To Target P1 and P4 Pockets
The modifications described above show that small hydrophobic groups at Ala1 and Ala3 positions are desirable for achieving high affinity binding to WDR5. We further explored these positions using a series of unnatural amino acids with small and constrained hydrophobic side chains. The tripeptides VRA and ARV were used as the templates for these modifications, since Val is favorable at both positions. The results are summarized in Tables 2 and 3.
Table 2.
Structures and Binding Affinities of Ac-XRV-NH2 Analogues Designed To Further Investigate the P1 Pocket
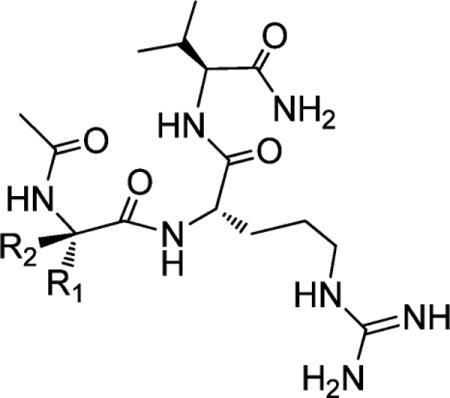
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | IC50 ± SD (μM) | Ki ± SD (μM) | |
| 3c | ARV |
|
−H | 0.11 ±0.02 | 0.02 ± 0.003 |
| 4a | ACPC-RV |
|
0.26 ±0.06 | 0.06 ± 0.013 | |
| 4b | CycVal-RV |
|
0.01 ± 0.008 | 0.002 ±0.001 | |
| 4c | CycLeu-RV |
|
0.04 ± 0.003 | 0.009 ± 0.001 | |
| 4d | homocycloLeu-RV |
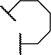
|
0.04 ± 0.01 | 0.007 ± 0.002 | |
| 4e | α-MeAla-RV |
|
|
0.01 ± 0.002 | 0.002 ± 0.0005 |
| 4f | Deg-RV |
|
|
0.02 ±0.01 | 0.004 ±0.002 |
| 4g | Tle-RV |
|
−H | 0.18 ±0.04 | 0.04 + 0.005 |
| 4h | Nva-RV |
|
−H | 0.18 ±0.01 | 0.04 ± 0.002 |
| 4i | Cpg-RV |
|
−H | 1.56 ±0.30 | 0.34 ± 0.06 |
| 4j | Phg-RV |
|
−H | 5.40 ± 1.10 | 1.20 ± 0.25 |
| 4k | D-Phg-RV | −H |
|
68 ± 4 | 14.6 ± 0.80 |
Table 3.
Structures and Binding Affinities of Ac-VRX-NH2 Analogues Designed To Further Investigate the P4 Pocket
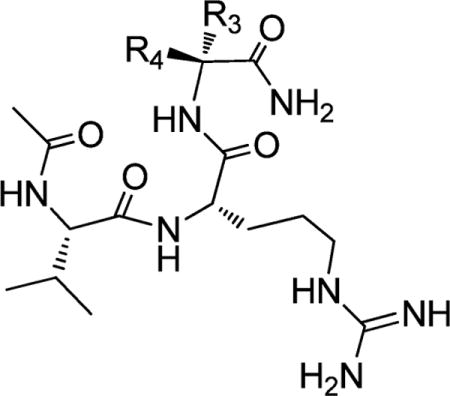
| |||||
|---|---|---|---|---|---|
| Compound | R3 | R4 | IC50 ± SD (μM) | Κi ± SD (μM) | |
| 1c | VRA | −H |
|
0.24 ± 0.03 | 0.05 ± 0.005 |
| 5a | VR-ACPC |
|
2.71 ± 0.34 | 0.6 ±0.07 | |
| 5b | VR-CycVal |
|
2.43 ± 0.55 | 0.52 ±0.12 | |
| 5c | VR-CycLeu |
|
0.09 ±0.01 | 0.018 ±0.001 | |
| 5d | VR-homocycloLeu |
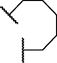
|
2.23 ± 0.67 | 0.48 ±0.14 | |
| 5e | VR-α-MeAla |
|
|
7.44 ± 0.15 | 1.6 ± 0.15 |
| 5f | VR-Tle | −H |
|
41 ± 7 | 8.7 ± 1.5 |
| 5g | VR-Cpg | −H |
|
41 ±5 | 8.8 ± 1.1 |
Similar binding affinities are observed when the Ala in ARV (3c) is replaced with either tert-leu (Tle, 4g) or norvaline (Nva, 4h) (Table 2). Cyclopentylglycine (Cpg, 4i) at this position decreases the binding affinity by 15 times as compared to 3c, whereas phenylglycine (4j) reduces the affinity by a factor of 50. Changing the chiral center in 4j, yielding 4k, further decreases the binding by >10-fold. Introduction of another methyl group in the Cα carbon in 3c yielded 4e, which is 10 times more potent than 3c. Changing the dimethyl groups in 4e to diethyl groups (4f) slightly decreases the binding affinity but 4f is still 5 times more potent than 3c. We next synthesized a series of compounds using the conformationally constrained cyclopropyl (4a), cyclobutyl (4b), cyclopentyl (4c), and cyclohexyl (4d) groups. While 4a with a cyclo-propyl group is 2 times less potent than 3c, the other three compounds all have improved binding affinities over 3c. Compound 4b with a cyclobutyl group is 10 times more potent than 3c and as potent as 4e with dimethyl groups at this site.
We next made similar modifications at the Ala3 position in the VRA peptide (1c) and the binding data are summarized in Table 3. Compound 5c with a constrained cyclopentyl side chain is 2 times more potent than 1c, while 5d with a cyclohexyl group is 2 times less potent than 1c. All the other modifications resulted in a decrease in binding affinity of at least 10 times.
Finally, we combined the most favorable modifications identified at the Ala1 and Ala3 positions. For the Ala1 position, we selected those used in 4b, 4e, and 4f, and for the Ala3 position, we chose those used in 3b, 3i, and 5c. These led to compounds 6a–6g (Table 4). Compounds 6a, 6c, 6d, and 6f have IC50 = 5–9 nM with Ki ≤ 1 nM. In direct comparison, compounds 6a and 6d are 100 times more potent than the initial ARA peptide (IC50 = 540 nM and Ki = 120 nM).
Table 4.
Binding Affinities of Peptidomimetics Combining Favorable Groups at the Ala1 and Ala3 Positions
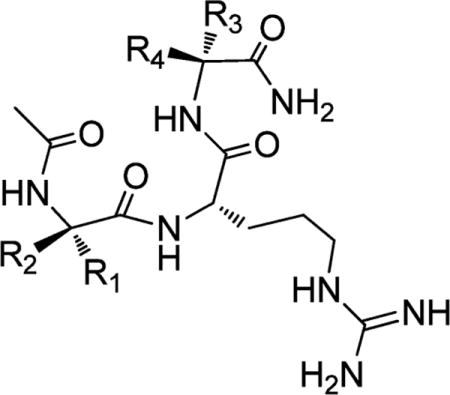
| ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | IC50 ± SD (nM) | Ki ± SD (nM) |
| ARA |
|
−H | −H |
|
540 ± 30 | 120 ± 10 |
| 6a |
|
|
−H |
|
5 ± 1 | <1 |
| 6b |
|
|
|
36 ± 5 | 8 ± 2 | |
| 6c |
|
|
−H |
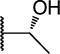
|
6 ± 0.4 | <1 |
| 6d |
|
−H |
|
5 ± 1 | <1 | |
| 6e |
|
|
32 ± 7 | 6 ± 2 | ||
| 6f |
|
−H |
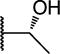
|
9 ± 1 | 1 ± 0.3 | |
| 6g |
|
|
|
80 ± 14 | 16 ± 3 | |
Further Modifications at the N- and C-Terminus
Based upon our predicted binding model of Ac-ARA-NH2 peptide with WDR5, the N-terminal portion of the molecule is projected toward a small hydrophobic pocket (P5 pocket, Figure 2) formed by G89 and A65. We thus modified the methyl group at this site with larger hydrophobic groups to further explore this site. As shown in Table 5, small groups such as isopropyl (7a, Ki = 0.04 μM) and cyclopropyl (7b, Ki = 0.11 μM) are much preferred over large (7g, Ki > 65 μM) or branched (7f, Ki = 4.7 μM) groups. The most favorable group at this position is isopropyl (7a, Ki = 0.04 μM), which improves the binding affinity by 3 times compared to methyl in Ac-ARA-NH2.
The C-terminal amide in Ac-ARA-NH2 is projected close to a small hydrophobic patch (P3 site) formed by hydrophobic portions of Y260 and K259 side chains (Figure 2). Similar to the N-terminal modifications, we substituted this portion of the molecule with different hydrophobic groups. The binding data for the resulting compounds are summarized in Table 6. All modifications larger than the methyl group (8b–8g) improve the binding affinity. In particular, the biphenylmethyl group in 8g enhanced the binding affinity by >10-fold.
Design of High-Affinity Peptidomimetics
Next we combined the most favorable groups for N- and C-termini with those of Ala1 and Ala3 positions for the design of highly potent peptidomimetics. We chose isopropyl and biphenylmethyl groups for N- and C-terminal modifications, respectively, and selected diethylglycine (Deg) for Ala1 and cycloleucine (CycLeu) for Ala3 positions. A combination of all these favorable modifications led to the design of MM-101 (MLL1 mimetic 101), which binds to WDR5 with IC50 = 2.9 nM and Ki < 1 nM (Table 7). Moreover, fluoro-substituted (MM-102) and chloro-substituted (MM-103) analogues were designed. Both MM-102 and MM-103 have a binding affinity to WDR5 similar to that of MM-101. However, MM-103 was found to have very low solubility and was not studied further.
Determination of Crystal Structures of MM-101 and MM-102 in Complex with WDR5
To provide the structural basis for the high-affinity binding of MM-101 and MM-102 to WDR5 and for further structure-based optimization, we determined their co-crystal structures in complex with WDR5 at a resolution of 3.4 Å (Figure S2) and 2.6 Å (Figure 3), respectively. Data collection and refinement statistics for WDR5/MM-101 and WDR5/MM-102 complexes are provided in the Supporting Information (Table S1).
Figure 3.
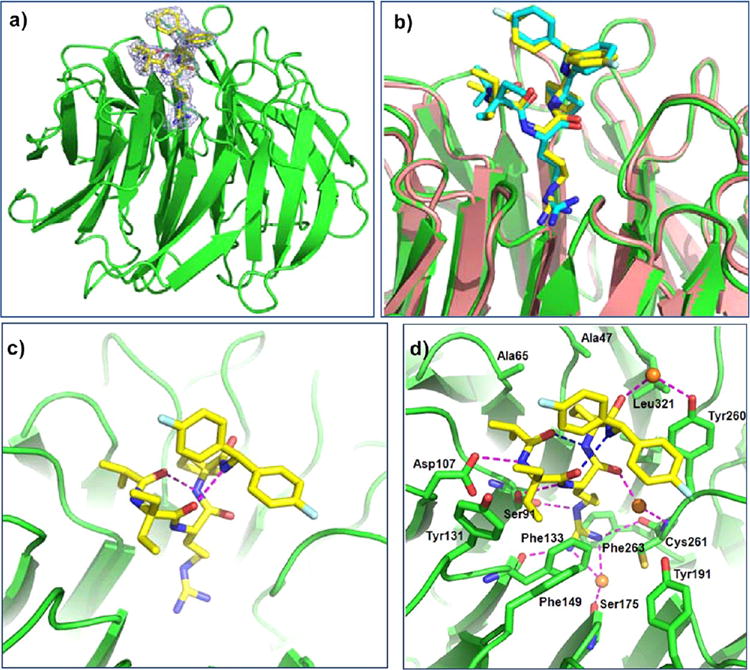
Crystal structure of WDR5/MM-102 complex. WDR5 is colored in green and MM-102 is colored in yellow. (a) Electron density (2Fo – Fc) map, contoured to 1δ, for MM-102. (b) Comparison of MM-101 and MM-102 in WDR5 complexes. In WDR5/MM-101 complex, WDR5 is colored in salmon and MM-101 is in cyan. (c) Intramolecular hydrogen bonds in MM-102 are shown in magenta dotted lines. (d) Interface between MM-102 (yellow) and WDR5 (green). MM-102 and WDR5 pack across an extensive interface, involving both hydrophobic packing and hydrogen bonds (magenta dotted lines). Intramolecular hydrogen bonds and water molecules are shown as blue dotted lines and orange spheres, respectively.
Consistent with our design, both compounds bind to the central channel (P2 pocket) of the WD40 propeller where MLL1 peptides bind (Figure 3a,b). As expected, MM-101 and MM-102 have very similar binding modes in their crystal structures (Figure 3b). In agreement with that observed in the MLL1 WIN peptide,12 two intramolecular hydrogen bonds are formed and stabilize the bound conformation for both MM-101 and MM-102 (Figure S2b for MM-101 and Figure 3c for MM-102).
Analysis of the higher resolution (2.6 Å) structure of the WDR5/MM-102 complex reveals the detailed map of the interaction network (Figure 3d). The Arg side chain of MM-102 inserts deeply into the central channel of WDR5, and is sandwiched between two aromatic rings from F133 and F263 (Figure S3a). All the nitrogen atoms in the molecule engage in either intra- or inter-molecular hydrogen bonds (Figure 3d). The N-terminal amide nitrogen and the Arg backbone nitrogen atoms interact with D107 and S91, respectively. The guanido group hydrogen bonds with S91, F133, and C261 directly and with S175 through a water molecule. The remaining amide nitrogen atoms at the C-terminus of the Arg residue form intramolecular hydrogen bonds. The first and second carbonyl groups from the N-terminus engage in intramolecular hydrogen bonds with the third and fourth amido groups from the N-terminus, respectively. The carbonyl groups at the C-terminus of Arg in MM-102 take part in water-mediated hydrogen bonds with C261 and Y260 of WDR5. MM-102 also makes extensive hydrophobic contacts with Y131, F149, F133, I305, Y260, K259, L321, A47, and A65 from WDR5, which are much more than those of the MLL1 peptide with WDR5. For example, the diethyl groups in the Ala1 position have extensive interactions with side chains of Y131 and F133 (Figure 3d); the cyclopentyl group at the Ala3 position has extensive interactions with the side chains of A47, S49 and L321 (Figure S3b). Furthermore, the diethyl groups at the Ala1 position and one of the phenyl groups shield the hydrophobic region constituted by F133, Y260, and C261 in WDR5 (Figure S4). Hence, MM-102 has much enhanced hydrophobic interactions to WDR5 as compared to the MLL1 WIN peptide, which explains its much higher affinity than the MLL1 WIN peptide.
Inhibition of H3K4 Methyltransferase Activity of the MLL1 Core Complex
Since the interaction of WDR5 and MLL1 is critical for H3K4 methyltransferase activity of the MLL1 complex,7,10–13 we predicted that our compounds designed to block this interaction should effectively inhibit the catalytic activity of the MLL1 complex.
To directly test this hypothesis, we developed and optimized a fully reconstituted in vitro HMT assay using recombinant MLL1, WDR5, RbBP5, and ASH2L proteins to form the core complex, an H3 10mer peptide as the substrate, and radio-labeled 3H-S-adenosyl methionine (3H-SAM) as the methyl donor. H3K4 methyltransferase activity was determined by incorporation of the radioactivity (3H-labeled methyl) into the Lys4 residue of the H3 peptide with a scintillation counter. Employing this assay, we have evaluated the inhibitory activity for a number of compounds with a wide range of binding affinities to WDR5, as determined in our FP-based assay. The results are summarized in Table 8, and representative curves for selected compounds in binding and HMT assays are shown in Figure 4 for comparison.
Table 8.
Binding Affinities and HMT Inhibition Activities of Selected Compounds
| compound | binding affinity to WDR5 | HMT inhibition |
|---|---|---|
| IC50 ± SD (nM) | IC50 ± SD (μM) | |
| 2d | >300 (μM) | >400 |
| 1a | 77.4 ± 13.2 (μM) | >400 |
| 1h | 3.1 ± 0.3 (μM) | >400 |
| ARA | 540 ± 30 | 141 ± 52 |
| 1c | 240 ± 30 | 12.1 ± 0.6 |
| 6g | 80 ± 14 | 2.8 ± 1.6 |
| 3b | 30 ± 8 | 1.8 ± 0.2 |
| 6b | 36 ± 5 | 5.2 ± 1.8 |
| 6a | 5 ± 1 | 0.5 ± 0.1 |
| 6c | 6 ± 0.4 | 0.9 ± 0.1 |
| 6f | 9 ± 1 | 0.7 ± 0.3 |
| MM-102 | 2.4 ± 1.7 | 0.4 ± 0.1 |
Figure 4.

(a) Inhibition of HMT activity of reconstituted MLL1 core complex as measured with scintillation counter assay in the presence of selected inhibitors. (b) Competitive binding curves of the same set of compounds for WDR5 as determined using FP-based binding assay.
Compounds 2d, 1a, and 1h, which have IC50 > 3 μM in the WDR5 binding assay, all fail to inhibit the MLL1 HMT activity up to 400 μM. The ARA peptide, which has IC50 = 0.54 μM to WDR5 in our binding assay, minimally inhibits the MLL1 HMT activity with IC50 = 141 μM. On the other hand, compounds with higher binding affinities to WDR5 effectively and dose-dependently inhibit the MLL1 HMT activity and have improved inhibitory activity over the ARA peptide. Four of the compounds, 6a, 6c, 6f, and MM-102, with the highest binding affinities to WDR5, also show the most potent inhibitory activity in the HMT assay with IC50 = 0.4–0.9 μM. The rank-order of the inhibitory activity for these compounds in the functional assay is in good agreement with that of their binding affinities to WDR5 (Table 8 and Figure 4). Of note, the apparent discrepancy between the IC50 values in the binding assay and HMT assay for these compounds is due to the fact that we used a 125-fold higher concentration of WDR5 protein (500 nM) in the functional assay than that (4 nM) in the binding assay.
Inhibition of HoxA9 and Meis-1 Expression by MM-102 in Cells
We next evaluated a number of potent compounds in binding and HMT assays, including MM-101 and MM-102, for their potential activity in modulation of MLL1 targeted gene expression in cells harboring MLL1 fusion proteins. Our initial screening data showed that MM-102 has the best activity in cells (data not shown). We therefore focused on MM-102 for further investigation.
HoxA9 and Meis-1 are two critical genes for MLL1 mediated leukemogenesis.5,24,25 Both genes are highly expressed in myeloblasts, the leukemia progenitors obtained upon transduction of murine bone marrow cells with the MLL1-AF9 fusion gene.26 One advantage of using MLL1-AF9 transduced myeloblasts in the gene expression assay is that the cells are homogeneous in terms of both differentiation status and HoxA9 expression level. Using these myeloblast cells, we tested the ability of MM-102 to inhibit expression of HoxA9 and Meis-1 genes to gain an insight into its cellular mechanism. As a control, we also tested a house-keeping gene GAPDH, whose expression is not regulated by MLL1.15
Treatment of the MLL1-AF9 transduced murine cells with MM-102, followed by gene expression analysis with quantitative RT-PCR (qRT-PCR), showed that MM-102 indeed reduces the expression of HoxA9 mRNA in a dose dependent manner (Figure 5, left panel). Approximately 50% and 70% reduction in gene expression was observed with 25 and 50 μM of MM-102, respectively, when compared with the Mock treatment. We also observed a clear decrease (40%) in Meis-1 expression with MM-102 at 50 μM (Figure 5). In contrast, MM-102 did not affect the expression of GAPDH, a house keeping gene (Figure 5, right panel), suggesting that the reduction of gene expression of HoxA9 and Meis-1 by MM-102 was not due to loss of cell viability.
Figure 5.
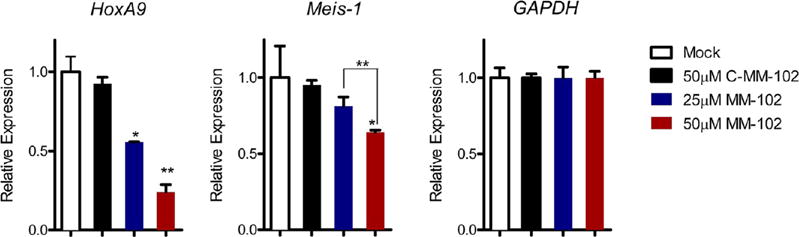
Inhibition of HoxA9 and Meis-1 expression in bone marrow cells transduced with MLL1-AF9 fusion gene upon MM-102 treatment for 96 h. The data were normalized to GAPDH expression, which is not affected by MM-102. *, p < 0.05; **, p < 0.01 (unpaired t test) compared to the Mock treatment.
To test the specificity of MM-102, we designed and synthesized an analogue of MM-102, C-MM-102. C-MM-102 is identical to MM-102 with the exception that the L-arginine was replaced with a D-arginine. C-MM-102 has Ki = 40 ± 16 nM in our binding assay and has the same physicochemical properties as MM-102. As shown in Figure 5 (left panel), C-MM-102 does not reduce HoxA9 expression at a concentration of 50 μM, suggesting that inhibition of HoxA9 expression by MM-102 is through its binding to WDR5 in cells.
These data show that MM-102 can specifically reduce expression of two critical MLL1 target genes required for MLL1 mediated leukemogenesis, supporting our hypothesis that inhibition of MLL1-WDR5 interaction can be used to regulate expression levels of MLL1 target genes.
Selective Inhibition of Cell Growth in Leukemia Cells Harboring MLL1 Fusion Proteins with MM-102
A previous study has shown that shRNA knockdown of HoxA9 in a panel of acute leukemia cell lines selectively inhibits growth of cells carrying MLL1 fusion proteins.27 We therefore evaluated MM-102 for its activity and selectivity in three leukemia cell lines with or without MLL1 fusion proteins.
As shown in Figure 6a, MM-102 dose-dependently inhibits cell growth in the MV4;11 and KOPN8 leukemia cell lines, which carry MLL1-AF4 and MLL1-ENL fusion proteins, respectively. MM-102 has IC50 = 25 μM in both cell lines and completely inhibits cell growth in these cell lines at 75 μM. In contrast, in the K562 leukemia cell line without MLL1 fusion protein but with BCR-ABL fusion protein, MM-102 has no effect for up to 50 μM and minimal effect at 75 μM.
Figure 6.
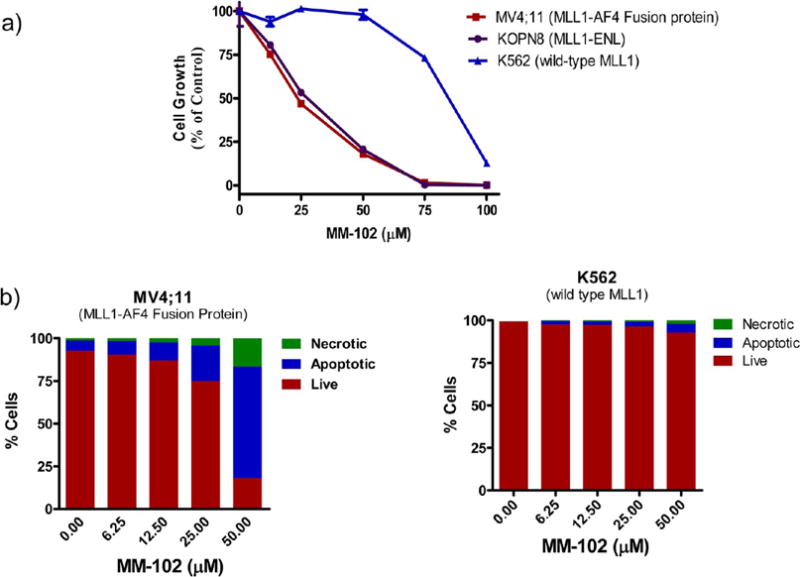
(a) Selective inhibition of growth of leukemia cell lines with MM-102 treatment for 7 days. (b) Induction of apoptosis by MM-102 in AML cell lines. Cells were treated for 96 h.
To further evaluate the activity and selectivity of MM-102, we tested its ability to induce apoptosis in the MV4;11 (MLL1-AF4) and K562 (wild-type MLL1) leukemia cell lines (Figure 6b). MM-102 effectively induces apoptosis in MV4;11 cells harboring MLL1-AF4 fusion protein in a dose-dependent manner and >75% of the cells undergo cell death with treatment of MM-102 at 50 μM for 96 h. In comparison, MM-102 has a minimal effect in K562 cells containing only wild-type MLL1 and <10% of the cells undergo cell death when treated with MM-102 up to 50 μM for 96 h.
Taken together, our data show that MM-102 effectively and selectively inhibits cell growth and induces apoptosis in leukemia cells harboring MLL1 fusion proteins and has minimal effect in leukemia cells with wild-type MLL1 protein.
SUMMARY
The H3K4 methyltransferase activity of MLL1 is tightly regulated by a core complex comprising MLL1, WDR5, RbBP5, and ASH2L (Figure 1a), and the interaction of WDR5 with MLL1 is essential for the structural integrity of this core complex and for its H3K4 methyltransferase activity. Our previous study has established that –CO-ARA-NH– in MLL1 is the minimal motif for the high-affinity binding between MLL1 and WDR5.14 In this study, we sought to further probe the MLL1 binding site in WDR5 with the goal to establish an extensive structure–activity relationship and to obtain highly potent peptides and peptidomimetics. Our second objective was to determine if high-affinity peptides and peptidomimetics designed to target the WDR5/MLL1 interaction can indeed function as potent antagonists of the MLL1 H3K4 methyltransferase activity in a fully reconstituted functional assay. Our third objective was to obtain cell-permeable compounds as probes with which to gain critical insights into their cellular mechanism, activity, and selectivity.
We performed extensive modifications on Ala1, Arg2, and Ala3 using both natural and unnatural amino acids starting from the ARA peptide. Modifications of Ala1 showed that small hydrophobic residues are preferred for high-affinity binding to WDR5. In particular, introduction of an additional methyl group into the Cα position enhances the binding affinity by >10 times. We found that unnatural amino acids with dimethyl, diethyl groups or a cyclobutyl group are the best for enhancing the binding affinity at this position. All the modifications on Arg2 residue led to complete loss of binding affinity, indicating the essential role of this residue. For Ala3, residues with small hydrophobic groups are also desirable for binding, and residues with an ethyl or a cyclopentyl side chain are the most preferred.
In the next step, we focused our efforts on modifications at N- and C-termini to probe two additional subpockets in WDR5 and have identified favorable hydrophobic groups for further enhancement of binding to WDR5. A combination of the most favorable groups for binding in all the five sites led to the successful design of MM-102. In our WDR5 binding assay, MM-102 achieves IC50 = 2.4 nM with an estimated Ki < 1 nM, which is >200 times more potent than the ARA peptide.
To determine if our designed compounds function as antagonists of the MLL1 HMT activity, we have developed and optimized a fully reconstituted in vitro functional assay using recombinant MLL1, WDR5, RbBP5 and ASH2L proteins, an H3 peptide, and a radio-labeled cofactor, 3H-S-adenosyl methionine. Using this functional assay, we evaluated the inhibitory activity of several compounds with a wide range of binding affinities to WDR5. Our functional experiments showed that our designed compounds with high binding affinities to WDR5 can indeed effectively and dose-dependently inhibit the MLL1 H3K4 methyltransferase activity in this fully reconstituted functional assay. Furthermore, their rank-order in inhibition of the MLL1 methyltransferase activity in the functional assay is in good agreement with that obtained from the WDR5 binding assay.
We determined the co-crystal structures of two potent compounds (MM-101 and MM-102) complexed with WDR5. Our co-crystal structures show that MM-101 and MM-102 bind to WDR5 with a similar binding mode as MLL1 peptides. While MM-101 and MM-102 maintain a similar intra- and intermolecular hydrogen bonding network as MLL1 peptides, additional hydrophobic contacts are observed between these peptidomimetics and WDR5. These co-crystal structures provide a solid foundation for further structure-based optimization of MM-101 and MM-102.
In our initial cell-based screening, we found that MM-102 is the most potent compound and we therefore employed MM-102 to gain further insights into its cellular mechanism. Using murine cells transduced with the MLL1-AF9 fusion gene, we showed that MM-102 effectively reduces the expression of HoxA9 and Meis-1, two essential MLL1 targeted genes for MLL1 mediated leukemogenesis. Importantly, MM-102 effectively and dose-dependently inhibits cell growth and induces apoptosis in leukemia cells harboring MLL1 fusion proteins and shows selectivity over leukemia cells containing only wild-type MLL1 protein. Although the cellular potency of MM-102 needs to be further improved for therapeutic applications, our data provide the first proof-of-concept that targeting MLL1/WDR5 protein–protein interaction using potent small-molecule inhibitors represents an attractive therapeutic strategy for acute leukemia carrying MLL1 fusion proteins.
Supplementary Material
Acknowledgments
Funding from the Leukemia and Lymphoma Society and American Association for Cancer Research and Stand-Up to Cancer is highly appreciated.
Footnotes
Supporting Information
Additional experimental data, and characterization of compounds by HRMS (ESI+), 1H NMR, and 13C NMR. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare the following competing financial interest(s): Asentage has licensed the technology related to this manuscript. Shaomeng Wang is a co-founder for Ascentage, owns stocks in Ascentage, and serves as a consultant for Ascentage.
References
- 1.Marmorstein R. Nat Rev Mol Cell Biol. 2001;2:422. doi: 10.1038/35073047. [DOI] [PubMed] [Google Scholar]
- 2.Kouzarides T. Cell. 2007;128:693. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Wang GG, Allis CD, Chi P. Trends Mol Med. 2007;13:363. doi: 10.1016/j.molmed.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 4.Chi P, Allis CD, Wang GG. Nat Rev Cancer. 2010;10:457. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J, Allis CD, Chait BT, Hess JL, Roeder RG. Cell. 2005;121:873. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 6.Ayton PM, Cleary ML. Oncogene. 2001;20:5695. doi: 10.1038/sj.onc.1204639. [DOI] [PubMed] [Google Scholar]
- 7.Dou Y, Hess JL. Int J Hematol. 2008;87:10. doi: 10.1007/s12185-007-0009-8. [DOI] [PubMed] [Google Scholar]
- 8.Thiel AT, Blessington P, Zou T, Feather D, Wu X, Yan J, Zhang H, Liu Z, Ernst P, Koretzky GA, Hua X. Cancer Cell. 2010;17:148. doi: 10.1016/j.ccr.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milne TA, Martin ME, Brock HW, Slany RK, Hess JL. Cancer Res. 2005;65:11367. doi: 10.1158/0008-5472.CAN-05-1041. [DOI] [PubMed] [Google Scholar]
- 10.Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. Nat Struct Mol Biol. 2006;13:713. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 11.Patel A, Vought VE, Dharmarajan V, Cosgrove MS. J Biol Chem. 2008;283:32162. doi: 10.1074/jbc.M806317200. [DOI] [PubMed] [Google Scholar]
- 12.Patel A, Dharmarajan V, Cosgrove MS. J Biol Chem. 2008;283:32158. doi: 10.1074/jbc.C800164200. [DOI] [PubMed] [Google Scholar]
- 13.Song JJ, Kingston RE. J Biol Chem. 2008;283:35258. doi: 10.1074/jbc.M806900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karatas H, Townsend EC, Bernard D, Dou Y, Wang S. J Med Chem. 2010;53:5179. doi: 10.1021/jm100139b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Couture JF, Collazo E, Trievel RC. Nat Struct Mol Biol. 2006;13:698. doi: 10.1038/nsmb1116. [DOI] [PubMed] [Google Scholar]
- 16.Han Z, Guo L, Wang H, Shen Y, Deng XW, Chai J. Mol Cell. 2006;22:137. doi: 10.1016/j.molcel.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 17.Otwinnowski Z, Minor W, editors. Methods in Enzymology. Academic Press; New York: 1997. [Google Scholar]
- 18.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Crystallogr. 2007;40:658. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr D: Biol Crystallogr. 2010;66:213. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emsley P, Cowtan K. Acta Crystallogr D: Biol Crystallogr. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 21.Hubbard SJ, Thornton JM. Naccess. Department of Biochemistry and Molecular Biology, University College London; 1993. [Google Scholar]
- 22.Tan J, Jones M, Koseki H, Nakayama M, Muntean AG, Maillard I, Hess JL. Cancer Cell. 2011;20:563. doi: 10.1016/j.ccr.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X, Li L, Pandey R, Byun JS, Gardner K, Qin Z, Dou Y. Cell Stem Cell. 2012;11:163. doi: 10.1016/j.stem.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thiel AT, Blessington P, Zou T, Feather D, Wu X, Yan J, Zhang H, Liu Z, Ernst P, Koretzky GA, Hua X. Cancer Cell. 2010;17:148. doi: 10.1016/j.ccr.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milne TA, Kim J, Wang GG, Stadler SC, Basrur V, Whitcomb SJ, Wang Z, Ruthenburg AJ, Elenitoba-Johnson KS, Roeder RG, Allis CD. Mol Cell. 2010;38:853. doi: 10.1016/j.molcel.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Somervaille TC, Cleary ML. Cancer Cell. 2006;10:257. doi: 10.1016/j.ccr.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 27.Faber J, Krivtsov AV, Stubbs MC, Wright R, Davis TN, van den Heuvel-Eibrink M, Zwaan CM, Kung AL, Armstrong SA. Blood. 2009;113:2375. doi: 10.1182/blood-2007-09-113597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.