

SUMMARY
Mutation of highly conserved residues in transcription factors may affect protein-protein or protein-DNA interactions leading to gene network dysregulation and human disease. Human mutations in GATA4, a cardiogenic transcription factor, cause cardiac septal defects and cardiomyopathy. Here, iPS-derived cardiomyocytes from subjects with a heterozygous GATA4-G296S missense mutation showed impaired contractility, calcium handling and metabolic activity. In human cardiomyocytes, GATA4 broadly co-occupied cardiac enhancers with TBX5, another transcription factor that causes septal defects when mutated. The GATA4-G296S mutation disrupted TBX5 recruitment, particularly to cardiac super-enhancers, concomitant with dysregulation of genes related to the phenotypic abnormalities, including cardiac septation. Conversely, the GATA4-G296S mutation led to failure of GATA4 and TBX5-mediated repression at non-cardiac genes and enhanced open chromatin states at endothelial/endocardial promoters. These results reveal how disease-causing missense mutations disrupt transcriptional cooperativity, leading to aberrant chromatin states and cellular dysfunction, including those related to morphogenetic defects.
Keywords: GATA4, TBX5, heart development, cardiomyopathy, congenital heart defects, disease modeling, systems biology, gene regulation, epigenetics, birth defects
Graphical abstract
A human missense mutation that causes congenital heart defects disrupts the cooperation between transcription factors at cardiac super-enhancers and gives rise to aberrant expression of endothelial genes
INTRODUCTION
Combinatorial interactions between transcription factors (TFs) result in tissue-specific gene expression that dictates cell identity and maintains homeostasis. TFs activate or repress gene transcription by recruiting other TFs, co-activators or co-repressors. Super-enhancers (SEs), clusters of putative enhancers densely occupied by Mediator complex and TFs, are implicated as regulators of cell identity in development and disease (Heinz et al., 2015; Whyte et al., 2013). SEs differ from typical enhancers (TEs) in size, motif density and transcriptional activation, rendering them more sensitive to changes in molarity of TF complexes. Dysregulation at SEs may contribute to human developmental disorders in embryogenesis and postnatal disease.
Developmental malformations occur in >5% of human births. Congenital heart defects (CHD) are most common (~0.8% live births) and are often due to haploinsufficiency of developmentally regulated cardiac TFs (Srivastava, 2006). Heterozygous mutations in TFs GATA4 and TBX5 cause familial CHD with overlapping phenotypes and we showed they co-immunoprecipitate when overexpressed. They are mutated in ~5% of sporadic CHD and are associated with cardiomyopathies (Rajagopal et al., 2007; Zhao et al., 2014). We reported a heterozygous disease-causing GATA4 glycine-to-serine missense mutation (G296S) that impaired in vitro interaction of GATA4 and TBX5 (Garg et al., 2003). Mice with compound heterozygous Gata4 and Tbx5 mutations develop atrioventricular septal defects (AVSD), providing genetic evidence for their interaction (Maitra et al., 2009).
Gata4—a TF with WGATAR-recognizing zinc fingers—is expressed in developing myocardial, endocardial, and endodermal cells (Heikinheimo et al., 1994). Gata4 deletion causes extraembryonic and foregut endoderm malformations (Kuo et al., 1997; Molkentin et al., 1997), and it is essential in regulating cardiomyocyte (CM) proliferation and septal development (Misra et al., 2012; Rojas et al., 2008). Deleting Gata4 in CMs causes cardiac decompensation and Gata4+/− mice have cardiac hypoplasia and reduced hypertrophic response to pressure overload (Bisping et al., 2006; Oka et al., 2006). Thus, Gata4 is essential in a dose-sensitive fashion for heart development and homeostasis.
Although Gata4 and Tbx5 are critical for mouse cardiogenesis, the gene targets or signaling pathways they co-regulate in human CMs and how they regulate human septal formation are unclear (Stefanovic et al., 2014; Xie et al., 2012). Complete loss of Tbx5 or Nkx2.5, Gata4-interacting partners, showed that these TFs interdependently modulate each other’s genomic occupancy in mouse cardiac differentiation (Luna-Zurita et al., 2016). Yet, it is unknown if this depends on protein-protein interactions and if dose-dependent perturbations in co-occupancy underlie heart disease.
We used patient-derived induced pluripotent stem (iPS) cells to dissect GATA4 regulatory mechanisms in human cardiac development and function. We found the heterozygous GATA4 G296S mutation impaired expression of the cardiac gene program and sonic hedgehog (SHH) signaling, while up-regulating genes of alternative fates, particularly the endothelial lineage and those related to cardiac septation. GATA4-dependent recruitment of TBX5 was disrupted at SE elements associated with genes for heart development and muscle contraction, and chromatin closure failed at loci involved in endothelial differentiation. This work reveals how a single missense mutation in a key cardiac TF leads to disease by dose-dependently regulating recruitment of TF complexes to enhancers and reveals potential nodes for therapeutic intervention.
RESULTS
Generation of Patient-Specific iPS Cells and Functional CMs
We reported a heterozygous c.886G>A mutation in human GATA4 linked to 100% penetrant atrial or ventricular septal defects (ASD; VSD), AVSD and pulmonary valve stenosis (PS) (Figure 1A, S1A) (Garg et al., 2003). Mutant-GATA4 translated into a G296S missense substitution flanking the second zinc-finger domain, involved in DNA-binding and protein-protein interactions (Figure 1A, bottom). Our previous study found abnormalities in cardiac morphogenesis, but we now found GATA4 G296S patients with delayed-onset cardiomyopathy. This was characterized by decreased left ventricular systolic function and an unusual echocardiographic appearance of the right ventricle with deep trabeculations and thickening of papillary muscles in the left ventricle (Figure 1B, Movie S1–2). Deep trabeculation is typical of non-compaction, thought to reflect failure of ventricular CMs to mature.
Figure 1. Pluripotent GATA4 iPS Cells and Differentiation to CMs.

(A) Top, GATA4 pedigree. Numbers in circles (females) and squares (males) are de-identified patient labels. Bolded border denotes CRISPR-corrected iPS line. WT, wildtype familial control. G296S, red, GATA4 mutants. cmy. cardiomyopathy. ASD, atrial septal defect. VSD, ventricular septal defect. AVSD, atrioventricular septal defect. PS, pulmonary valve stenosis. Bottom, schematic of GATA4 protein domains. TAD, transactivation domain. ZF, zinc-finger domain. NLS, nuclear localization signal.
(B) Still frames from transthoracic apical four-chamber view echocardiograms from a normal child and GATA4 G296S subject. Arrow indicates dense trabeculation in the right ventricle (RV). Right atrium, RA. Left ventricle, LV. Left atrium, LA.
(C) CRISPR-correction strategy.
(D) Sequence chromatograms show c.886G>A, G296S mutation in G296S 4, WT6 and CRISPR-corrected iWT4.
(E) Calcium flux measurements of hiPS-derived CM show expected responses to indicated agonists.
(F) Electron micrograph of representative iPS-derived CM. See also Figure S1.
We reprogrammed dermal fibroblasts from four subjects with the GATA4 G296S mutation and four family members without it into patient-specific iPS cells (Figure 1A). We used CRISPR/Cas9 nickases to edit the point mutation (A) back to its wildtype sequence (G) in iPS cells of patient 4 to yield isogenic controls (iWT) (Figure 1C–D). All cell lines had ES cell-like gene expression, morphologies and normal karyotypes (Figure S1B–E). RNA sequencing (RNA-seq) confirmed a genome-wide correlation in gene expression signature between ES and iPS cells (Figure S1F). All iPS lines differentiated into the three germ layers (Figure S1G).
We used a step-wise differentiation protocol to generate purified CMs from the iPS cell lines (Figure S2A) (Lian et al., 2012; Tohyama et al., 2013). RNA-seq at various times showed stage-specific gene signatures for mesoderm, cardiac progenitor cells (CPCs) and CMs with expected gene ontologies (GO) (Figure S2B–D). iPS-CMs spontaneously contracted, expressed sarcomeric markers, and had membrane electrophysiology and gene expression similar to human CMs; 30% were binucleated (Figure S2E–H). Calcium flux showed proper drug responses. Electron microscopy indicated abundant mitochondria with defined Z-lines and sarcomeres (Figure 1E–F).
Impaired Contractility, Calcium Handling, and Metabolic Activity in Mutant CMs
We generated >90% pure cTnT+ day 32 (D32)-CMs from WT, G296S, and CRISPR-corrected isogenic iPS cells (Figure 2A), although mutant lines showed slight delays in onset of spontaneous contraction (Figure S2I–J). We built a micropatterning platform to measure contraction of single iPS-derived CMs (Figure 2B) (Ribeiro et al., 2015). Only 50% of patterned G296S CMs responded accurately to electrical pacing at 1Hz, compared to 70% of WT CMs. While WT cells did not respond to pacing at frequencies over 1Hz, 20% of G296S CMs beat at a faster rate (Figure 2C). G296S CMs had reduced contractile force generation per cell movement with decreased contraction time (Figure 2C, S2K), consistent with the cardiomyopathic phenotype in patients. Upon further differentiation at D70, G296S CMs were dysfunctional in response to electrical pacing and relaxation velocity, but force generation improved (Figure S2L–N).
Figure 2. GATA4 G296S CMs Have Impaired Cardiac Function.

(A) FACS analysis of cTnT+ CMs from representative WT and G296S differentiation after lactate purification.
(B) CMs micropatterned in arrays of single cells (top) and immunostained for αActinin or F-actin (bottom).
(C) Contractile measurements on micro-patterns. % of single-CM responding to 1 Hz pacing in WT and G296S (left). Traction force microscopy measurements of force production as a function of cell movement of CMs responding to 1Hz pacing (right). All measurements were done in triplicate with CM generated independently from two patient lines. Data for patient 4 are shown.
(D) Action potential measurements of WT and G296S CM. Overshoot potential (OSP) indicates highest membrane potential reached. Data shown are mean ± SEM from 2 WT and 2 G296S lines. *, p<0.05 (Mann-Whitney test).
(E) Calcium flux measurement on microclusters. F/F0 (Max), peak amplitude relative to baseline fluorescence between action potentials. Data shown are mean ± SEM from 2 WT and 2 G296S lines. *, p<0.05 (t test).
(F) Calcium flux measurement on patterned microtissues. CMs patterned on hydrogels of 10kPa-stiffness; 1-mm-long lines (left) and calcium flux measured as F/F0 (center). Rates of rise and fall (right) between action potentials. Data are mean ± SEM. *, p<0.05 (t test).
(G) Percentage of CMs of individual sarcomeric classes observed by α-Actinin staining. Class IV represents the most disarrayed sarcomeric organizations. n= >150 CMs.
(H) Mitochondria staining intensity of single-CM micropatterns (top). Mitotracker red intensity relative to cell area was quantified (bottom). Data shown are mean ± SEM from 2 G296S lines. **, p<0.005 (t test).
(I) Seahorse measurements of glycolytic functions. Isogenic CM data are mean ± SEM. **, p<0.005, ***, p<0.0005 (t test). See also Figure S2.
In patch-clamp studies, G296S CMs had increased overshoot potential without altered maximum upstroke velocity or action potential duration (Figure 2D, S2O), suggesting a more depolarized membrane. Calcium transients in cell clusters had increased relative peak amplitude, suggesting defects in calcium ion handling (Figure 2E). When CMs were patterned onto 1-mm lines to induce uniaxial cell-cell communication, calcium flux in G296S CMs was higher (Figure 2F). A larger percentage of G296S CMs had disorganized sarcomeres (Figure 2G, S2P).
We hypothesized that the reduced contractile force came from defects in mitochondrial function or metabolic activity. Indeed, G296S CMs had decreased mitochondrial staining (Figure 2H), glycolytic capacity and glycolytic reserve (Figure 2I). Although mitochondrial DNA (mtDNA) heteroplasmy is linked to neuropathogenicity, sequencing showed no increased de novo mutations of G296S mtDNA (Figure S2Q).
Attenuated Cardiac Gene Program in Mutant CPCs and CMs
We performed RNA-seq on isogenic iPS cells during differentiation into CPCs on day 7, contracting CMs before (D15-CMs) and after (D32-CMs) lactate purification (Figure 3A, Table S1). LASSO-regression algorithm predicted the iWT CM data represented the heart transcriptome (0.6–1). Transcriptomes of GATA4 G296S cells from each stage had lower cardiac scores (<0.6) (Figure 3B). In G296S cells, 2228 genes were differentially expressed in at least one of the three stages with dynamic changes going from CPCs to mature CMs (Figure S3A–B). At all stages, 38 genes in Wnt-planar cell polarity pathway or vasculature-, endocardial-, heart-development or cardiac progenitor differentiation were dysregulated (Figure S3C–D).
Figure 3. Transcriptome Aberrations in G296S CPCs and CMs.

(A) Heatmap shows hierarchical clustering of Spearman correlation scores for all differentiation time course samples based on RNA-seq profiles. Red, GATA4 mutants. Score of 1 (yellow) denotes perfect correlation.
(B) Human fetal tissue prediction matrix for all differentiation time course samples based on RNA-seq profiles. Red, GATA4 mutants. Score of 1 (green) denotes highest similarity.
(C). GSEA (top) and heatmap (bottom) shows downregulation of SHH signaling response genes in G296S CPCs. NES, normalized enrichment score. Values are row-scaled to show relative expression. Blue and red are low and high levels respectively.
(D–F) Heatmap shows hierarchical clustering of differentially expressed genes in CPCs (D), D15-CMs (E) or D32-CMs (F). Values are row-scaled to show relative expression. Blue and red are low and high levels respectively. Representative down- (blue box) and up-regulated (red box) genes are listed.
(D′–F′) GO analyses of down- (blue box) and up-regulated (red box) genes in CPCs (D′), D15-CMs (E′) or D32-CMs (F′). Significance shown as −Log10 Bonferroni p-value after multiple hypothesis correction. See also Figure S3.
In G296S CPCs, Gene Set Enrichment Analyses (GSEA) showed decreased expression of genes typically present in cells receiving the SHH signal, including the PTCH1/PTCH2 receptors and GLI2/GLI3 transcriptional effectors (Figure 3C). In development, SHH secreted by pulmonary endoderm is received by neighboring atrial myocardium, resulting in growth of the atrial septum (Hoffmann et al., 2009); disrupting this in mice yields ASDs and AVSDs. Thus, down-regulating genes required for SHH response is consistent with septal defects of GATA4-G296S patients. More broadly, down-regulated genes were involved in heart development, cardiac chamber morphogenesis, myofibril assembly, heart contraction and cardiac progenitor differentiation, suggesting incomplete activation of the myocardial gene program (Figure 3D, 3D′). Upregulated genes (e.g., TAL1, ETS1, ROBO4, SOX17, TIE1, KDR and KLF5) were involved in vasculature development, angiogenesis, extracellular matrix organization, integrin interactions and calcineurin-NFAT transcription. Many are signaling or transcriptional regulators of the endocardial/endothelial program.
The percentage of G296S CPCs expressing high levels of GATA4, NKX2.5 and TBX5 were reduced, validating the gene expression decrease. Reciprocally, abundance of the endothelial-specific protein, KDR, was increased in GATA4/NKX2.5/ISL1-positive CPCs (Figure S3E, F). The percentage of CD31-positive endothelial cells did not increase in unpurified D15 cultures (Figure S3G). Thus, the upregulated endothelial/endocardial program was likely due to failure in gene silencing, rather than more cells adopting an endothelial fate. When iPS cells were differentiated to promote the endothelial lineage (Theodoris et al., 2015), G296S cells were only marginally increased in propensity to commit into endothelial cells (Figure S3H).
In G296S D15-CMs, down-regulated genes were critical in organ morphogenesis, heart development and glycolysis (Figure 3E, 3E′), consistent with the phenotypic abnormalities and impairment in glycolysis (Figure 2I). Like the CPC stage, up-regulated genes at D15 participated in blood vessel development, cell-cell communication, and Integrin and PI3K-Akt signaling pathways. Persistent expression of cardiac progenitor genes, such as ISL1, and upregulation of smooth muscle genes suggested alternative fate genes were abnormally activated as CMs matured. Increased expression of CAMK2D and CASQ2 was consistent with the increased calcium transients (Figure 2E–F).
In D32-CMs purified by lactatehi glucoselo media, differentially expressed genes continued to show an attenuated cardiac gene program and persistent up-regulation of the endothelial/endocardial gene program (Figure 3F, 3F′). GO terms for heart development, muscle contraction, cardiomyopathy, and cardiac septum development were enriched in downregulated genes. Up-regulated genes were enriched for vasculature development, angiogenesis and PI3K-Akt signaling. Genes in vascular and neuronal pathfinding were most upregulated in the neurogenesis category. Also, G296S CMs down-regulated chamber myocardium genes, and up-regulated atrioventricular canal myocardium and smooth muscle-associated genes, suggesting a broader mis-specification in cell identity (Figure S3I). Up-regulating TBX2 was notable, given that it represses “working” myocardium genes and regulates atrioventricular canal development (Aanhaanen et al., 2011). Cellular respiration genes was reduced in G296S CMs (Figure S3J), consistent with decreased metabolic activity observed (Figure 2H–I). Quantitative PCR validation of the RNA-seq results showed a strong correlation for all three stages (Figure S3K).
Open Chromatin Anomalies in GATA4 Mutant CPCs
Chromatin accessibility is linked to TF occupancy and transcriptional output (Zaret and Carroll, 2011). To examine changes in open chromatin status (Figure 4A), we analyzed transposase-accessible chromatin by deep sequencing (ATAC-seq) in iWT and G296S CPCs (Table S2) (Buenrostro et al., 2013). In iWT CPCs, 14,532 ATAC-seq loci had 88% overlap with ENCODE DNase-hypersensitivity sites (DHSs) from human-CMs or ES-derived CPCs and at loci expected to be transposase-accessible (Figure 4B–C). Furthermore, >75% had histone marks of activation (H3K4me3) but not repression (H3K27me3), and localized to introns (43%) of protein-coding genes (82%) (Figure 4C–D). In G296S CPCs, open chromatin status was broadly reduced at cardiac genes (Figure 4B, E), consistent with their decreased expression (Figure 3D). Open chromatin status was increased at SOX17, a key regulator of hemogenic-endothelium (Clarke et al., 2013). This trend was also seen at 86 cardiac and 99 endothelial genes that were differentially expressed (Figure 4F).
Figure 4. Chromatin Accessibility Aberrations in G296S CPCs.

(A) GSEA analyses of genesets for cardiac (top) and endothelial/endocardial (bottom) development. NES, normalized enrichment score. FDR, false discovery rate. Positive and negative NES indicate higher and lower expression in iWT respectively.
(B) IGV browser tracks at chr14:23693015-24168059 show normalized ATAC-seq signal from WT (black) and G296S (red) matches normalized signal from ENCODE-DHS (blue) (grey regions).
(C) Heatmap of normalized read counts from ENCODE DHSs, H3K4me3, H3K27me3 (D5CPC) around ATAC-seq loci identified in iWT CPCs. White and blue are low and high signal intensity, respectively.
(D) Pie-chart shows gene-body, upstream, downstream distribution (top) and coding and non-coding gene distribution (bottom) of 14532 iWT ATAC-seq loci.
(E) IGV browser tracks at TBX5 (top) and SOX17 (bottom) loci show decreased and increased (grey regions) ATAC-seq signal between WT (black) and G296S (red). Y-axis shows reads/million/25 bp. Blue track, normalized GATA4 ChIP-seq signal in WT1 CMs.
(F) Metagenes plot of iWT (black) and G296S (red) normalized ATAC-seq signal ± 5 kb around the TSS of genesets for cardiac (top) and endothelial (bottom) development.
(G) Known consensus motifs enriched in ATAC-seq loci up-regulated in G296S CPC.
(H) GO analyses of down- (blue box) and up-regulated (red box) ATAC-seq loci after generic loci were filtered out using a fibroblast DHS dataset. Nearest gene to a peak was defined within a 100kb window. Significance shown as −Log10 Bonferroni p-value after multiple hypothesis correction.
(I) FPKM values of select, differentially expressed NOTCH and NFAT target genes in iWT and G296S CPCs or CMs. Data are mean ± SEM. *, FDR<0.05.
Genomic loci with increased ATAC-seq signal were enriched for DNA motifs of core transcriptional regulators of endothelial cells (SOX17, KLF5, FOXO1, STAT6) and ETS-factors (GABPA, ELF5, ERG), suggesting the endocardial/endothelial program was not effectively silenced in G296S CPCs (Figure 4G). These loci mapped to genes involved in AV valve morphogenesis, coronary vasculogenesis and endocardial cushion development (Figure 4H), consistent with the AVSD diagnosis in the individual with the GATA4 mutation (Figure 1A). Hey1 and Hey2 are GATA4-interacting co-repressor proteins (Kathiriya et al., 2004) that were down-regulated in G296S CMs and may contribute to failure of chromatin closure at endothelial/endocardial genes, while genes dependent on NFATc, an endocardial regulator, were upregulated (Figure 4I).
Genome-Wide Co-Occupancy of GATA4 and TBX5 in Human CMs
Open chromatin anomalies in mutant CPCs and GATA4’s known function as a “pioneer factor” (Cirillo et al., 2002) led us to survey the genome-wide occupancy of GATA4 and TBX5 and histone marks of active-promoters (H3K4me3), repressed-promoters (H3K27me3), transcription elongation (H3K36me3) and active-enhancers (H3K27ac) (Table S3, S4). In WT CMs, chromatin immunoprecipitation with antibodies to the endogenous protein and deep sequencing (ChIP-seq) validated many direct targets of GATA4 or TBX5 identified in mouse studies (Figure 5A, S4A). These gene targets were co-bound by GATA4 and TBX5 (G4T5), had high levels of H3K27ac, H3K4me3 and H3K36me3, but undetectable H3K27me3. GATA4 and TBX5 ChIP-seq signals positively correlated with gene expression (Figure S4B). GATA4, TBX5 and H3K27ac shared the strongest overlap in genome occupancy (Figure 5B), with nearly half of GATA4 sites co-bound by TBX5 (Figure S4C–D). The 2,428 sites co-bound by human G4T5 had higher ChIP-seq signals than sites bound by GATA4 or TBX5 alone (Figure 5C). Co-bound sites mapped to intronic (48%) and intergenic (35%) sites of genes for myofibril assembly, cardiac muscle development and contraction, CHD and cardiomyopathy (Figure S4E–F). GATA4 and TBX5 motifs ranked at the top in motif analyses of G4T5 sites (Figure 5D). Motifs for TEAD4, MEF2C, NKX2.5, ISL1, SRF and SMAD2/3 were enriched at these loci, indicating a TF code that maintains the cardiac gene program. Enrichment of motifs for endothelial regulators, FOXO1 and HOXB4, indicate a potential repressive role for G4T5 at these sites.
Figure 5. TF Mis-Localizations in G296S CMs.

(A) IGV browser tracks of indicated ChIP-seq signals at known GATA4 target loci (NPPA, NPPB) in WT CM. Grey boxes, significant peaks identified by MACS2. Y-axis shows reads/million/25 bp.
(B) Metagenes plot of normalized ChIP-seq signals for indicated factors at 2428 G4T5 co-bound sites (±5kb) identified in WT CM.
(C) Normalized GATA4 (left) or TBX5 (right) signal at sites that are G4T5 co-bound versus single TF bound. Boxplot and whiskers show mean, 25th and 75th percentile followed by 5th and 95th percentile. ****, p<0.00005, (Kolmogorov-Smirnov test).
(D) Known consensus motifs enriched in 2428 G4T5 co-bound sites in WT CM.
(E) Venn diagram shows changes in GATA4, TBX5 or G4T5 bound sites between WT and G296S CMs. Number of sites lost in WT (L), gained in G296S (E) and unchanged (U) are shown (top row). Legend for metagenes of relative (G296S/WT) ChIP-seq occupancy at sites that are L (blue line), U (green) or E (red) (top row, far-right). 2nd to 4th rows show relative changes in GATA4, TBX5, and H3K27ac occupancy at these L, U or E sites.
(F) FPKM values of genes mapped ± 20 kb of 1186 G4T5L sites in iWT and G296S cells at 3 differentiated stages. Boxplot and whiskers show mean, 25th and 75th percentile followed by 5th and 95th percentile. *, p<0.05, **, p<0.005, ***, p<0.0005, (Wilcoxon signed-rank test).
(G) Gap distances between GATA4 and TBX5 motifs within G4T5U vs.G4T5L sites on the same (blue) or different (red) DNA strand. Boxplot and whiskers show mean, 25th and 75th percentile followed by 5th and 95th percentile. *, p<0.05 (Fisher’s exact test).
(H) Bar graph showing number of sites with ≥1 GATA4-TBX5 motif pairs (left) and number of motif pairs on same or different DNA strands (right) within G4T5U vs. G4T5L sites. *, p<0.05, ****, p<0.00005 (Fisher’s exact test).
(I) GO analyses of 1186 G4T5L sites. Significance shown as −Log10 Bonferroni p-value after multiple hypothesis correction.
(J) Heatmap shows hierarchical clustering of 414 putative G4T5 target genes in D15-CMs/D32-CMs and changes to GATA4 and TBX5 binding. RNA-seq expression is row-scaled to show relative expression (left). ChIP-seq shows relative (Log2FC) GATA4, TBX5 occupancy (right). One ChIP-seq peak with the largest fold difference was selected for each gene. Rows between GATA4 and TBX5 are approximately matched. Blue and red are low and high levels respectively.
(K) Heatmap shows clustering of 82 endothelial genes and changes to GATA4 and TBX5 binding within endothelial TADs. RNA-seq (left) and ChIP-seq (right) show relative (Log2FC) gene expressions and GATA4, TBX5 occupancy (right). Blue and red are down- and up-regulation respectively. Rows between RNAseq and ChIPseq results are approximately matched. See also Figure S4 and S5.
We systematically compared sites bound by GATA4, TBX5 or G4T5 in G296S and WT cells (Figure 5E; top). For GATA4 sites, 54% of sites were lost (L), 46% were unchanged (U), and 16% were ectopic sites gained (E) in mutants, suggesting dose-sensitivity for DNA-binding at many sites and redistribution to others. For TBX5 sites, 26% were lost (L), 74% were unchanged (U), and 24% were ectopically gained (E). G296S had 34% fewer G4T5 co-bound sites than WT CMs (Figure S4D, S5A–B), with 48% lost (L), 52% unchanged (U), and 21% gained (E). Next, we parsed the L, U and E sites for the relative occupancy of GATA4, TBX5 and H3K27ac (Figure 5E; bottom). Consistent with the reduced DNA binding affinity of G296S GATA4, GATA4 occupancy was decreased particularly at G4L and G4T5L sites and correlated with increased TBX5 occupancy particularly at T5E and G4T5L. A broad increase in TBX5 occupancy suggested that loss of TBX5 occupancy at other sites was unlikely due to decreased TBX5 gene expression. The active enhancer mark H3K27ac was increased most at G4E, T5E, and G4T5E sites. Nearly all of the changes were significant (Figure S5C) and GATA4, TBX5 and H3K27ac were not mis-localized at random genomic sites. From our RNA-seq data, genes mapping to G4T5L sites were largely down-regulated in CPC, D15-CMs and D32-CMs (Figure 5F, S5D).
To gain insights into a motif grammar that may explain why some loci were more sensitive to loss of G4T5 co-binding in the presence of the GATA4 G296S mutation, we compared the distance between GATA4 and TBX5 motifs within sites that lost G4T5 co-binding and sites unchanged in co-binding. The distance in GATA4 and TBX5 motifs was greater in G4T5L than G4T5U sites, regardless of strandedness (Figure 5G). G4T5L sites had fewer GATA4-TBX5 motif pairs than G4T5U sites, and motif pairs within G4T5U sites were preferentially located on the same strand, compared to those in G4T5L sites (Figure 5H). Thus, protein-DNA interactions may compensate for disrupted protein-protein interactions. Also, motif analyses identified PRDM1, NR5A2, IRF1, PBX1 and HNF4A motifs in G4T5L sites, and TEAD4, EGR1, HIF1A, MEIS1/3p-TBX5 motifs in G4T5E sites (Figure S5E). Cross-referencing the G4T5L sites to binding sites of >200 ENCODE transcriptional regulators revealed closest proximity to p300-, CTCF-bound neuronal enhancers (Figure S5F). These results indicate that G4T5 cooperation is most robust when underlying cis-sequences are closely linked on the same DNA strand, ~75 bp apart, but may be most sensitive to perturbation at enhancers active in non-cardiac cells, perhaps due to weaker DNA interactions that require tethering of the TFs.
Consistent with an impaired cardiac gene program (Figure 2–4), G4T5L genes were involved in cardiac muscle contraction, cardiac septal defect, and cardiomyopathy (Figure 5I). To determine putative GATA4 and TBX5 targets, we examined all differentially expressed genes with a G4T5 site within 20 kb (Figure S5G). Genes with decreased GATA4 and TBX5 binding (G4DOWN_T5DOWN) were down-regulated (Figure S5H), suggesting differential gene expression is directly due to DNA binding aberrations by GATA4 and TBX5. GATA4 binding was decreased and TBX5 binding concomitantly increased at 414 putative targets (Figure 5J). Importantly, GATA4 binding was reduced at 49% of sites near 82 up-regulated endothelial genes (Figure 5K). Consistent with TBX5-motif enrichment in G296S CPCs (Figure 4G), TBX5 binding was increased at 64% of 207 TBX5 sites within these endothelial topologically associating domains, suggesting anomalous transcriptional activation by mis-localized TBX5 and perhaps other coactivators. This correlated with increased H3K4me3 and decreased H3K27me3 marks at endothelial TSS in G296S CMs (Figure S5I). Proximal promoters of up-regulated endothelial genes were enriched in binding sites for GATA-, FOXO-, and ETS-family proteins (Figure S5J).
GATA4 binding sites within endothelial TADs and PI3K genes mapped closely with binding sites of multiple co-repressors (Figure S5K) whose proteins were expressed at detectable levels (Figure S5L). Since the GATA4/HDAC complex mediates gene repression in mouse AV canal (Stefanovic et al., 2014), we performed HDAC2 ChIP-seq in D15-CMs. GATA4 and HDAC2 binding sites overlapped and sites that were TBX5-HDAC2 co-bound were enriched in genes for cardiovascular development, muscle cell differentiation, insulin and integrin signaling (Figure S5M–N). At endothelial TADs in G296S CMs, ~30% of sites had less HDAC2 binding than WT-CMs, suggesting endothelial gene upregulation was partially attributed to decreased HDAC2-repression (Figure S5O).
GATA4 and TBX5 Co-Regulate Human Cardiac SEs
Regions of high MED1 (Mediator Complex) occupancy across several kilobases mark SEs (Whyte et al., 2013), but MED1-classified SEs have not been described in human CMs. Here, we identified 213 SEs (top 4%) by MED1 ChIP-seq in WT CMs (Figure 6A) (Table S4). These were proximal to cardiac-enriched genes with multiple constituents of G4T5 and robust H3K27ac enhancer marks (Figure 6B, S6A). MED1 ChIP-seq signal positively correlated to gene expression levels (Figure S6B). SEs had 11-fold more MED1 binding than TEs, were longer (3–80 kb; average, 10kb), and induced four-fold more gene expression (Figure 6C, S6C). As expected, GATA4 and TBX5 binding was enriched in SE elements (Figure 6D, S6D), as were motifs for MEF2C, SRF, TEAD4, SMAD2/3, MEIS1 and NKX2.5, all critical for cardiac development (Figure 6E). SE elements were near genes involved in striated muscle development, cardiomyopathy, heart development and cardiac muscle contraction (Figure 6F).
Figure 6. Aberrant Cardiac SE Regulation in G296S CMs.

(A) Distribution of MED1 ChIP-seq signal across 5,040 putative enhancers in WT CM. 213 SEs show highest MED1 intensity. Representative genes within 20 kb are labeled.
(B) IGV browser tracks of ChIP-seq signals at MYH6 and MYH7 loci show 47 kb SE element. A 1.3 kb STAU2 TE also shown. Y-axis shows reads/million/25 bp.
(C) Enhancer length (left) and nearest (20 kb) gene expression (right) of TEs vs. SEs. Boxplot and whiskers show mean, 25th and 75th percentile followed by 5th and 95th percentile. ****, p<0.00005 (t test).
(D) Metagenes plot of normalized ChIP-seq signals at 4827 TE and 213 SE identified in WT CMs.
(E) Known consensus motifs enriched at constituent enhancers within SE elements in WT CM.
(F) GO analyses of 213 SE elements. Significance shown as −Log10 Bonferroni p-value after multiple hypothesis correction.
(G) Distribution of MED1 ChIP-seq signal in G296S CMs. 172 SEs show highest MED1 intensity. Representative genes within 20 kb are labeled.
(H) Venn diagram (top) shows changes in MED1-bound SE elements between WT (black circle) and G296S (red). Number of sites lost in WT (L), gained in G296S (E) or unchanged (U) are shown.
(I) Metagenes plot of normalized GATA4 and TBX5 ChIP-seq signal within SE that are L, U or E in WT (black line) and G296S (red) CM.
(J) Example genes within 20 kb of the SE elements that are L, U or E in G296S CMs.
(K) FPKM values of genes mapped ± 20 kb around SEs in iWT and G296S cells at 3 differentiated stages. Boxplot and whiskers show mean, 25th and 75th percentile followed by 5th and 95th percentile. *, p<0.05, ***, p<0.0005, ****, p<0.00005 (Wilcoxon signed-rank test).
(L) Sub-network extracted from global network diagram analyzed by TDA shows enrichment for genes regulated by SE regions and co-bound by GATA4-TBX5. Red and blue colors represent high and low enrichment respectively. Blue colors in GATA4-siRNA expression network show down-regulation of this SE-regulated geneset upon GATA4 knockdown. See also Figure S6 and S7.
In contrast, MED1 ChIP-seq in G296S CMs identified 172 SE elements (Figure 6G). Comparison of SE elements showed loss of 34% (SEL), with 66% being unchanged (SEU) and 12% being ectopically gained (SEE) in mutant CMs (Figure 6H). TBX5 binding in the SEL and SEU elements were markedly reduced, despite comparable GATA4 DNA-binding (Figure 6I, S6E), most likely from disruption of the GATA4–TBX5 interaction and failure of GATA4 to recruit TBX5 to cardiac SEs. Key cardiac genes with lost SE elements included RBM20, SMYD1 and SRF (Figure 6J). In line with a primed endothelial gene program in mutants, HES1 gained SE elements, as did several members of WNT signaling. RNA-seq showed altered expression of genes with SE elements in mutant CPCs, D15-CMs, and D32-CMs (Figure 6K). SEL elements were enriched in MEF2A, TEAD4 and NFATC2 motifs and SEE elements were enriched in motifs of endothelial regulators, such as HIF1A and FOXP1, as well as MEIS1 and GATA4 motifs (Figure S6F). Down-regulated genes from the RNA-seq data were disproportionally enriched for SE elements (Figure S6G).
To identify multivariate relationships between GATA4 and TBX5 binding with cardiac SE gene regulation, we used topological data analysis (TDA), which applies principal component analysis by singular value decomposition (Lum et al., 2013). Related genes are clustered into nodes, and clusters that share >1 gene are connected via an edge. TDA accurately grouped SE genes into a distinct smaller network that was highly enriched for MED1, TBX5, GATA4, H3K27ac, H3K4me3, H3K36me3 but not H3K27me3 (Figure S6H) (Table S5). This predicted SE network was attenuated by GATA4 knockdown, which supports its biological importance (Figure 6L). Interestingly, TBX5 binding was better correlated in this SE network than GATA4 binding, suggesting TBX5 is a better predictor of cardiac SE genes than GATA4.
SE elements mapped to several long-non-coding RNAs and TFs with undetermined cardiogenic functions. We hypothesized that they may be required to maintain CM function. Indeed, their depletion in CMs mostly induced abnormalities in contractility, calcium flux, and mitochondria mass (Figure S7A–C). Depleting MALAT1 and KLF9 induced a collapse of the cardiac transcriptional network (Figure S7D).
Regulatory Hubs in a GATA4-TBX5 Network Centered on PI3K Signaling
We used a systems-biology approach to construct a GATA4-TBX5 gene regulatory network (GRN), by integrating down- and up-regulated genes in G296S CMs (Figure 4), G4T5 bound genes in WT or G296S CMs (Figure 5), genes with SE elements (Figure 6), with STRING datasets (Table S5). We predicted a “scale-free” network of 716 nodes connected by 2,353 edges with an average 6.6 neighbors and path length of 4.3 (Figure 7A). Nodes were connected by edges representing physical (protein-protein) or functional (genetic, co-expression, co-occurrence) interactions. At least five sub-networks connected through 20 regulatory “hubs” were identified. When we extracted the top-20 hubs as a subnetwork connected by 70 edges, each had 27–53 neighbors—4- to 8-fold more than the average node in the GRN (Figure 7B). This sub-network had a significant interaction of p<6.5e-11. Interestingly, the top-4 hubs were G4T5 co-bound genes linked to PI3K signaling: PIK3CA (α-catalytic subunit), PIK3R1 (regulatory subunit), and PTK2 and EGFR, the upstream signal transduction components. In PTK2, G4T5 co-occupancy was lost in GATA4 mutants (Figure 7B). ITGA2, ITGA9 and KDR were also hubs and involved in PI3K signaling. GO analysis showed enrichment for integrin, PI3K-Akt, Phosphatidylinositol and EGF signaling (Figure S7E). When CMs were further treated with a PI3K inhibitor (LY294002), iWT CMs had a decrease in force generation, but G296S CMs were insensitive (Figure 7C, left). Treatment with an insulin-receptor-substrate (IRS) synthetic peptide to activate PI3K signaling reduced force generation in G296S CMs (Figure 7C, right). While the PI3K inhibitor had some effect on beat rates, the IRS peptide increased beating rates in G296S CMs 3-fold greater than iWT CMs, suggesting a hyper-sensitivity to PI3K pathway activation (Figure 7C–D, S7F). The evidence that mutant CMs exhibit dysregulated PI3K signaling provides a potential node for correcting the diseased GRN.
Figure 7. GATA4-TBX5 GRN Revealed Hubs Centered on PI3K Signaling.

(A) GATA4-controlled GRN. Nodes are genes that are differentially expressed or G4T5 co-bound or have MED-1 SE elements. Edges are physical or functional interactions between nodes as extracted from STRING. Yellow, top-20 hubs with the most direct neighbors. Hubs are grouped into 5 subnetworks (pink circle).
(B) Sub-network plot of extracted top-20 hubs named by gene symbol. Number of edges from entire GRN shown beside each node. Blue or red are gene expressions down- or up-regulated, respectively. Diamond, square, or circle represents genes that gained, lost or were unchanged for G4T5 binding. Bolded border represents genes with SE elements.
(C) Relative change in force generation between iWT (black) and G296S (red) CMs after inhibition (circle) or activation (triangle) of PI3K signaling. Traction force microscopy (TFM) measurements of CMs responding accurately to 1 Hz pacing. Data are mean ± SEM, *, p<0.05, **, p<0.005, ***, p<0.0005 (Mann-Whitney test).
(D) Beat rate measurements between iWT (black) and G296S (red) CMs after inhibition (circle) or activation (triangle) of PI3K signaling. TFM measurements of CMs responding accurately to 1Hz pacing. Data are mean ± SEM, *, p<0.05, **, p<0.005, ***, p<0.0005 (Mann-Whitney test).
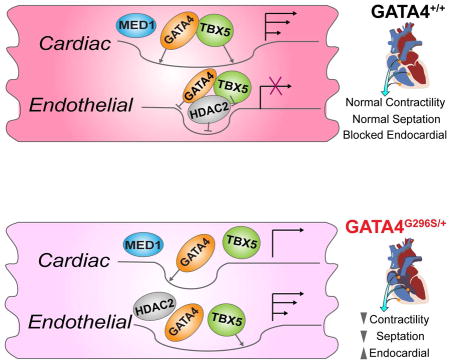
(E) Proposed model. Top, cardiac gene loci in WT are open and permissive to G4T5 binding at MED1-bound SE elements, which activates transcription; G4T5 and HDAC2 repress aberrant endothelial gene transcription. Bottom, transcriptional and epigenetic consequences of GATA4 G296S. Cardiac gene loci have reduced open chromatin and TBX5 binding to SE elements which reduces transcription; aberrantly open chromatin is depleted of GATA4-HDAC2 but enriched for TBX5, along with motifs for ETS factors resulting in failure to silence endothelial gene transcription and other sites involved in septal development not depicted. See also Figure S7.
DISCUSSION
Here we show that proper cardiac development and function require GATA4-TBX5 co-occupancy in MED1-bound, H3K27ac-marked SE elements to maintain an open chromatin state and activate cardiogenic gene transcription (Figure 7E). In GATA4 heterozygosity with a missense mutation that affects protein-protein interactions, a loss of TBX5 recruitment to SE elements is associated with failure to maintain open chromatin and diminished transcription of cardiac genes. The GATA4 G296S mutation allows mis-localization of TBX5 and perhaps other transcriptional activators, resulting in a failure to recruit HDAC2, and achieve a more closed chromatin signature at endothelial promoters. The result is aberrant activation of endothelial gene expression and alternative lineages. Furthermore, dysregulation of genes involved in the reception of SHH signals and cardiac septation provides a molecular basis for three-dimensional septal defects of GATA4 G296S patients, despite the two-dimensional model. These studies show how TF complexes cooperatively regulate genome-wide localization of trans-acting factors to control activation and repression of gene expression, and how diseases occur when cooperativity is disrupted.
GATA4 Maintains Homeostatic CM Function
GATA4 is a well-known master regulator of early heart development, cardiac specification, and hypertrophy (Bisping et al., 2006). With MEF2C and TBX5, it reprograms fibroblasts to a CM-like fate, and the cooperativity shown here partially explains the induction of a cardiogenic program (Ieda et al., 2010; Qian et al., 2012). That mutant CMs have impaired contractility, calcium handling, sarcomeric organization and metabolic activity is in line with GATA4 mutations associated with familial cardiomyopathy (Zhao et al., 2014), including those involving GATA4 G296S. Dysregulation of sarcomeric and metabolic genes explains many of the defects in human CMs. Our findings that GATA4 and putative co-repressors function in a negative feedback loop to limit PI3K signaling that becomes dysregulated in GATA4 mutants are consistent with reports of Gata4 mediating PI3K-dependent hypertrophic responses to physiological stress in mouse hearts (McMullen et al., 2004).
GATA4 Promotes Cardiomyocyte and Represses Alternative Fate Gene Expression
Our results show that GATA4 is critical for cardiac vs endothelial gene regulation in CPCs. GATA4’s function as a positive driver of cardiogenesis is unambiguous, but its potential as a repressor of endocardial/endothelial gene expression in CMs has been unknown. Scl/Tal1 promotes the hematopoietic gene program in hemogenic endothelium and prevents mis-specification into the cardiomyogenic fate by a combinatorial mechanism (Van Handel et al., 2012). Our data support this concept from the reciprocal angle where a disease-causing mutation of a TF that normally promotes cardiogenesis induces an ectopic endothelial gene program during CM differentiation. TAL1 was upregulated in G296S CPCs and may contribute to aberrant endothelial gene expression. G4T5 sites in CMs were enriched for motifs of key regulators of hemogenic endothelium, FOXO1 and HOXB4, and G4T5 occupancy normally was associated with gene repression at these sites. However, in GATA4 G296S mutants, loci of inappropriately open chromatin were enriched for motifs of endothelial regulators such as FOXO1 and numerous ETS factors, suggesting loss of G4T5 repression. The reduction in HDAC2 recruitment and downregulation of the GATA4-interacting repressors, HEY1 and HEY2, provide a potential mechanism for de-repression of endothelial gene targets that may contribute to septal defects.
Even in a monolayer system, genome-wide analyses revealed gene expression and chromatin dysregulation of genes required for atrioventricular canal development, endocardial cushion formation and septal morphogenesis in GATA4 G296S CMs. These observations suggest iPS cells can be used as an in vitro model to understand cellular events leading to morphogenetic defects. Specifically, TBX2, a regulator of AV canal myocardium (Aanhaanen et al., 2011), was upregulated, and genes necessary for receiving the SHH signal in myocardium were downregulated. This was particularly interesting because exogenous SHH signals from pulmonary endoderm are received by the developing atrium, resulting in expansion of the posterior second heart field-derived dorsal-mesenchymal-protrusion that forms part of the atrial septum. Failure to respond to the SHH signal results in septal defects in mice (Hoffmann et al., 2009). Evidence for GATA4 regulating SHH signaling suggests a potential mechanism for septal defects observed in mice and humans haploinsufficient for GATA4.
Combinatorial Regulation of Human Cardiac Enhancers
Our results show a combinatorial TF binding code for activating the human cardiac gene program, similar to mouse CMs (Luna-Zurita et al., 2016), and reveal how disrupting this code by a missense mutation leads to epigenetic and transcriptional dysregulation and human disease. ATAC-seq analyses of open chromatin signature and genome-wide profiling of GATA4 and TBX5 binding sites provide a detailed catalog of TF-bound enhancers in humans and complement the sparse ENCODE data on cardiac cell types, which we leveraged in identifying a putative G4T5 co-repressor. We found that GATA4 and TBX5 cooperation was robust when underlying cis-sequences were closely linked on the same DNA strand and in the same 5′-3 orientation; in such situations, protein-DNA interactions may overcome a lack of protein-protein interaction between GATA4 and TBX5 in the mutant setting.
Until now, human cardiac SEs had not been identified by MED1 ChIP-seq. Our cataloging of SEs pinpoints transcriptional regulators and long noncoding RNAs that may be crucial in human cardiac development and function. TDA with machine learning distinguished genes with SE elements from other genes and placed TBX5 at a higher hierarchical level than GATA4 in mapping cardiac SEs. TEs seem to have a different TF binding code than SEs. In GATA4 mutants, TBX5 binding was decreased at SEs, but increased at many TEs. This difference suggests cardiac TFs operate via diverse rules at various enhancer sites, perhaps dictated by underlying cis-sequence and/or local chromatin configuration.
In conclusion, this study reveals a combinatorial TF code that ensures a robust cardiac gene program, illustrates how human disease occurs when this code is altered by disrupting TF cooperativity, and highlights potential nodes for therapeutic intervention.
STAR ★ METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact Dr. Deepak Srivastava at dsrivastava@gladstone.ucsf.edu.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Family members with and without GATA4-G296S heterozygous mutation were previously identified and diagnosed by clinicians (Garg et al., 2003). Eight skin biopsy samples were obtained from this family following IRB protocol (“Induced Pluripotent Stem Cells for Cardiovascular Research” UCSF H51338-32135-02) approved by University of California San Francisco with patient informed consent. All genomic DNA used for genotyping were isolated from subsequent Human dermal fibroblasts (HDFs) or iPS cells.
| Fibroblasts | Clinical description at time of biopsy | Age | Sex |
|---|---|---|---|
| 1 | Normal | 45 | Female |
|
| |||
| 2 | ASD, VSD, PS and cardiomyopathy | 17 | Male |
| 3 | ASD, VSD, PS | 16 | Female |
| 4* | ASD, AVSD, PS and cardiomyopathy | 14 | Male |
|
| |||
| 5 | Normal | 9 | Male |
| 6 | Normal | na | Male |
| 7 | Normal | na | Female |
|
| |||
| 8 | ASD, VSD, PS | na | Male |
Human embryonic stem cell line H7 (NIHhESC-10-0061) was used for cardiac differentiation and SNL feeder (CBA-316) cells were used to generate iPS cells.
CB17 SCID mice (Charles River, NOD.CB17-Prkdcscid/NcrCrl) were used for teratoma formations in accordance with the UCSF’s Institutional Animal Care and Use Committee guidelines.
METHOD DETAILS
iPS Cells Derivation and Cell Culture
HDFs were reprogrammed into iPS clones as previously described (Okita et al., 2011). Briefly, 3×105 HDF were NEON®-transfected (Invitrogen) with three pCXLE episomal, non-integrating plasmids encoding human OCT4, SOX2, KLF4, LIN28, L-MYC and p53-shRNA, and seeded onto inactivated SNL feeder layers. Small ES-like colonies start to emerge after 14 days and individual TRA-1-81-positive colonies were picked into 48-well plates between Days 20–25. Electroporated HDFs were initially grown on hES-media containing Knockout-serum-replacement and bFGF but subsequently cultured in mTeSR® on hESC-qualified (BDsciences) matrigel-coated plates once stable ES-like colonies emerged (StemCell Technologies). HDFs were cultured on DMEM containing 10% fetal-bovine-serum with supplements. Terminally differentiated, lactate purified human CMs were cultured in RPMI1640 with B27 supplements (Life Technologies) on plates coated with fibronectin (Sigma, 12.5 ug/ml in 0.02% gelatin) and passaged with accutase as needed (StemCell Technologies). Non-purified CM cultures >30 days were passaged with TrypLE (Invitrogen) and mild scraping. All cell cultures were maintained at 37 °C with 5% CO2.
CRISPR Dual Nickase Editing
The c.886G>A mutation was corrected back to its wildtype sequence using low off-target CRISPR dual nickase (H840A), guide-RNAs targeting chr8:11607673-11607763 (hg19) plus a donor DNA for homology-directed-repair (Figure S1B). Guide-RNA sequences (5′:CGCAGGACCCACGTACCCC; 3′: CACGCTGTGGCGCCGCAAT) were designed at http://crispr.mit.edu/ and cloned as described at http://www.genome-engineering.org/crispr/. The donor DNA contained respectively 440 bp and 438 bp of left and right homology arms around the point mutation (chr8:11607722) with a loxP-flanked selection cassette downstream of the mutation. The selection cassette contained mCherry and Puromycin-resistance driven by the CAG promoter meant to be inserted into the intron 3′ of the mutated exon. 1.5×106 cells of an iPS clone generated from Patient 4 were transfected using the Human Stem Cell Nucleofection (Lonza) with 4 μg of each gRNA-Nickase plasmid and 6 ug donor DNA. Nucleofected, mCherry-positive iPS cells were puromycin (0.5 μg/ml) selected after 3 days, for only 30 hrs, then selected again after 5 days at 1 μg/ml for long term maintenance. Single mCherry-positive colonies were picked and verified with Digital Droplet PCR (Biorad) using LNA probes (IDT) specific for the G>A mutation to measure allelic frequency. Nonviral TAT-Cre (Excellgen) was used to excise the selection cassette followed by single-cell FACS sorting for mCherry-negative cells. Sanger sequencing confirmed the gene-correction, ensured no spurious indels ± 500bp around mutation on the WT allele and a single loxP scar inside the intron on the mutant allele. We derived one CRISPR-corrected isogenic iWT line and one CRISPR-targeted but uncorrected G296S line to control against idiosyncrasies during the CRISPR-editing process. Short-tandem-repeat genotyping at Genetic Resources Core Facility (http://grcf.med.jhu.edu/) verified that all HDF, iPS, isogenic iPS and CM were derived from the same genome.
EB and Teratoma Formation
Potential to generate the three germ layers was confirmed by aggregating embryoid bodies (EB) in ultra-low attachment plates (Corning #3262) in differentiation medium (KO DMEM, 20% FBS, non-essential amino acids, glutamine, 100 μM BME) for 21 days followed by immunocytochemistry for endoderm marker AFP (R&D, MAB1368), mesoderm marker SMA (Abcam, ab5694) and ectoderm marker GFAP (Dako, Z0334).
All animal procedures were performed in accordance with the UCSF’s Institutional Animal Care and Use Committee guidelines. Approximately 1–2×106 cells were injected subcutaneously into immuno-compromised CB17 SCID mice (Charles River). Teratomas were excised 4–6 weeks post-injection, fixed overnight in formalin, embedded in paraffin, sectioned and stained with hematoxylin and eosin by the Gladstone Histology Core (http://labs.gladstone.ucsf.edu/histology/home). Histological evaluation was performed using a Keyence BZ-9000 with reference to Atlas of Pathology (Robbins & Cotran).
Real-Time qPCR, Single-cell qPCR, Immunocytochemistry, Western Blot, Electron Microscopy
For gene expression analyses, total RNA was Trizol-extracted (Invitrogen), column-purified with RNeasy kits (Qiagen), and reverse transcribed using the High Capacity reverse transcription kit (Applied Biosystems). All quantitative PCR analyses were performed using the Fast Taqman Master Mix and Gene Expression Taqman Assay probes (Applied Biosystems) following manufacturers’ protocol on the ABI7900HT Fast Real-Time PCR System (Applied Biosystems). No PCR products were observed in the absence of template. Taqman probe IDs are available upon request. In RT-qPCR analysis by ΔΔCt method, all data were normalized to GAPDH and represented relative to a control sample (set at 1). PRISM software was used for analysis, graphing and statistical analyses (http://www.graphpad.com/scientific-software/prism/).
Single human iPS/ES/adult CM were captured using the 10–17 uM chip in a C1 Single-Cell Auto Prep System (Fluidigm) following manufacturer’s protocol. Resultant single cell cDNAs were diluted 5-fold prior to amplification using a Universal PCR Master Mix and inventoried TaqMan gene expression assays (Applied Biosystems) in 96×96 Dynamic Arrays on a BioMark HD System (Fluidigm). Amplification included a 10 min, 95 °C hot-start followed by 40 cycles of a two-step program consisting of 15 sec at 95 °C and 60 sec at 60 °C. Taqman probe IDs are available upon request. Ct-values were calculated using BioMark Real-Time PCR Analysis Software v2.0 (Fluidigm). Spearman correlation matrix was calculated from GAPDH-normalized Ct values in R and clustered as a heatmap.
Immunocytochemistry and FACS were done as described before (Ang et al., 2011). hiPS cells were stained for TRA-1-60 (Millipore, MAB4360), TRA-1-81 (Millipore, MAB4381), NANOG (Abcam, ab21624), OCT4 (Cell Signaling, 2750S) and SSEA4 (Abcam, ab16286). Human CPC/CM were stained for ISL1 (ab20670), cTnT (MS-295-PO), GATA4 (sc1237), TBX5 (sc17866), NKX2.5 (sc8697), CD31 (BD-558068), cTnI (sc15368), MLC2v (ab92721), MLC2a (ab68086), α Actinin (A7811), VIM (sc6260), HCN4 (AGP-004). Immunostained samples were counterstained with DAPI and visualized in Zeiss Axio Inverted microscope and associated ZEN software. Sarcomeric organization was quantified from 20 fields of images (n=>150) by 2 researchers according to classification scheme similar to Heidersbach et al., Elife 2013. Quantification was performed in Volocity-v6.3 using object finding parameters consistently across all samples. FACS samples were measured using MACSQuant Analyzer10 (Miltenyl Biotec) or LSRII (BD) and further analyzed using Flowjo-v10 (http://www.flowjo.com/).
Immunoblotting was done as described previously (Ang et al., 2011). hIPS derived CMs were lysed and whole cell lysate was separated on a gradient gel, transferred onto PVDF membranes and probed with the following antibodies for co-repressors: HDAC2 (ab12169), HEY1 (ab22614), HDAC9 (ab18970), HEY2 (ab167280), TRIM28 (PA1-9059), RCOR1 (sc30189), REST (PA5-34583), TLE (sc13373), GAPDH (sc25778).
Transmission electron microscopy was done on a JEOL JEM-1230 transmission electron microscope equipped with a Gatan high resolution CCD camera after fixation of tissues, dehydration and resin embedding of specimens, ultramicrotomy and contrast staining (http://labs.gladstone.ucsf.edu/electron_microscopy/home).
Human Cardiac Differentiation
For human cardiac differentiation into CPC and CM, we modified the protocols originally developed by Lian et al. and Tohyama et al. to achieve stage-specific, high yield, high-purity cardiac commitment in vitro (Lian et al., 2012; Tohyama et al., 2013). Briefly ES/iPS cells were detached from matrigel with accutase and reseeded on matrigel at 0.4–1.2×106 cells per 6well in mTeSR with Rock inhibitor (Stem Cell Tech., Y27632, final:5uM) for 3 days. On the day of cardiac induction (day0), 8 uM CHIR99021 (Stemgent) was added for 48 hr or 12 uM CHIR99021 was added for 24 hr in 3 ml of B27-supplemented (without insulin) RPMI1640 media. The lower cell densities required a lower dose of CHIR99021 while the higher cell densities tolerated a higher but shorter dose of CHIR99021. We optimized each iPS clone individually to identify the best condition that resulted in high ISL1-positive CPC at day 5 and high cTnT-positive CM at day 15. At day 3, 5 uM IWP4 (Stemgent) in B27-supplemented (without insulin) RPMI1640 media was added to activate Wnt signaling. At day 5, IWP4 was removed, cells were detached with accutase and replated on fibronectin coated plates at a 1:3 dilution. This replating significantly increased the yield of beating CM and improved the uniformity of cell-cell adhesion within each well. The cells between day 5–7 are routinely at least 75%-pure multipotent progenitors; as defined by ISL1 and GATA4 FACS analyses. At day 10 the media was changed to regular B27-supplemented RPMI1640 hereafter. Typically in parallel differentiations under identical conditions, WT cells started spontaneous contraction as early as day11 while mutant cells tend to be slightly delayed by 48–96 hr, as in the case for the isogenic corrected and uncorrected clones (90 hr delay in G296S). Between day 18–22, to obtain >90% pure CM, we purify the contracting CM cultures using a glucose-depleted (Invitrogen, 11966-025) but lactate-supplemented (Sigma L7022, final:4mM) media for 4–7days. Every 48 hr, cells were gently flushed during new media change. CM may experience morphological changes and contract faster in this media and non-contracting cells will become detached and aspirated. For cells to recover, we add 12% FBS containing media for 2 days and then switched back into the B27-supplemented RPMI1640 media for long term culture (>30 day). At this point (day 30), purified-CM cultures can be frozen, harvested for functional assays and molecular profiling or Lipofectamine-transfected with siRNA (Sigma).
Micro-patterning and Image Analyses
CM contractile function was analyzed after seeding cells on polyacrylamide hydrogel devices containing matrigel micropatterns that were fabricated as previously detailed (Ribeiro et al., 2015). Matrigel micropatterns were transferred with microcontact printing onto a glass coverslip from the top of microfabricated polydimethylsiloxane microstamps. Polydimethylsiloxane 182 (Dow Corning) was used to fabricate microstamps from SU-8 (Microchem) micromolds developed on silicon wafers (ø10 cm, University Wafer). Before microncontact printing, stamps were incubated with Matrigel diluted 1:10 in L-15 medium. Matrigel patterns transferred from microstamps were rectangular and had an aspect ratio of 7:1 (length:width) and an area of 2000 μm2. These specific micropatterns were used to analyze single CMs. Matrigel micropatterns were also designed to fabricate lines of CM microtissues with a 1-mm length and 40-μm width. Matrigel micropatterns on glass coverslips were transferred to the surface of polyacrylamide hydrogels by inducing hydrogel gelation under the coverslips containing micropatterns. Polyacrylamide hydrogels are incubated in PBS after gelation is complete and the top coverslip is removed with a razor blade. To ensure that polyacrylamide substrates do not float and move in an aqueous environment, polyacrylamide gels on top of glass coverslips or glass-bottom dishes that were previously treated with 0.4% 3-(trimethoxysilyl)propyl methacrylate (Sigma-Aldrich) in Milli-Q water, pH 3.5, for 1 h, washed six times with Milli-Q water and dried with N2 gas. After this treatment, glass surfaces are functionalized with a methacrylate group that binds polyacrylamide during gelation and therefore support the hydrogel substrates. The aqueous solution that was gelled was composed of acrylamide (Sigma-Aldrich)(10% w/v), bisacrylamide (Sigma-Aldrich) (0.1% w/v), ammonium persulfate (Sigma-Aldrich) (0.01% w/v) and N,N,N′,N′-tetramethylethylenediamine (Sigma-Aldrich) (0.1% v/v), HEPES (Life Technologies) (35 mM) and Milli-Q water. To calculate the forces generated by cells attached to polyacrylamide surfaces with traction force microscopy, green fluorescent microbeads were also dispersed in the gel solution to yield a final concentration of 6.25 × 109 microbeads/mL.
Calculation of forces generated by single CMs with traction force microscopy
Quantification of forces generated during the contractile cycle of CMs was done as described previously (Ribeiro et al., 2015). Briefly, 4–5 s videos of beating live single CMs on patterns were acquired with microscopy at a speed of 26 frames/s with a Zeiss Axiocam MRm camera mounted on a Zeiss Axiovert 200 M inverted microscope with fluorescent capabilities. Specifically, bright field videos of moving contractile CMs and videos of green fluorescent microbeads moving due to cellular contractile forces were acquired. Brightfield videos were used to calculate the cellular movement of each contractile cycle. Frames within videos of fluorescent microbeads were submitted to a traction force microscopy algorithm to calculate contractile force. Brightfield videos of moving cells were processed with Ncorr to calculate the average displacements of single cells during contractions. To test response of CMs to PI3K-Akt pathway activation and inhibition, we used Insulin receptor substrate peptide (SCBT, sc3036) and LY294002 (Selleckchem, S1105) respectively. Both compounds were added to CM culture media for identical amount of time (15–30mins) at final concentrations of 10uM (LY294002) and 10ug/ml (IRS-1). TFM was performed before and after addition of compounds and data were matched for the same cell.
Analysis of movement in cell clumps
Movement of beating clumps was analyzed from brightfield videos acquired with a Zeiss Axiocam MRm camera mounted on a Zeiss Axiovert 200 M inverted microscope at 14 frame/s. Regions of interest in the acquired videos were delineated around beating cell clumps and movement was quantified with 2D digital image correlation using Ncorr.
Patch Clamp, Calcium Flux and Metabolic Assays
Current-clamp recordings were performed to measure action potentials (Aps) of differentiated ES/iPS-CM (>day30) at the single cell level by whole-cell patch-clamp with an Axopatch 200B amplifier and the pClamp10.2 software (Axon Instruments Inc., Foster City, CA). Patch pipettes were prepared from 1.5 mm thin-walled borosilicate glass tubes using a Sutter micropipette puller P-97 and had typical resistances of 4–6 MΏ when filled with an internal solution containing (mmol/L): 110 K+ aspartate, 20 KCl, 1 MgCl2, 0.1 Na-GTP, 5 Mg-ATP, 5 Na2-phospocreatine, 1 EGTA, 10 HEPES, pH adjusted to 7.3 with KOH. The external Tyrode’s bath solution consisted of (mmol/L): 140 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 D-glucose, 10 HEPES, pH adjusted to 7.4 with NaOH. For recording APs, cells were given a stimulus of 0.5 nA for 5 ms to elicit a response or not (for spontaneously-firing cells). CM were categorized into pacemaker, atrial, or ventricular phenotypes according to parameters such as the maximum rate of rise of the AP and APD and the ratio of APD50 vs. APD90. Calcium flux was measured by Fluo-4 staining followed by time lapse imaging at 14–25 fps. Specific regions of spontaneously contracting microclusters were selected and the peak amplitude of the calcium transient expressed relative to the baseline fluorescence measured between action potentials (F/F0) was quantified using pClamp-v10 software. To calculate calcium flux in 1mm lines, videos of cells labeled with Fluo-4 were acquired for 4–5 seconds with an inverted microscope at 10–15 frame/s. Intensity of Fluo-4 was calculated for each pixel within a region of interest. Cells were electrically paced to beat at 1 Hz. Under these conditions, Fluo-4 intensity oscillates, presenting a spike every second as the cell beats. For each beat, the rate of signal rise and the rate of signal decay were calculated for each pixel from the derivative of the variation of fluorescence intensity with time. Peak amplitude of the calcium transient expressed relative to the baseline fluorescence is calculated (F/F0). Values were averaged for all pixels within the region of interest.
CM on single-cell patterns were fixed with 4% paraformaldehyde and their mitochondria were labelled using the Mitotracker Deep Red FM (Invitrogen) at 100nM for 15mins. This dye passively diffuses across the plasma membrane and accumulates in active mitochondria which are visible through the Cy5 filter on a Zeiss Axio Inverted microscope. Quantification was done in Volocity-v6.3 using objects finding parameters consistently across all samples. Intensity per cell is calculated by normalizing the mean intensity of each mitochondria object over the number of DAPI-containing cells in that object. To measure glycolytic function in CM, we used the Glycolysis Stress Test Kit measured on a Seahorse XF96 Analyzer (Seahorse Bioscience) following manufacturer’s protocol. Equal number (3×104) of iWT and G296S CM were seeded in the same Seahorse plate and measured before and after addition of glucose, oligomycin and 2-deoxy-glucose. Glycolytic reserve and capacity values were exported from Seahorse software and plotted using PRISM with statistical tests.
Targeted Sequencing for Mitochondrial DNA
100 ng of DNA was used in the preparation of each library. For targeted sequencing, the Agilent SureSelect XT2 Target Enrichment system was used. Briefly, samples were sheared to 250 bp fragments using a Covaris E220. Samples were then indexed and hybridized to a custom bait set specifically designed for mtDNA enrichment (sequences available upon request). Samples were multiplexed in pools containing 30 samples and then sequenced on a single Illumina HiSeq 2000 lane (paired-end 101 bp).
RNA-sequencing (RNA-seq) Assay
Only CM >90% purity determined by cTnT FACS were used as biological replicates. Total RNA was Trizol-extracted (Invitrogen) and further purified using the Qiagen RNAeasy Micro Kit with DnaseI in-column treatment. cDNA was prepared with the NuGen Ovation RNA-seq System V2 Kit. Total RNA (50 ng) was reverse transcribed to synthesize the first-strand cDNA using a combination of random hexamers and a poly-T chimeric primer. The RNA template was then partially degraded by heating, and the second strand cDNA was synthesized using DNA polymerase. The double-stranded DNA was then amplified using single primer isothermal amplification (SPIA). In this linear cDNA amplification process, RNase H degraded RNA in DNA/RNA heteroduplex at the 5′-end of the double-stranded DNA, after which the SPIA primer bound to the cDNA and the polymerase started replication at the 3′-end of the primer by displacement of the existing forward strand. Random hexamers were then used to amplify the second-strand cDNA linearly. Finally, libraries from the SPIA amplified cDNA were made using the NuGen Ovation Ultralow DR Kit. The mRNA-seq libraries were analyzed by Agilent Bioanalyzer and quantified using an Illumina Library Quantification Kit (KAPA Biosystems). Libraries were prepared by the Gladstone Genomics Core (http://labs.gladstone.ucsf.edu/genomics/home). Four mRNA-seq libraries were pooled per lane of paired-end 100 bp sequencing (High output) on an Illumina HiSeq 2500 instrument (http://humangenetics.ucsf.edu/genomics-services/sample-processing/).
Assay for Transposase-Accessible Chromatin with deep sequencing (ATAC-seq) Assay
Only CM >90% purity determined by cTnT FACS were used as biological replicates. We prepared isogenic WT and G296S CPC samples for ATAC-seq based on the protocol previously described (Buenrostro et al, 2013). Aliquots of 50,000 cells were spun down (310 RCF for 3 min) and washed with 200 μl chilled PBS. Samples were spun down once more and lysed with 200 μl chilled lysis buffer (20mM Tris-HCl (pH 8.0), 85mM KCl, 0.5% NP-40). The lysates were spun down at a higher speed (500 RCF for 5 min) to aid cell lysis and to pellet the nuclei. The nuclear pellets were then each transposed with 25 μl Tagment DNA Buffer, 2.5 μl Tagment DNA Enzyme (Nextera Sample prep Kit from Illumina), and 22.5 μl Nuclease-Free H2O. The samples were then incubated at 37° C for 20 minutes, and stored at −20 °C. Transposed DNA were purified using the Qiagen MinElute Reaction Cleanup Kit (cat #28204). Samples were then amplified using 25 μl Nextera PCR Master Mix, 1.25 μM Nextera custom primer (common to all samples), 1.25 μM Nextera custom primers with unique barcodes (different for each sample/pool), and Nuclease-Free H2O. Samples were amplified using the following PCR conditions: 72 °C for 5 minutes; 98 °C for 30 seconds; and thermocycling at 98 °C for 10 seconds, 63 °C for 30 seconds and 72 °C for 1 minute. Half of each sample was amplified for 15 cycles, MinElute purified, and run on the Bioanalyzer to check library quality and diversity. The other half of the sample was run for 12 cycles, MinElute purified and run on the bioanalyzer once more for concentration value and quality control. Two ATAC-seq libraries were pooled per lane of paired-end 100bp (High output) sequencing on an Illumina HiSeq 2500 instrument by the UCSF Genomic Core (http://humangenetics.ucsf.edu/genomics-services/sample-processing/).
Chromatin Immunoprecipitation and sequencing (ChIP-seq) Assay
Only CM >90% purity determined by cTnT FACS were used as biological replicates. Like performed previously (Ang et al., 2011), cells were grown to an approximate final count of 2.4×107 cells for each TF ChIP and 4×106 cells for each modified-histone ChIP. Cells were detected off plates and chemically cross-linked with 1% formaldehyde solution for 10 minutes at room temperature with gentle agitation and quenched with 0.125M glycine. Cells were rinsed twice with 1xPBS, flash frozen and stored at −80 °C. Pellets were resuspended, lysed, and sonicated to solubilize and shear crosslinked DNA. To ensure consistent sonication between samples, we used Bioruptor Plus (Diagenode) to simultaneously process up to 6 samples in TPX tubes. Sonication was done at Power:3.0 for 9 cycles x 3min (30s-ON, 30s-OFF) with 1.5min rests between each cycle. The resulting chromatin extract was incubated overnight at 4 °C with 100ul Dyna Protein G magnetic beads preincubated with 4–7 ug of the appropriate antibody for at least 3 hrs. Beads were washed 5 times with RIPA buffer, once with TE containing 50 mM NaCl and complexes were eluted from beads in elution buffer by heating at 65 °C and shaking in a Thermomixer. Reverse crosslinking was performed overnight at 65°C. Input DNA (reserved from sonication) was concurrently treated for crosslink reversal. DNA was treated with RNaseA, proteinase K and purified using the Qiagen PCR purification kit. Primary antibodies used for ChIP were: GATA4 (sc1237, lot:E0912), TBX5 (sc17866), MED1 (A300-793A, lot:A300-793A), HDAC2 (ab12169), H3K27ac (ab4729, lot:GR104852-2), H3K4me3 (CS200580, lot:NG1848343), H3K27me3 (07-449, lot:2194165), H3K36me3 (ab9050, lot:GR114293-2). The specificity of these antibodies were validated in previous publications (Mikkelsen et al., 2007; Whyte et al., 2013). Barcoded ChIP-seq libraries were made from ChIP DNA using the NuGen Ovation Ultralow DR or Diagenode Microplex Kit, checked for adapter artifacts by Agilent Bioanalyzer using DNA high sensitivity reagents and chips, and quantified by qPCR using an Illumina Library Quantification kit (KAPA Biosystems) by the Gladstone Genomics Core (http://labs.gladstone.ucsf.edu/genomics/home). Four ChIP-seq libraries were pooled per lane of single-end 50bp (Rapid mode) sequencing on an Illumina HiSeq 2500 instrument by the UCSF Genomic Core (http://humangenetics.ucsf.edu/genomics-services/sample-processing/).
Computational Analyses
RNA-seq
Contaminants were removed and raw reads were trimmed for low quality reads using the default settings of fastq-mcf from ea-utils 1.1.2-537 (http://code.google.com/p/ea-utils). Trimmed reads were then aligned to the hg19 genome and transcriptome using Tophat (Kim et al., 2013) with the following settings: “--segment-mismatches 2 -m 2 -r 192 --mate-std-dev 200 --microexon-search”. Unmapped reads (“-F 0x4”), non-primary aligned reads (“-F 0x100”), and low quality reads (MAPQ less than 30, “-q 30”) were filtered using samtools 0.1.19 (Li et al., 2009). PCR duplicates were removed using the default settings of MarkDuplicates from picard-tools 1.98 (http://broadinstitute.github.io/picard). USeq application DefinedRegionDifferentialSeq (Nix et al., 2008) was used to normalize raw read counts, calculate FPKM (counts/exonicBasesPerKB/millionTotalMappedReadsToGeneTable), and analyze differential expression using DESeq’s negative binomial p-value following the Benjamini and Hochberg multiple testing correction. The gene mapping file contained 20115 unique protein-coding genes from the hg19 Human Reference 37 NCBI build. HOPACH was used for clustering with the correlation metric in Figure 1. Differentially expressed genes were identified based on the following criteria: Log2 fold change > 0.585 for up-regulated, < −0.585 for down-regulated; adjusted p-value (FDR) <0.05; and counts>5. Variance corrected values, estimated using DESeq’s blind method per gene variance corrected counts in log space, were hierarchically clustered in R using custom scripts with package pheatmap and parameters scale=“row”, clustering_distance_rows = “euclidean”, clustering_method=“average”. Spearman correlation matrix was calculated from Variance Corrected values of all detectable genes in R and clustered as a heatmap. FPKM values of individual gene were plotted in PRISM or EXCEL. Keygenes predictor tool (Roost et al., 2015) was used to classify our time course RNA-seq data using the provided ‘fetal2t’ training set.
ATAC-seq
Data was processed in a similar manner to (Buenrostro et al., 2013). Briefly, Nextera adaptor sequences were trimmed using cutadapt (version 1.8.1; custom parameters: (-O 5, -m 30 –q 15)) from all FASTQ files and then aligned to the hg19 genome using bowtie (version 1.1.1; custom parameters: (-X 2000, -m 1)). Next, duplicate reads were filtered using Picard MarkDuplicates (version 1.92) and reads in repetitive regions (ENCODE Blacklist) were filtered using samtools (version 0.1.19). Finally, peaks were called using MACS2 (version 2.1.0; custom parameters: (--nomodel –shift -100 –extsize 200) and signal track data were generated using align2rawsignal (custom parameters: (-n=5 –l=1, -w=200). All peaks reported passed an FDR threshold of 5%. ATAC-seq signal was converted into .bigwig files and visualized using IGV browser along with ENCODE DHSs from HCM (Tier 3). To profile the intensity of DHSs, H3K4me3 and H3K27me3 in the iWT ATAC peaks, we extracted the corresponding .bam files for day5 cardiac progenitors (Stergachis et al., 2013) and plotted its signal within ± 1kb of our iWT ATAC-seq regions using ngsplot (Shen et al., 2014). Genome distribution of ATAC-seq peaks were analyzed using PAVIS (http://manticore.niehs.nih.gov/pavis2/) with a ± 20 kb upstream and downstream window. Normalized ATAC-seq signal at TSS of specific genesets were plotted as averaged profiles in ngsplot using .bam files from both iWT and G296S samples and option: ngs.plot.r -G hg19 -R tss.
ChIP-seq
Contaminants were removed and raw reads were trimmed for low quality reads using the default settings of fastq-mcf from ea-utils 1.1.2-537 (http://code.google.com/p/ea-utils). Trimmed reads were then aligned to the hg19 genome using default settings of bowtie 2.1.0 (Langmead et al., 2012). Low quality alignments were removed using samtools 0.1.19 (Li et al., 2009) with a MAPQ score cutoff of 30 (“-q 30”) and duplicates removed using the default settings of MarkDuplicates from picard-tools 1.98 (http://broadinstitute.github.io/picard). Peaks were called, in comparison to input, using MACS2 (version 2.1.0; custom parameters: (--keep-dup 1 –g 2.7e9 –q 0.01 –bw 498). HDAC2 peaks were called without the use of input. All peaks reported passed an FDR threshold of 1% or 5% (HDAC2 only). Individual peak files were merged to create a WT- or MUT-specific list of peaks if each peak is found in 2 or more replicates. Bigwig files from ChIP-seq alignments were visualized in IGV browser. Normalized ChIP-seq signal at specific regions were plotted as averaged profiles in ngsplot using replicate-merged .bam files from both WT and G296S samples and option: ngs.plot.r -G hg19 -R bed. To determine WT-unique (L) or G296-unique (E) regions, bedtools intersect was used with reciprocal commands: bedtools intersect -a WT_peaks -b MUT_peaks –v > WT_unique_peaks. The relative (G296S/WT) occupancy plot of various regions was generated as averaged profiles in ngsplot using replicate-combined .bam files from both WT and G296S samples but with option: G296S.bam:WT.bam. ChIP-seq signal for individual regions were systematically extracted from corresponding .bam files using bedtools coverage –counts and further normalized to the total number of counts per sample. This normalized ChIP intensity is then plotted and statistically tested in PRISM using one-way ANOVA and Dunn’s test for multiple hypothesis correction. Genome distribution of ChIP-seq peaks were analyzed using PAVIS (http://manticore.niehs.nih.gov/pavis2/) with a ± 20 kb upstream and downstream window. Corresponding FPKM values of the mapped genes were plotted and statistically tested in PRISM using Wilcoxon matched-pairs signed rank test. Log2 fold changes (G296S/WT) of GATA4, TBX5 ChIP-seq mapped closest to a gene was represented as a heatmap in R.
To call super-enhancers by MED1 intensity ranking, the ROSE python script (https://bitbucket.org/young_computation/rose) was used with default parameters (stitch distance=12.5kb without promoter exclusion) using peaks called by MACS2 and the replicate-merged .bam files for WT and G296S samples. Nearest gene was then mapped via PAVIS with a ± 20kb upstream and downstream window. Normalized ChIP-seq signals, comparison of Loss, Ectopic SE elements, coverage counts of specific regions and FPKM values of nearest gene were analyzed and plotted similarly to GATA4 and TBX5 ChIP-seq results.
Analysis of Differential Chip-Seq/ATAC-Seq Peaks
Normalized genome-wide signal track data were generated for all experiments using a two-step process: first, the estimated fragment length was assessed using cross-correlation analysis (phantompeakqualstools; ChIP-Seq experiments only). Second, align2rawsignal was used to produce normalized signal track data {custom parameters for ChIP-Seq: -l [fragment size inferred from cross-correlation analysis]; for ATAC-seq: -w 200}. Next, peaks seen in at least two samples (within each group of WT or mutant) were pooled and overlapping peaks were merged together (bedtools). We then used extract signal to compute the mean signal intensity under each peak, and then assessed differences between mutated and non-mutated samples using a moderated t-test (limma).
Targeted Sequencing for Mitochondrial DNA
Mutations were called using a modified version of the Huge-Seq pipeline. Briefly, (i) fastq files were trimmed for adaptor sequences using cutadapt (version 1.8.1) and then aligned to the hg19 genome using bwa-mem (version 0.7.7), (ii) duplicates were filtered using PicardTools MarkDuplicates, (iii) realigned using GATK, (iv) base scores re-calibrated using GATK, and finally (v) mutations were called using GATK HaplotypeCaller.
GO and GSEA analysis
Gene Ontology analysis of stage-specific gene signature and G296S differentially expressed genes were performed using ToppGene Suite (https://toppgene.cchmc.org/enrichment.jsp) using all Homo sapiens genes as background. Statistically significant (Bonferroni p-value<0.05) categories within the GO:Biological Process, Mouse/Human-Phenotype, Disease and Pathway sections were extracted and replotted. For all GATA4, TBX5, MED1 ChIP-seq binding sites, GO enrichment was done using the GREAT tool (http://bejerano.stanford.edu/great/public/html/) using ‘basal plus extension’ and Distal=20 kb. For ATAC-seq regions that were up- or down-regulated, generic/housekeeping open chromatin regions were filtered out by intersecting with DHSs of BJ fibroblasts from the ENCODE consortium. The 1722 cardiac-specific regions were then tested in GREAT using ‘basal plus extension’ and Distal=100kb.Gene Set Enrichment Analysis was performed using the GSEA software (http://www.broadinstitute.org/gsea/) with permutation=geneset, metric=Diff_of_classes, metric=weighted, #permutation=2500.
Consensus Motif Enrichment Analyses
Specific GATA4 and TBX5 ChIP-seq were used to find enriched known DNA motifs using the HOMER tool (http://homer.salk.edu/homer/ngs/). The findMotifsGenome.pl command using HOMER-randomized sequences as a background set was called with options: size 200 -mask -len 8,10,12 -mis 3 -S 25 -N 20000. Due to the long length of super-enhancers, the individual MED1-bound constituents inside a large piece of SE were instead used for motif enrichment with options: -len 7,9,11,13 -size 1000 -mask -mis 3 -S 30 -N 20000. Specific ChIP-seq or ATAC-seq regions that were altered (loss or ectopic) between WT and G296S mutants, the motif enrichment is performed by reciprocally using the loss or ectopic regions as the background set for the other. An example command looks like this: WT_G4T5peaks.bed hg19 Motifs_in_Loss/-size 200 -mask -len 8,10,12 -bg MUT_G4T5peaks.bed -mis 3 -S 40.
ChIP-seq peaks were scanned for motifs using the RTFBS package (http://biorxiv.org/content/biorxiv/early/2016/01/05/036053.full.pdf) using the default threshold. GATA4 and TBX5 motifs used were the number 1 de novo motif discovered by HOMER. For analysis of motif pairs, only pairs within the same ChIP-seq peak were included. If we include the same vs different strand analyses, the GATA4 motif position and strand were used to anchor all calculations.
ENCODE TF co-occupancy analysis
A list of unified peak calls (ENCODE) spanning 91 different cell types and 161 unique transcription factors was downloaded from the UCSC Genome Browser on May 10, 2016. Next, we evaluated the similarity between the ENCODE ChiP-Seq datasets and our data using a previously published method (Chikina and Troyanskaya, 2012). In brief, this method takes each peak (of length ‘l’) in a given query ChIP-Seq dataset (i.e., our dataset) and calculates the distance (‘d’) to the closest peak in a gold-standard dataset (i.e., one of the ENCODE datasets). Next, we compute a p-value which is defined as the ratio of number of intervals of length ‘l’ at most distance ‘d’ from a peak in the gold-standard set to the total number of intervals of length ‘l’ which can be placed on the query chromosome. Finally, the similarity between the query experiment and the gold-standard experiment is calculated as the fraction of peaks in the query set with P < 0.05. Note, query peaks which were >50 kbp away from any peak in a gold-standard set were automatically assigned P > 0.05.
Ayasdi network generation
Topological Data Analysis (TDA) which uses topology, a mathematical discipline, to create compact visualization of multi-dimensional datasets was used to cluster genes based on their similarity across all RNAseq and ChIPseq results gathered in our study (Lum et al., 2013). An ensemble machine learning algorithm performs millions of iterations to generate the most stable, consensus vote for a resulting “golden network” (Reeb graph), that may represent the multidimensional data shape. Two kinds of parameters are involved in calculating a TDA. First, is a measurement of similarity, also called a “metric”, which calculates the distance between two points in some space. Second, is lens, which represents different functions employed on data points. User-chosen lenses generate intersecting bins in the data set, where the bins are preimages under the lens of an interval. Intersecting groups of intervals will then generate intersecting bins in the data. In our case, clusters of genes will be grouped as nodes and similar relationships among clusters will be connected via an edge. The network was plotted largely using data points presented in Table-S8. Briefly, RNAseq data is imputed as continuous variables of G296S/WT Log fold change, whereas ATACseq and ChIPseq data are imputed as discrete, all-or-none, variables depending on whether a gene is bound in the wildtype condition. The Ayasdi web app v5.5.0 (Build# 1BE5868) was used to predict the network using metric: variance normalized euclidean, lens 1: L-infinity centrality (res:20, gain:2), lens 2: MDS coord 1 (res:20, gain:2), and lens 3: Metric PCA coord 1 (res:20, gain:2). The resulting topological network was color-coded for each of the dataset that was imputed where red and blue signifies strong and weak similarity between the predictor and outcome variables. A non-parametric Kolmogorov-Smirnov test was then used to determine what were the underlying variables which could distinguish two sub-cluster of the TDA network.
GATA4-TBX5 GRN Prediction
To predict a GATA4-TBX5 GRN using our RNA-seq and ChIP-seq datasets, we extracted genes that were differentially expressed in RNA-seq from d32 CM or co-bound by GATA4-TBX5 or had MED1-superenhancer elements. These 2,173 genes served as input node data into the STRING database. An interaction network was compiled by selecting an association if the genes had physical (protein-protein) or functional (genetic, co-expression, co-occurrence) interactions. The options to include associations based on computational prediction and literature-basis were excluded to derive more stringent interactions. The network was then replotted (force-directed layout) and analyzed in Cytoscape 3.2.1 (http://www.cytoscape.org/). Hubs were defined as the top-20 nodes with the most degrees (direct neighbors) and further extracted and replotted as a circular layout sub-network. These 20-nodes were also used for GO enrichment analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters including the exact value of n, precision measures (mean ± SEM) and statistical significance are reported in the Figures and the Figure Legends. Statistical analysis was performed by unpaired, parametric two-tailed Student’s t-test for samples that tested significant for normality, or non-parametric Mann-Whitney test for samples not significant for normality; unless otherwise noted. For all bar graphs, data are represented as mean ± SEM. *, p<0.05; **, p<0.005; ***, p<0.0005; ****, p<0.00005; were considered significant. All calculations were performed using R or GraphPad Prism software. No randomization was used and no cells were selectively excluded from the analysis.
DATA AND SOFTWARE AVAILABILITY
RNA-seq, ATAC-seq and ChIP-seq data are deposited at GEO database with project number GSE85631.
Supplementary Material
Gene expression signature of WT and G296S iPS cells during cardiac differentiation.
Open chromatin signature of iWT and G296S derived human CPCs.
GATA4, TBX5 and MED1 binding sites from WT and G296S derived human CMs.
H3K27ac, H3K4me3, H3K27me3, H3K36me3 marked sites from WT and G296S derived human CMs.
Compilation of RNA-seq, ATAC-seq and ChIP-seq results for Ayasdi and GRN predictions.
(A) Genotype-phenotype information of GATA4 pedigree with associated iPS cell lines. Colored rows represent GATA4 heterozygous mutants.
(B) Morphology of mCherry-positive, puromycin-resistant CRISPR-iPS clones before (left) and after (right) TAT-Cre excision of mCherry-puromycin selection casette.
(C) Karyotype analyses show all patient-iPS clones have normal karyotypes.
(D) Heatmap shows hierarchical clustering of gene expressions of HDF and self-renewal markers measured by RT-qPCR. Values are row-scaled to show their relative expression. Blue and red are low and high levels respectively.
(E) Immunostaining of all iPS lines for pluripotency markers. Red, GATA4 mutants.
(F) Heatmap shows hierarchical clustering of Spearman correlation scores of iPS and ES (H7) cells based on RNA-seq profiles. Score of 1 (yellow) denotes perfect correlation. Red, GATA4 mutants.
(G) Immunostaining of all iPS lines after spontaneous EB differentiation (left group) and teratoma formation (right group). GFAP and neural rosettes indicate ectoderm differentiation, SMA and cartilage indicate mesoderm differentiation, AFP and gut-tube indicate endoderm differentiation. Red, GATA4 mutants.
(A) Immunostaining of CM-specific markers during purification in lactatehi glucoselo media. Percentage of cTnT+ cells increased from 31–70% and total cell numbers decreased from 674–187 over 4 days.
(B) Heatmap shows gene expression of select mesoderm and CM markers during step-wise cardiac differentiation as measured by RNA-seq. Values are row-scaled to show their relative expression. Blue and red are low and high levels respectively.
(C) RNA-seq expression values (FPKM) of representative stage-specific genes during ES-H7 (blue box), MES (green), CPC (grey) CM (pink) differentiation. Known markers validate differentiation. Unknown transcriptional regulators and markers are highlighted.
(D) Heatmap shows HOPACH clustering of stage-specific genes. FPKM values are row-scaled to show relative expression. Blue and red are low and high levels respectively. Select GO and signaling categories are highlighted for each stage with matching colors.
(E) Immunostaining of 4 CM-enriched proteins in iPS-derived CM. Parentheses, >90% of CM are positive for all markers.
(F) Patch clamp recordings of single iPS-derived CM show action potential morphologies resembling pacemaker, ventricular and atrial CM.
(G) Heatmap shows hierarchical clustering of Spearman correlation scores of adult primary CMs, ES and iPS differentiated CMs based on single-cell RT-qPCR of 61 CM marker genes. Score of 1 (yellow) denotes perfect correlation.
(H) Percentage of binucleated vs. mononucleated CMs assessed on single-cell patterns.
(I) Immunostaining of CM-enriched proteins in all iPS-derived CM vs ES-derived CM after lactate purification. Parentheses, >90% of CM are positive for cTnT, MLC2v, αActinin and cTnI. Red, GATA4 mutants.
(J) Immunostaining of cTnT in isogenic CRISPR-corrected iWT vs mutant G296S derived CM after purification. Both lines were differentiated in parallel under identical conditions. Percentage of cTnT cells shown at bottom right.
(K) Contractile measurements on micro-patterns. Time (s) spent in each contraction cycle is shown for all CM responding accurately to 1 Hz pacing. Data are mean ± SEM. ****, p<0.0001 (t test).
(L–N) Contractile measurements on micro-patterns at early (D35) vs. late (D70) stages of CM differentiation. (L) % of single-CM responding accurately to 1 Hz electrical pacing in iWT and G296S. Blue arrow shows reduced % of G296S-CMs responding accurately to stimulation at both stages. (M) Traction force microscopy measurements of force production of all CMs responding accurately to 1 Hz pacing. (N) Traction force microscopy measurements of relaxation velocity of all CMs responding accurately to 1 Hz pacing. All measurements were done on isogenic CMs generated in parallel differentiations.
(O) Action potential measurements of WT and G296S CM. dV/dtmax, maximum upstroke velocity; APD90, duration of action potential at 90% repolarization. Data shown are mean ± SEM from 2 WT and 2 G296S lines. *, p<0.05 (Mann-Whitney test).
(P) Description of sarcomere scoring scheme. Class IV represents the most disarrayed sarcomeric organization.
(Q) Sequencing of mitochondrial-DNA for heteroplasmy mutations. Each data point represents CM from 1 patient-iPS-CM sample. Data shows mean ± SEM. G296S CM (siblings) showed highly similar number of de novo mutations in mtDNA and the WT (unrelated), reflecting pattern of inheritance.
(A) Heatmap shows hierarchical clustering of 2228 genes differentially expressed at any time point. Blue and red represent decreased and increased expressions (G296S/iWT Log2FC) respectively.
(B) Venn diagram showing overlaps of down- (left) and up-regulated (right) genes by G296S at CPC (black circle), D15-CM (red) and D32-CM (blue) stages of cardiac differentiation. 38 genes were consistently down or upregulated at all 3 time points.
(C) Heatmap shows gene expressions of 38 genes consistently down- (blue) or up-regulated (red) during cardiac differentiation of iWT and G296S CM as measured by RNA-seq. Values are row-scaled to show their relative expression. Blue and red are low and high levels respectively.
(D) Gene Ontology analyses (BioPro/Disease/Pathway) of 38 genes consistently down or upregulated during cardiac differentiation of iWT and G296S CM. Significance shown as −Log10 Bonferroni p-value after multiple hypothesis correction.
(E–F) FACS analyses of cardiogenic markers (GATA4, NKX2.5, TBX5, ISL1) and hemogenic-endothelial marker (KDR) in iWT vs. G296S D7-CPCs during CM differentiation. Reduced protein expression of GATA4, NKX2.5 and TBX5 was observed in G296S. Increased ISL1 was observed.
(G) FACS analyses of CM marker (cTnI) and endothelial marker (CD31) in iWT vs. G296S D15-CMs during CM differentiation. No marked increase in CD31+ cells were observed in G296S.
(H) FACS analyses of endothelial marker (CD31) in iWT vs. G296S during endothelial cell differentiation. Marginal increase in CD31+ cells was observed in G296S.
(I) Heatmap shows expressions of genes associated with chamber or atrioventricular canal myocardiums and smooth muscle are down- (blue) and up-regulated (red) in G296S CMs respectively. Values are row-scaled to show their relative expression. Blue and red are low and high levels respectively.
(J) GSEA analyses of geneset representing cellular respiration showed reduction in G296S CMs. NES, normalized enrichment score. FDR, false discovery rate.
(K) RT-qPCR validation of RNAseq results at D7-CPC, D15-CM and D32-CM. High R2 value indicate strong correlation between RNAseq and RT-qPCR.
(A) IGV browser tracks of ChIP-seq signal for GATA4, TBX5, H3K27ac, H3K4me3, H3K36me3, H3K27me3 at known gene target loci (TNNT2, TNNI1) in WT CM. Grey boxes denote significantly enriched (over input) peaks identified by MACS2. Y-axis represents reads/million/25bp.
(B) Metagenes plot of normalized ChIP-seq signal for H3K4me3, H3K27ac, H3K27me3, H3K36me3, GATA4 and TBX5 at genes that are high (green), mid (orange) and low (skyblue) expressed in WT CMs. TSS, transcription start site. TES, transcription end site. All ChIP intensities, except H3K27me3’s, were positively correlated to gene expression levels.
(C) Heatmap shows ChIP-seq signal at sites (± 5 kb) bound by GATA4 only, TBX5 only and G4T5 co-bound in WT CM. ChIP-seq signal of modified-histone marks also shown at these sites. Sites are ordered by decreasing intensity of GATA4 signal.
(D) Venn diagram of sites bound by GATA4 (black circle) and/or TBX5 (purple) in WT CM. 2,428 sites are G4T5 co-bound in human CMs.
(E) Pie-chart shows gene-body, upstream, downstream distribution of 2,428 G4T5 co-bound sites.
(F) Gene Ontology analyses (BioPro/Disease/Pathway) of 2,428 G4T5 co-bound sites. Significance shown as −Log10 Bonferroni p-value after multiple hypothesis correction.
(A) Venn diagram of sites bound by GATA4 (purple circle) and/or TBX5 (red) in G296S CMs. Only 1,605 sites are G4T5 co-bound. Total number of G4T5 sites was reduced in G296S.
(B) Metagenes plot of normalized ChIP-seq signal for GATA4, TBX5 and H3K27ac at G4T5 co-bound sites (± 5 kb) in WT and G296S CM.
(C) Normalized ChIP-seq signal of GATA4, TBX5, H3K27ac specifically at G4T5L, G4T5U, G4T5E, in WT (black) and G296S (red) CMs. Boxplot and whiskers show mean, 25th and 75th percentile followed by 5th and 95th percentile. ***, p<0.0005, ****, p<0.00005, ns, not significant (one-way ANOVA and Dunn’s test for multiple hypothesis correction).
(D) FPKM values of genes mapped ± 20 kb of all G4T5E sites during iWT (black) and G296S (red) cardiac differentiations. Boxplot and whiskers show mean, 25th and 75th percentile followed by 5th and 95th percentile. *, p<0.05, ns, not significant (Wilcoxon signed-rank non-parametric test)..
(E) Known consensus motifs enriched at G4T5L (left) and G4T5E (right) sites.
(F) Bar chart showing average distance (bp) between G4T5L sites to any ENCODE-TF binding site. The TF and the cell type profiled are shown on the Y-axis. For example, G4T5L sites are located proximally (<50bp) to EP300 sites within SKNSH neuronal cells.
(G) Putative direct targets of GATA4 and TBX5 as defined by ChIP-seq binding in WT and RNA-seq differential expression. Venn diagrams show differentially expressed genes (black circle) overlapped with G4T5 binding (blue) within 20 kb. Differential gene expressions were further separated into 3 classes: D15-CM or D32-CM stage (top), D15-CM stage (bottom left), D32-CM stage (bottom right).
(H) FPKM values of genes mapped ± 20 kb of sites that have decreased G4 and increased T5 binding (G4DOWN_T5UP) versus genes mapped ± 20 kb of sites that have decreased G4 and decreased T5 binding (G4DOWN_T5DOWN). The genes with concomitant reduction in G4T5 binding are expressed at a lower level. Boxplot and whiskers show mean, 25th and 75th percentile followed by 5th and 95th percentile.
(I) Metagenes plot of normalized ChIP-seq signal for H3K4me3 and H3K27me3 at TSS of endothelial specific genes (± 5 kb) in WT (black) and G296S (red) CM.
(J) Promoter motif enrichment analyses at 81 endothelial genes that were up-regulated. Significance shown as −Log10 Bonferroni p-value after multiple hypothesis correction.
(K) Scatter plot of GATA4 sites within endothelial or PI3K genes with respect to any ENCODE-TF binding site shown as a function of average distance (bp) against percentage. The top ENCODE-TF and the cell type profiled are highlighted in red. For example, GATA4 sites in endothelial TADs are located proximally (<100bp) and frequently (>25%) to RCOR1 sites within K562 cells.
(L) Western blot of putative co-repressors in iWT and G296S D15-CMs. GAPDH is used as a loading control.
(M) Venn diagram of sites bound by GATA4 or TBX5 (black circle) and HDAC2 (blue) in iWT D15-CMs.
(N) Gene Ontology analyses (BioPro/Disease/Pathway) of 1524 TBX5-HDAC2 co-bound sites. Significance shown as −Log10 Bonferroni p-value after multiple hypothesis correction.
(O) Heatmap shows relative (G296S/iWT; LogFC) HDAC2 occupancy at 298 binding sites within endothelial TADs. Blue and red are decreased and increased HDAC2 occupancy respectively. ~30% of HDAC2 sites have decreased occupancy in G296S D15-CMs.
(A) IGV browser tracks of ChIP-seq signal for GATA4, TBX5, MED1, H3K27ac, at select gene loci with a ROSE-predicted putative SE element (grey box). Y-axis represents reads/million/25 bp.
(B) Metagenes plot of normalized ChIP-seq signal for MED1 at genes that are high (green), mid (orange) and low (skyblue) expressed in WT CMs.
(C) Normalized MED1 ChIP-seq signal of TEs and SEs in WT CMs. Boxplot and whiskers show mean, 25th and 75th percentile followed by 5th and 95th percentile. ****, p<0.00005 (t test).
(D) Heatmap shows ChIP-seq signal of MED1, TBX5, GATA4, H3K27ac, H3K27me3 and H3K36me3 around (± 20 kb) 213 SE elements.
(E) Normalized ChIP-seq signal of GATA4, H3K27ac and TBX5 specifically at SEL, SEU, SEE, between WT (black) and G296S (red) CM. Boxplot and whiskers show mean, 25th and 75th percentile followed by 5th and 95th percentile. *, p<0.05, ***, p<0.0005, ****, p<0.00005, ns, not significant (one-way ANOVA and Dunn’s test for multiple hypothesis correction).
(F) Known consensus motifs enriched at SEL (left) and SEE (right) elements.
(G) Bar graph showing relative proportion of genes that are down- (green) or up-regulated (red) and contain a SE element within 20 kb. Grey bar shows all genes that are differentially expressed. The down-regulated genes are largely cardiogenic genes and hence contain a higher proportion of putative SE elements.
(H) Global network diagram analyzed by Topological Data Analysis. Organization of network was performed using RNAseq and ChIPseq results and red or blue gradient colors represent high and low enrichment for each class identifier.
(A–C) Functional validation of previously unrecognized cardiac factors regulated by putative SE elements. (A) Contraction velocity, (B) Calcium flux and (C) Mitochondria mass were quantified after siRNA knockdown of long-non-coding RNAs (MALAT1, HECTD2as, LIN00881, NEAT1) and TFs (HES1, MEIS1, KLF9). SCR, scrambled control, grey line. TBX5 siRNA used as a positive control.
(D) Gene expression of transcriptional network after 48 hr depletion of MALAT1 (a long-non-coding RNA) and KLF9 (a TF) as measured by real-time PCR. Log2 fold change relative to SCR control. Blue and red represents down- and up-regulations respectively.
(E) Gene Ontology analyses (BioPro/Disease/Pathway) of top-20 hubs from GATA4-controlled GRN. Significance shown as −Log10 Bonferroni p-value after multiple hypothesis correction.
(F) Example plots of contractile measurements on micro-patterns. Contractile force plotted as a function of time before (black line) and after (blue line) LY294002 (top) or IRS-1 peptide (bottom) treatment in iWT (black box) and G296S (red box) CMs. Peak amplitude and periodicity are associated with force and beat rate respectively.
HIGHLIGHTS.
Systems-level approach reveals GATA4 roles in human cardiac development and function
Heterozygous GATA4 missense mutation impairs cardiac gene program
GATA4 G296S mutation disrupts TBX5 genome occupancy at cardiac super-enhancers
PI3K signaling is a key “hub” in the GATA4 gene regulatory network
Acknowledgments
We thank all family members for their participation, D.S. lab members for technical assistance and helpful suggestions, G. Howard for scientific editing and B. Taylor for assistance with manuscript preparation. Generous software support from Ayasdi Inc. was provided for topological data analysis. This work was supported by NIH P01 HL098707 (D.S.), U01 HL098179 (D.S.), R01 HL057181 (D.S.), U01 HL100406 (D.S.); R01 EB006745 (B.P.); by AHA 14POST18360018 (A.R.) and 13POST17390040 (YS.A.); by NSF MIKS-1136790 (B.P.); and by Damon Runyon Foundation DRG-2187-14 (R.S.). D.S. was supported by the Roddenberry Foundation and the Younger Family Fund. This work was also supported by NIH/NCRR grant C06 RR018928 to the Gladstone Institutes. This work is dedicated to Dr. Ian Spencer.
Footnotes
AUTHOR CONTRIBUTIONS
YS.A. and R.R. initially conceived this project, generated and characterized iPS cell lines, CRISPR-corrected patient line and performed cardiac differentiations. Metabolic assay was performed by R.R. A.R. conducted single cell micro-patterning and calcium flux assays, supervised by B.P. J.R. assisted in CRISPR-editing, cardiac differentiations and sarcomeric scorings. YS.A. performed all RNA-seq and ChIP-seq assays. K.P. and N.S. performed ATAC-seq assay. YS.A. integrated RNA-seq, ATAC-seq and ChIP-seq results, with important input from R.S. and M.L. JD.F. performed patch clamp analysis and single cell qPCR. C.S. performed patch clamp and calcium flux. N.D. and H.Y. performed motif analyses. T.M. and E.R provided modified mRNA molecules. mtDNA sequencing performed by R.S. and A.N., supervised by M.S.. A.M performed echocardiography of patients. YS.A. and D.S. designed and coordinated experiments and co-wrote the manuscript with input from all co-authors.
Supplemental Information includes seven figures, two movies and five tables available with the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aanhaanen WT, Boukens BJ, Sizarov A, Wakker V, de Gier-de Vries C, van Ginneken AC, Moorman AF, Coronel R, Christoffels VM. Defective Tbx2-dependent patterning of the atrioventricular canal myocardium causes accessory pathway formation in mice. J Clin Invest. 2011;121:534–544. doi: 10.1172/JCI44350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang YS, Tsai SY, Lee DF, Monk J, Su J, Ratnakumar K, Ding J, Ge Y, Darr H, Chang B, et al. Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell. 2011;145:183–197. doi: 10.1016/j.cell.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisping E, Ikeda S, Kong SW, Tarnavski O, Bodyak N, McMullen JR, Rajagopal S, Son JK, Ma Q, Springer Z, et al. Gata4 is required for maintenance of postnatal cardiac function and protection from pressure overload-induced heart failure. Proc Natl Acad Sci U S A. 2006;103:14471–14476. doi: 10.1073/pnas.0602543103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikina MD, Troyanskaya OG. An effective statistical evaluation of ChIPseq dataset similarity. Bioinformatics. 2012;28:607–613. doi: 10.1093/bioinformatics/bts009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Molecular cell. 2002;9:279–289. doi: 10.1016/s1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- Clarke RL, Yzaguirre AD, Yashiro-Ohtani Y, Bondue A, Blanpain C, Pear WS, Speck NA, Keller G. The expression of Sox17 identifies and regulates haemogenic endothelium. Nature cell biology. 2013;15:502–510. doi: 10.1038/ncb2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- Heikinheimo M, Scandrett JM, Wilson DB. Localization of transcription factor GATA-4 to regions of the mouse embryo involved in cardiac development. Developmental biology. 1994;164:361–373. doi: 10.1006/dbio.1994.1206. [DOI] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nature reviews Molecular cell biology. 2015;16:144–154. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann AD, Peterson MA, Friedland-Little JM, Anderson SA, Moskowitz IP. sonic hedgehog is required in pulmonary endoderm for atrial septation. Development. 2009;136:1761–1770. doi: 10.1242/dev.034157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Loganantharaj R, Schroeder B, Fargo D, Li L. PAVIS: a tool for Peak Annotation and Visualization. Bioinformatics. 2013;29:3097–3099. doi: 10.1093/bioinformatics/btt520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375–386. doi: 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathiriya IS, King IN, Murakami M, Nakagawa M, Astle JM, Gardner KA, Gerard RD, Olson EN, Srivastava D, Nakagawa O. Hairy-related transcription factors inhibit GATA-dependent cardiac gene expression through a signal-responsive mechanism. The Journal of biological chemistry. 2004;279:54937–54943. doi: 10.1074/jbc.M409879200. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CT, Morrisey EE, Anandappa R, Sigrist K, Lu MM, Parmacek MS, Soudais C, Leiden JM. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997;11:1048–1060. doi: 10.1101/gad.11.8.1048. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QY, Newbury-Ecob RA, Terrett JA, Wilson DI, Curtis AR, Yi CH, Gebuhr T, Bullen PJ, Robson SC, Strachan T, et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nature genetics. 1997;15:21–29. doi: 10.1038/ng0197-21. [DOI] [PubMed] [Google Scholar]
- Li RG, Li L, Qiu XB, Yuan F, Xu L, Li X, Xu YJ, Jiang WF, Jiang JQ, Liu X, et al. GATA4 loss-of-function mutation underlies familial dilated cardiomyopathy. Biochemical and biophysical research communications. 2013;439:591–596. doi: 10.1016/j.bbrc.2013.09.023. [DOI] [PubMed] [Google Scholar]
- Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, Palecek SP. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A. 2012;109:E1848–E1857. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum PY, Singh G, Lehman A, Ishkanov T, Vejdemo-Johansson M, Alagappan M, Carlsson J, Carlsson G. Extracting insights from the shape of complex data using topology. Scientific reports. 2013;3:1236. doi: 10.1038/srep01236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna-Zurita L, Stirnimann CU, Glatt S, Kaynak BL, Thomas S, Baudin F, Samee MA, He D, Small EM, Mileikovsky M, et al. Complex Interdependence Regulates Heterotypic Transcription Factor Distribution and Coordinates Cardiogenesis. Cell. 2016;164:999–1014. doi: 10.1016/j.cell.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra M, Schluterman MK, Nichols HA, Richardson JA, Lo CW, Srivastava D, Garg V. Interaction of Gata4 and Gata6 with Tbx5 is critical for normal cardiac development. Developmental biology. 2009;326:368–377. doi: 10.1016/j.ydbio.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nature biotechnology. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen JR, Shioi T, Huang WY, Zhang L, Tarnavski O, Bisping E, Schinke M, Kong S, Sherwood MC, Brown J, et al. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110alpha) pathway. The Journal of biological chemistry. 2004;279:4782–4793. doi: 10.1074/jbc.M310405200. [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra C, Sachan N, McNally CR, Koenig SN, Nichols HA, Guggilam A, Lucchesi PA, Pu WT, Srivastava D, Garg V. Congenital heart disease-causing Gata4 mutation displays functional deficits in vivo. PLoS Genet. 2012;8:e1002690. doi: 10.1371/journal.pgen.1002690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Lin Q, Duncan SA, Olson EN. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11:1061–1072. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]
- Nix DA, Courdy SJ, Boucher KM. Empirical methods for controlling false positives and estimating confidence in ChIP-Seq peaks. BMC bioinformatics. 2008;9:523. doi: 10.1186/1471-2105-9-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–845. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, Hong H, Nakagawa M, Tanabe K, Tezuka K, et al. A more efficient method to generate integration-free human iPS cells. Nature methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu JD, Srivastava D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012;485:593–598. doi: 10.1038/nature11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal SK, Ma Q, Obler D, Shen J, Manichaikul A, Tomita-Mitchell A, Boardman K, Briggs C, Garg V, Srivastava D, et al. Spectrum of heart disease associated with murine and human GATA4 mutation. J Mol Cell Cardiol. 2007;43:677–685. doi: 10.1016/j.yjmcc.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro AJ, Ang YS, Fu JD, Rivas RN, Mohamed TM, Higgs GC, Srivastava D, Pruitt BL. Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proc Natl Acad Sci U S A. 2015;112:12705–12710. doi: 10.1073/pnas.1508073112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A, Kong SW, Agarwal P, Gilliss B, Pu WT, Black BL. GATA4 is a direct transcriptional activator of cyclin D2 and Cdk4 and is required for cardiomyocyte proliferation in anterior heart field-derived myocardium. Mol Cell Biol. 2008;28:5420–5431. doi: 10.1128/MCB.00717-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roost MS, van Iperen L, Ariyurek Y, Buermans HP, Arindrarto W, Devalla HD, Passier R, Mummery CL, Carlotti F, de Koning EJ, et al. KeyGenes, a Tool to Probe Tissue Differentiation Using a Human Fetal Transcriptional Atlas. Stem cell reports. 2015;4:1112–1124. doi: 10.1016/j.stemcr.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, Nestler E. ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC genomics. 2014;15:284. doi: 10.1186/1471-2164-15-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava D. Genetic regulation of cardiogenesis and congenital heart disease. Annu Rev Pathol. 2006;1:199–213. doi: 10.1146/annurev.pathol.1.110304.100039. [DOI] [PubMed] [Google Scholar]
- Stefanovic S, Barnett P, van Duijvenboden K, Weber D, Gessler M, Christoffels VM. GATA-dependent regulatory switches establish atrioventricular canal specificity during heart development. Nature communications. 2014;5:3680. doi: 10.1038/ncomms4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergachis AB, Neph S, Reynolds A, Humbert R, Miller B, Paige SL, Vernot B, Cheng JB, Thurman RE, Sandstrom R, et al. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell. 2013;154:888–903. doi: 10.1016/j.cell.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoris CV, Li M, White MP, Liu L, He D, Pollard KS, Bruneau BG, Srivastava D. Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell. 2015;160:1072–1086. doi: 10.1016/j.cell.2015.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y, et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell stem cell. 2013;12:127–137. doi: 10.1016/j.stem.2012.09.013. [DOI] [PubMed] [Google Scholar]
- Van Handel B, Montel-Hagen A, Sasidharan R, Nakano H, Ferrari R, Boogerd CJ, Schredelseker J, Wang Y, Hunter S, Org T, et al. Scl represses cardiomyogenesis in prospective hemogenic endothelium and endocardium. Cell. 2012;150:590–605. doi: 10.1016/j.cell.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Hoffmann AD, Burnicka-Turek O, Friedland-Little JM, Zhang K, Moskowitz IP. Tbx5-hedgehog molecular networks are essential in the second heart field for atrial septation. Developmental cell. 2012;23:280–291. doi: 10.1016/j.devcel.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome biology. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Xu JH, Xu WJ, Yu H, Wang Q, Zheng HZ, Jiang WF, Jiang JF, Yang YQ. A novel GATA4 loss-of-function mutation responsible for familial dilated cardiomyopathy. International journal of molecular medicine. 2014;33:654–660. doi: 10.3892/ijmm.2013.1600. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene expression signature of WT and G296S iPS cells during cardiac differentiation.
Open chromatin signature of iWT and G296S derived human CPCs.
GATA4, TBX5 and MED1 binding sites from WT and G296S derived human CMs.
H3K27ac, H3K4me3, H3K27me3, H3K36me3 marked sites from WT and G296S derived human CMs.
Compilation of RNA-seq, ATAC-seq and ChIP-seq results for Ayasdi and GRN predictions.
(A) Genotype-phenotype information of GATA4 pedigree with associated iPS cell lines. Colored rows represent GATA4 heterozygous mutants.
(B) Morphology of mCherry-positive, puromycin-resistant CRISPR-iPS clones before (left) and after (right) TAT-Cre excision of mCherry-puromycin selection casette.
(C) Karyotype analyses show all patient-iPS clones have normal karyotypes.
(D) Heatmap shows hierarchical clustering of gene expressions of HDF and self-renewal markers measured by RT-qPCR. Values are row-scaled to show their relative expression. Blue and red are low and high levels respectively.
(E) Immunostaining of all iPS lines for pluripotency markers. Red, GATA4 mutants.
(F) Heatmap shows hierarchical clustering of Spearman correlation scores of iPS and ES (H7) cells based on RNA-seq profiles. Score of 1 (yellow) denotes perfect correlation. Red, GATA4 mutants.
(G) Immunostaining of all iPS lines after spontaneous EB differentiation (left group) and teratoma formation (right group). GFAP and neural rosettes indicate ectoderm differentiation, SMA and cartilage indicate mesoderm differentiation, AFP and gut-tube indicate endoderm differentiation. Red, GATA4 mutants.
(A) Immunostaining of CM-specific markers during purification in lactatehi glucoselo media. Percentage of cTnT+ cells increased from 31–70% and total cell numbers decreased from 674–187 over 4 days.
(B) Heatmap shows gene expression of select mesoderm and CM markers during step-wise cardiac differentiation as measured by RNA-seq. Values are row-scaled to show their relative expression. Blue and red are low and high levels respectively.
(C) RNA-seq expression values (FPKM) of representative stage-specific genes during ES-H7 (blue box), MES (green), CPC (grey) CM (pink) differentiation. Known markers validate differentiation. Unknown transcriptional regulators and markers are highlighted.
(D) Heatmap shows HOPACH clustering of stage-specific genes. FPKM values are row-scaled to show relative expression. Blue and red are low and high levels respectively. Select GO and signaling categories are highlighted for each stage with matching colors.
(E) Immunostaining of 4 CM-enriched proteins in iPS-derived CM. Parentheses, >90% of CM are positive for all markers.
(F) Patch clamp recordings of single iPS-derived CM show action potential morphologies resembling pacemaker, ventricular and atrial CM.
(G) Heatmap shows hierarchical clustering of Spearman correlation scores of adult primary CMs, ES and iPS differentiated CMs based on single-cell RT-qPCR of 61 CM marker genes. Score of 1 (yellow) denotes perfect correlation.
(H) Percentage of binucleated vs. mononucleated CMs assessed on single-cell patterns.
(I) Immunostaining of CM-enriched proteins in all iPS-derived CM vs ES-derived CM after lactate purification. Parentheses, >90% of CM are positive for cTnT, MLC2v, αActinin and cTnI. Red, GATA4 mutants.
(J) Immunostaining of cTnT in isogenic CRISPR-corrected iWT vs mutant G296S derived CM after purification. Both lines were differentiated in parallel under identical conditions. Percentage of cTnT cells shown at bottom right.
(K) Contractile measurements on micro-patterns. Time (s) spent in each contraction cycle is shown for all CM responding accurately to 1 Hz pacing. Data are mean ± SEM. ****, p<0.0001 (t test).
(L–N) Contractile measurements on micro-patterns at early (D35) vs. late (D70) stages of CM differentiation. (L) % of single-CM responding accurately to 1 Hz electrical pacing in iWT and G296S. Blue arrow shows reduced % of G296S-CMs responding accurately to stimulation at both stages. (M) Traction force microscopy measurements of force production of all CMs responding accurately to 1 Hz pacing. (N) Traction force microscopy measurements of relaxation velocity of all CMs responding accurately to 1 Hz pacing. All measurements were done on isogenic CMs generated in parallel differentiations.
(O) Action potential measurements of WT and G296S CM. dV/dtmax, maximum upstroke velocity; APD90, duration of action potential at 90% repolarization. Data shown are mean ± SEM from 2 WT and 2 G296S lines. *, p<0.05 (Mann-Whitney test).
(P) Description of sarcomere scoring scheme. Class IV represents the most disarrayed sarcomeric organization.
(Q) Sequencing of mitochondrial-DNA for heteroplasmy mutations. Each data point represents CM from 1 patient-iPS-CM sample. Data shows mean ± SEM. G296S CM (siblings) showed highly similar number of de novo mutations in mtDNA and the WT (unrelated), reflecting pattern of inheritance.
(A) Heatmap shows hierarchical clustering of 2228 genes differentially expressed at any time point. Blue and red represent decreased and increased expressions (G296S/iWT Log2FC) respectively.
(B) Venn diagram showing overlaps of down- (left) and up-regulated (right) genes by G296S at CPC (black circle), D15-CM (red) and D32-CM (blue) stages of cardiac differentiation. 38 genes were consistently down or upregulated at all 3 time points.
(C) Heatmap shows gene expressions of 38 genes consistently down- (blue) or up-regulated (red) during cardiac differentiation of iWT and G296S CM as measured by RNA-seq. Values are row-scaled to show their relative expression. Blue and red are low and high levels respectively.
(D) Gene Ontology analyses (BioPro/Disease/Pathway) of 38 genes consistently down or upregulated during cardiac differentiation of iWT and G296S CM. Significance shown as −Log10 Bonferroni p-value after multiple hypothesis correction.
(E–F) FACS analyses of cardiogenic markers (GATA4, NKX2.5, TBX5, ISL1) and hemogenic-endothelial marker (KDR) in iWT vs. G296S D7-CPCs during CM differentiation. Reduced protein expression of GATA4, NKX2.5 and TBX5 was observed in G296S. Increased ISL1 was observed.
(G) FACS analyses of CM marker (cTnI) and endothelial marker (CD31) in iWT vs. G296S D15-CMs during CM differentiation. No marked increase in CD31+ cells were observed in G296S.
(H) FACS analyses of endothelial marker (CD31) in iWT vs. G296S during endothelial cell differentiation. Marginal increase in CD31+ cells was observed in G296S.
(I) Heatmap shows expressions of genes associated with chamber or atrioventricular canal myocardiums and smooth muscle are down- (blue) and up-regulated (red) in G296S CMs respectively. Values are row-scaled to show their relative expression. Blue and red are low and high levels respectively.
(J) GSEA analyses of geneset representing cellular respiration showed reduction in G296S CMs. NES, normalized enrichment score. FDR, false discovery rate.
(K) RT-qPCR validation of RNAseq results at D7-CPC, D15-CM and D32-CM. High R2 value indicate strong correlation between RNAseq and RT-qPCR.
(A) IGV browser tracks of ChIP-seq signal for GATA4, TBX5, H3K27ac, H3K4me3, H3K36me3, H3K27me3 at known gene target loci (TNNT2, TNNI1) in WT CM. Grey boxes denote significantly enriched (over input) peaks identified by MACS2. Y-axis represents reads/million/25bp.
(B) Metagenes plot of normalized ChIP-seq signal for H3K4me3, H3K27ac, H3K27me3, H3K36me3, GATA4 and TBX5 at genes that are high (green), mid (orange) and low (skyblue) expressed in WT CMs. TSS, transcription start site. TES, transcription end site. All ChIP intensities, except H3K27me3’s, were positively correlated to gene expression levels.
(C) Heatmap shows ChIP-seq signal at sites (± 5 kb) bound by GATA4 only, TBX5 only and G4T5 co-bound in WT CM. ChIP-seq signal of modified-histone marks also shown at these sites. Sites are ordered by decreasing intensity of GATA4 signal.
(D) Venn diagram of sites bound by GATA4 (black circle) and/or TBX5 (purple) in WT CM. 2,428 sites are G4T5 co-bound in human CMs.
(E) Pie-chart shows gene-body, upstream, downstream distribution of 2,428 G4T5 co-bound sites.
(F) Gene Ontology analyses (BioPro/Disease/Pathway) of 2,428 G4T5 co-bound sites. Significance shown as −Log10 Bonferroni p-value after multiple hypothesis correction.
(A) Venn diagram of sites bound by GATA4 (purple circle) and/or TBX5 (red) in G296S CMs. Only 1,605 sites are G4T5 co-bound. Total number of G4T5 sites was reduced in G296S.
(B) Metagenes plot of normalized ChIP-seq signal for GATA4, TBX5 and H3K27ac at G4T5 co-bound sites (± 5 kb) in WT and G296S CM.
(C) Normalized ChIP-seq signal of GATA4, TBX5, H3K27ac specifically at G4T5L, G4T5U, G4T5E, in WT (black) and G296S (red) CMs. Boxplot and whiskers show mean, 25th and 75th percentile followed by 5th and 95th percentile. ***, p<0.0005, ****, p<0.00005, ns, not significant (one-way ANOVA and Dunn’s test for multiple hypothesis correction).
(D) FPKM values of genes mapped ± 20 kb of all G4T5E sites during iWT (black) and G296S (red) cardiac differentiations. Boxplot and whiskers show mean, 25th and 75th percentile followed by 5th and 95th percentile. *, p<0.05, ns, not significant (Wilcoxon signed-rank non-parametric test)..
(E) Known consensus motifs enriched at G4T5L (left) and G4T5E (right) sites.
(F) Bar chart showing average distance (bp) between G4T5L sites to any ENCODE-TF binding site. The TF and the cell type profiled are shown on the Y-axis. For example, G4T5L sites are located proximally (<50bp) to EP300 sites within SKNSH neuronal cells.
(G) Putative direct targets of GATA4 and TBX5 as defined by ChIP-seq binding in WT and RNA-seq differential expression. Venn diagrams show differentially expressed genes (black circle) overlapped with G4T5 binding (blue) within 20 kb. Differential gene expressions were further separated into 3 classes: D15-CM or D32-CM stage (top), D15-CM stage (bottom left), D32-CM stage (bottom right).
(H) FPKM values of genes mapped ± 20 kb of sites that have decreased G4 and increased T5 binding (G4DOWN_T5UP) versus genes mapped ± 20 kb of sites that have decreased G4 and decreased T5 binding (G4DOWN_T5DOWN). The genes with concomitant reduction in G4T5 binding are expressed at a lower level. Boxplot and whiskers show mean, 25th and 75th percentile followed by 5th and 95th percentile.
(I) Metagenes plot of normalized ChIP-seq signal for H3K4me3 and H3K27me3 at TSS of endothelial specific genes (± 5 kb) in WT (black) and G296S (red) CM.
(J) Promoter motif enrichment analyses at 81 endothelial genes that were up-regulated. Significance shown as −Log10 Bonferroni p-value after multiple hypothesis correction.
(K) Scatter plot of GATA4 sites within endothelial or PI3K genes with respect to any ENCODE-TF binding site shown as a function of average distance (bp) against percentage. The top ENCODE-TF and the cell type profiled are highlighted in red. For example, GATA4 sites in endothelial TADs are located proximally (<100bp) and frequently (>25%) to RCOR1 sites within K562 cells.
(L) Western blot of putative co-repressors in iWT and G296S D15-CMs. GAPDH is used as a loading control.
(M) Venn diagram of sites bound by GATA4 or TBX5 (black circle) and HDAC2 (blue) in iWT D15-CMs.
(N) Gene Ontology analyses (BioPro/Disease/Pathway) of 1524 TBX5-HDAC2 co-bound sites. Significance shown as −Log10 Bonferroni p-value after multiple hypothesis correction.
(O) Heatmap shows relative (G296S/iWT; LogFC) HDAC2 occupancy at 298 binding sites within endothelial TADs. Blue and red are decreased and increased HDAC2 occupancy respectively. ~30% of HDAC2 sites have decreased occupancy in G296S D15-CMs.
(A) IGV browser tracks of ChIP-seq signal for GATA4, TBX5, MED1, H3K27ac, at select gene loci with a ROSE-predicted putative SE element (grey box). Y-axis represents reads/million/25 bp.
(B) Metagenes plot of normalized ChIP-seq signal for MED1 at genes that are high (green), mid (orange) and low (skyblue) expressed in WT CMs.
(C) Normalized MED1 ChIP-seq signal of TEs and SEs in WT CMs. Boxplot and whiskers show mean, 25th and 75th percentile followed by 5th and 95th percentile. ****, p<0.00005 (t test).
(D) Heatmap shows ChIP-seq signal of MED1, TBX5, GATA4, H3K27ac, H3K27me3 and H3K36me3 around (± 20 kb) 213 SE elements.
(E) Normalized ChIP-seq signal of GATA4, H3K27ac and TBX5 specifically at SEL, SEU, SEE, between WT (black) and G296S (red) CM. Boxplot and whiskers show mean, 25th and 75th percentile followed by 5th and 95th percentile. *, p<0.05, ***, p<0.0005, ****, p<0.00005, ns, not significant (one-way ANOVA and Dunn’s test for multiple hypothesis correction).
(F) Known consensus motifs enriched at SEL (left) and SEE (right) elements.
(G) Bar graph showing relative proportion of genes that are down- (green) or up-regulated (red) and contain a SE element within 20 kb. Grey bar shows all genes that are differentially expressed. The down-regulated genes are largely cardiogenic genes and hence contain a higher proportion of putative SE elements.
(H) Global network diagram analyzed by Topological Data Analysis. Organization of network was performed using RNAseq and ChIPseq results and red or blue gradient colors represent high and low enrichment for each class identifier.
(A–C) Functional validation of previously unrecognized cardiac factors regulated by putative SE elements. (A) Contraction velocity, (B) Calcium flux and (C) Mitochondria mass were quantified after siRNA knockdown of long-non-coding RNAs (MALAT1, HECTD2as, LIN00881, NEAT1) and TFs (HES1, MEIS1, KLF9). SCR, scrambled control, grey line. TBX5 siRNA used as a positive control.
(D) Gene expression of transcriptional network after 48 hr depletion of MALAT1 (a long-non-coding RNA) and KLF9 (a TF) as measured by real-time PCR. Log2 fold change relative to SCR control. Blue and red represents down- and up-regulations respectively.
(E) Gene Ontology analyses (BioPro/Disease/Pathway) of top-20 hubs from GATA4-controlled GRN. Significance shown as −Log10 Bonferroni p-value after multiple hypothesis correction.
(F) Example plots of contractile measurements on micro-patterns. Contractile force plotted as a function of time before (black line) and after (blue line) LY294002 (top) or IRS-1 peptide (bottom) treatment in iWT (black box) and G296S (red box) CMs. Peak amplitude and periodicity are associated with force and beat rate respectively.