

Abstract
Motivation: A perennial problem in the analysis of environmental sequence information is the assignment of reads or assembled sequences, e.g. contigs or scaffolds, to discrete taxonomic bins. In the absence of reference genomes for most environmental microorganisms, the use of intrinsic nucleotide patterns and phylogenetic anchors can improve assembly-dependent binning needed for more accurate taxonomic and functional annotation in communities of microorganisms, and assist in identifying mobile genetic elements or lateral gene transfer events.
Results: Here, we present a statistic called LCA* inspired by Information and Voting theories that uses the NCBI Taxonomic Database hierarchy to assign taxonomy to contigs assembled from environmental sequence information. The LCA* algorithm identifies a sufficiently strong majority on the hierarchy while minimizing entropy changes to the observed taxonomic distribution resulting in improved statistical properties. Moreover, we apply results from the order-statistic literature to formulate a likelihood-ratio hypothesis test and P-value for testing the supremacy of the assigned LCA* taxonomy. Using simulated and real-world datasets, we empirically demonstrate that voting-based methods, majority vote and LCA*, in the presence of known reference annotations, are consistently more accurate in identifying contig taxonomy than the lowest common ancestor algorithm popularized by MEGAN, and that LCA* taxonomy strikes a balance between specificity and confidence to provide an estimate appropriate to the available information in the data.
Availability and Implementation: The LCA* has been implemented as a stand-alone Python library compatible with the MetaPathways pipeline; both of which are available on GitHub with installation instructions and use-cases (http://www.github.com/hallamlab/LCAStar/).
Contact: shallam@mail.ubc.ca
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
The rise of next-generation sequencing technologies has generated a tidal wave of sequencing projects focused on natural and engineered ecosystems, resulting in a plethora of environmental sequence information. Several software pipelines, including MG-RAST (Meyer et al., 2008), HUMAnN (Abubucker et al., 2012) and MetaPathways (Hanson et al., 2014a,b; Konwar et al., 2013, 2015) have been developed to process environmental sequence information, provide taxonomic and functional annotations and assist in metabolic pathway reconstruction. Despite the availability of these pipelines, accurately assigning taxonomy to environmental sequence information remains a challenging enterprise given a general lack of reference genomes for most uncultivated microorganisms. From a bioinformatics perspective, current software tools lack statistical frameworks and hypotheses tests for the assignment of reads or assembled contigs to discrete taxonomic bins leaving developers and investigators with limited theoretical direction (Prosser, 2010; Thomas et al., 2012).
Due to the popularity of the MEGAN software, the lowest-common ancestor (LCA) method is routinely applied to individual open reading frame (ORF) annotations with a correction based on homology search quality statistics (Huson et al., 2007). While using LCA to assign individual ORF taxonomy seems straight-forward, it is unclear how to effectively apply this rule to contigs or scaffolds containing multiple ORFs. Consider an alternative perspective of electing a representative taxonomy where each qualifying ORF annotation ‘votes’ for overall contig assignment. In this election individual ORFs may have differing ‘taxonomic opinions’, projecting their vote differently onto the Tree of Life. Two popular Voting Theory results provide justification in choosing a majority as the correct response. Condorcet’s Jury Theorem considers an election of two opinions, one correct and one incorrect and voters each independently choose one of these two opinions with the assumption that they choose the correct response with probability (Estlund, 1994). The observed majority converges in probability to the correct decision as the election size grows to infinity. Alternatively, Feige and colleagues studied the depth of noisy decision trees where each query at a node produces the correct answer with some probability (Feige et al., 1994). They derived tight bounds on the number of queries required to compute threshold and parity functions, and analyze a noisy comparison model with tight bounds on comparison, sorting, selection and merging. However, applying these Voting Theory methods to taxonomic count data is complicated by the hierarchical definitions. For instance, individual ORFs predicted on the same contig may be assigned to different taxonomic levels within closely related lineages, e.g. species, sub-species, strain, or serovar. The approximate nature of popular homology search algorithms and idiosyncratic database annotation increases uncertainty in taxonomic estimation from functional genes, making accurate placement on the taxonomic hierarchy a challenge. Moreover, sparse observations of multiple related taxa can undermine confidence in the reported majority.
Here, we introduce LCA*, an entropy-based statistic and algorithm for declaring a sufficiently supported majority on the NCBI Taxonomic Database Hierarchy (NCBI Tree) (Federhen, 2012). The statistic offers a principled method of electing a majority taxon by applying results from Information and Voting Theory to contig or scaffold annotations, obtaining an acceptable majority while minimizing changes to the underlying taxonomic distribution. Moreover, order-restricted statistical results can be used to provide supremacy tests for an elected taxonomy as an alternative to traditional χ-squared uniformity tests. Using both simulated and real world datasets we demonstrate that in the presence of known reference alignments, voting-based methods of simple Majority and LCA* are consistently more accurate than the conventional LCA algorithm popularized by MEGAN, hereafter referred to as LCA2, and that LCA* taxonomy strikes a balance between specificity and confidence to provide an estimate appropriate to the available information in the data.
1.1 Motivation
Lets work LCA* through an illustrative example where taxonomic annotations are quite variable and dispersed (Fig. 1). A simple Majority method, choosing the taxonomy with the most annotations may be intuitive, but a majority of 3 out of 11 taxa is not very convincing. Alternatively, to combat this dispersion it might be a good idea to elect the LCA as the majority, but this very conservative estimate manifests limited resolving power (e.g. root, prokaryotes, etc.). In the case of individual ORF annotations, LCA estimates would be made less extreme by discarding annotations that do not meet certain quality thresholds, on the evolutionary assumption that hits to taxa phylogenetically further away from the origin will be less similar. However, in the case of assembled environmental sequence information, e.g. contigs or scaffolds, this is often not an option, because common practice summarizes annotations via some taxonomic estimation method (e.g. Best-BLAST, LCA). LCA* takes a bottom-up approach by expanding the specific simple majority estimate upwards in the taxonomic hierarchy, progressively collapsing annotations until a satisfying majority, an α-majority, is obtained. Here, we can leverage relevant Voting Theory results like Condorcet’s Jury Theorem and the work of Feige on noisy decision trees to justify a proportion . However, there remains the issue of how to collapse the tree automatically, as collapsing arbitrarily can introduce significant bias. Here, we will use the information-theoretic interpretation of entropy to motivate an algorithm of collapsing annotations in a principled way.
Fig. 1.

Illustrative example of taxonomic assignment methods: LCA*, Majority and LCA2. Node numbers indicate the number of annotations associated with each taxonomic position in the tree, and the double-circled node is the actual originating taxonomy. In a typical metagenomic assembly ORF, annotations can be variable, spanning a number of different positions in the tree. In this example, LCA2 provides a conservative estimate, while the simple Majority method provides a specific taxon without very much support from the data. The LCA* tempers the Majority estimate by collapsing annotations up the tree in a principled way until a sufficient α-majority is reached (), distributing the entropy of the underlying taxonomic distribution as little as possible
Since the application of information entropy is relatively rare in microbial ecology outside of diversity estimation (Legendre and Legendre, 2012), we will provide a brief introduction that will help in the understanding and motivation of LCA*. Entropy (Shannon’s Entropy) is the fundamental unit of information defined as the average amount of information needed to specify the state of a random variable. Intuitively it can be described as a measure of uncertainty. The uniform random variable, a situation where all outcomes are equally likely has the highest possible uncertainty, and therefore the highest entropy, while a more spiked random variable has less uncertainly, and therefore less entropy. Entropy is an extremely useful concept, and has been central to the development of coding theory (e.g. file compression), cryptography and signal processing (Wade, 1994). Moreover, differences in entropy (also known as Relative Entropy or the Kullback–Leibler divergence) are used as a measure for the divergence between two probability distributions (Kullback and Leibler, 1951). For example, in Machine Learning, Random Forest classifiers use entropy as a measure of separation quality, and it is used to identify the most discriminating separations in a hierarchy of classifier models (Breiman, 2001). The LCA* algorithm, uses the concept of entropy to measure the amount of change that collapsing annotations up the tree to a particular node will cause, calculating an α-majority while minimizing the change in entropy of the taxonomic distribution as much as possible.
2 LCA*: derivation and algorithm
In order to reason clearly about assembled environmental sequence information, annotations and taxonomy within the NCBI Tree, it is first necessary to construct a mathematical framework that defines them, their relationships, and describes any additional notation needed to perform the entropy calculations and implement the algorithm. First, we will describe notation for the inherent tree-structure of the NCBI Tree, and add some specific notations for child nodes and child sub-trees that will be useful when calculating the entropy of annotations within the tree. Next, we will describe assembled environmental sequence information from contigs or scaffolds, predicted ORFs and annotations, and define what it means for a particular set of annotations to have a sufficient α-majority. To facilitate the collapsing of annotations up the taxonomic hierarchy, we will define an annotation as having a taxonomic lineage in terms of partially ordered sets, which will allow us to define phylogenetically valid transformations among observed annotations. In particular, we will define consistent reductions to be a special kind of transformation for collapsing all annotations within a sub-tree up to its root node.
Having devised clear methods for moving annotations around the tree, we will then define the entropy of the tree in terms of its annotations collapsed at a particular node. From here we will make a key observation that the entropy of annotations collapsed at a given node can be decomposed to the sum of itself and its children. Using this new decomposition to formulate the difference in entropy between two annotations, we observe that minimizing the difference is equivalent to minimizing the entropy of the node we choose to move to, an observation that will be extremely useful in formulating an efficient algorithm. Reasoning that we can calculate the change in entropy of annotations collapsed at every node, there must be some node with annotations that has both a valid α-majority and a minimal entropy change compared to all other nodes in the tree. This node is the target LCA*. Finally, we formulate an algorithm to calculate LCA*. We first describe a brute-force method of finding the valid node, and then observe that a node-coloring scheme restricting calculations to observed annotations significantly reduces computational complexity.
2.1 Derivation
Let the NCBI Taxonomic Database Hierarchy be a tree TNCBI, where the nodes x represent taxa and edges represent phylogenetic relationships. Let X denote the set of all nodes in TNCBI, , where M is the total number of taxa in TNCBI (including taxa at internal nodes). Next, let Tx denote a sub-tree within TNCBI rooted at node x. Let the set of nodes in sub-tree Tx be denoted Xx, allowing a complete recursive notation for all trees and sub-trees of TNCBI (Fig. 2). As a special case we will denote the root node of TNCBI as , and it follows that .
Fig. 2.

The NCBI taxonomy tree structure used in our derivation. Nodes represent taxons and a line between two nodes shows taxonomic relationships. Tx denotes the sub-tree of TNCBI rooted at x and are the immediate children of x
It will be convenient to discuss the children of a given node , so let the set of immediate children of node x be , where s is the number of immediate child nodes of x. Further, the set of immediate children of x have respective sub-trees , where each child sub-tree has the set of nodes . A node x is a leaf node if it has no immediate children, i.e. and , otherwise x is a non-leaf node.
Next, let us describe assembled contigs, ORFs, and their taxonomic annotations. Let R be the set of ORFs in the metagenome, and every ORF in R is by way of some annotation associated with a taxonomic node on TNCBI. Let ORFs that came through the annotation procedure without a known taxonomy be set to ‘root’ at node . In other words, every predicted ORF in a contig has a corresponding taxonomic annotation, which is set to ‘root’ if it did not find an acceptable hit in the annotation database. Suppose contig C has the set of N ORFs with a corresponding n-tuple of annotations where annotation ai corresponds to ORF for . We use the notation to denote the set of annotations in the n-tuple A, with the set of annotations from contig C being detonated as .
We need to determine or elect a taxon from annotations to label contig C. One straight-forward method might be to use a simple majority vote procedure on the taxonomic assignments for each ORF on contig C, AC. However, there may not be a simple majority among taxa AC, or even a majority with a minimum proportion of the votes α, where , a so-called α-majority.
Definition 1 (α-majority). Given an n-tuple of annotations A, for any we say that A satisfies an α-majority if there is a taxon that constitutes at least α-fraction of the elements. Conversely, if no such taxon a exists then we say annotations A does not satisfy an α-majority.
Clearly, given two proportions α and such that , if some annotation n-tuple A has an α-majority, then by transitivity it implies that A also has an -majority.
It might be possible to obtain an α-majority by replacing annotations A with modified annotations , where each taxon a is replaced by one of its ancestral taxa , and define such a relationship as a partial order on taxa as , where . For example, if a is Alphaproteobacteria, could be Proteobacteria or some other ancestor of a all the way to the root . Clearly, it is always possible to create a majority by replacing each taxon with the root . However, this trivial result has limited resolving power, as we have lost almost all taxa-specific information about contig C other than ‘C came from LUCA.’ In fact, any modified set of taxa essentially represents some loss of taxon-specific information from A. Therefore, we would like to formulate a way to quantify this loss of information in a principled way such that we can design an algorithm to construct an α-majority while minimizing the amount of information loss required to attain it.
To formulate this problem, we need to extend the definition of the partial order ≼ to n-tuples as follows. We will now denote some specific transformations on an n-tuple of taxa that we call reductions:
For any two taxa we denote the reduction of a to as , such that, . If there exists an annotation such that then either is equal to a or . In other words, is either a itself or in its lineage. When a is reduced to through then we denote such a multistep reduction of a to as .
We define the partial order relation for n-tuples A and as: if for every pair of elements a and from A and , at the same index positions, satisfies the relation . Then we denote by to mean for every corresponding element a (in A) and (in ) we have ; and by we denote the fact that for every corresponding pair of elements a (in A) and (in ) we have some series of transformations . Note that for both and we have .
We define annotation n-tuple to be consistent if for every pair of annotations a and from A and we have and . Thus, we define a consistent reduction to be any reduction , and similarly, a set of consistent reductions as where this condition holds. This consistency condition is imposed in order to not bias a taxon in terms of its depth on the NCBI Tree (measured from the root node). For example, if for annotations , where Alphaproteobacteria and Proteobacteria, then A does not preserve consistency since . However, annotations , where Alphaproteobacteria and Betaproteobacteria, then annotations preserves consistency. Intuitively, we can view a consistent reduction as a reduction of all annotations descending from x to x, or in other words, the collapsing of all annotations corresponding to a sub-tree of x to x.
Let’s note some observations about the reduction of annotation n-tuples. Every reduction step for an annotation n-tuple A to another n-tuple is less specific with respect to A. It is important to realize that cannot convey any new information about A. Moreover, for any annotation n-tuple A, there exists a reduction where respects α-majority for some α in the interval ; note that , where is the n-tuple where every element is the root , can always provide a possible solution. Therefore, if annotation n-tuple A does not have a α-majority, there exists an that has α-majority and , i.e. a sequence of single step reductions . For a given A there may be multiple solutions to take the position of , and in such cases we would like to pick the candidate that loses the least amount of taxonomic information. In this case, we assume that information-theoretic entropy and biological ‘taxonomic information’ coincide. We must now define entropy of taxonomic annotations A.
Definition 2. Given annotation n-tuple A and node x in TNCBI, we define entropy as , where is the unique set of elements in A, is the number of annotations in that are taxon z, and N is the length of the annotation n-tuple A.
Having defined our reductions and tree entropy given annotation n-tuple A, given an acceptable majority proportion threshold , we can now formulate a minimal entropy reduction on A to an α-majority satisfying .
Definition 3 (Minimal Entropy Reduction). Given annotation n-tuple A from contig C and a majority proportion α, we would like to produce an annotation through reductions , such that satisfies an α-majority and minimizes the change in entropy for all such , i.e. , electing the α-majority taxon of as the origin of C.
In order to find an annotation n-tuple that has an α-majority and minimizes the change in entropy, it is sufficient to replace some subset of A, S, by the lowest common ancestor of all taxa a in S, i.e. for all . If there exists another such that for each s, this implies , (i.e. the lowest common ancestor of S is unique). Therefore, one brute-force way would be to compute the change in entropy for all valid transformations at every node x.
Next we will expand on and define some simplifications of entropy and change in entropy , that will prove useful in the actual construction of the LCA* algorithm. Notice that entropy can be written
where refers to the number of annotations assigned to taxon node z. Similarly, observing that the set of annotations in a sub-tree at x, Xx, can be partitioned as the union of itself and the nodes in its immediate children’s sub-trees , we can partition the entropy of a set of annotations as follows:
where if y is a leaf node in TNCBI and , otherwise. Note that we decomposed the entropy into two main terms, the entropy of node x, , and the sum of the entropy terms of its immediate children’s trees, (Fig. 3).
Fig. 3.
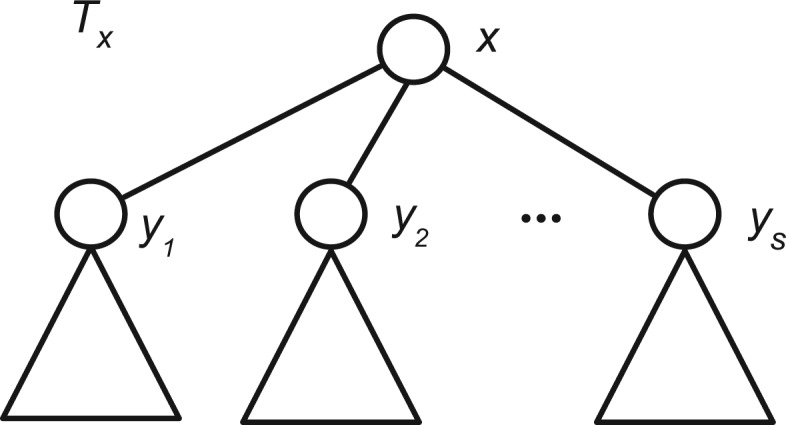
Decomposition of entropy into sub-trees. A key observation in our derivation is that the entropy of annotation A in a tree rooted at a given annotation node x, Tx, can be decomposed into the sum of node x and the nodes of its immediate children’s subtrees as . From here we can decompose the calculation of entropy H(A) in the same partition
From here we can express the change in entropy on a consistent reduction of annotations as
Since we are interested in an that minimizes , note that all terms corresponding to A in the above relation remain constant. We can simplify the calculation by focusing on finding an such that is a consistent reduction of A and minimizes and the recursive relation
| (1) |
based on which we will design our algorithm. We will now show that such a transformation to exists for any given starting annotations A.
Proposition 1. Suppose A is any n-tuple of annotations, then for any there exists a taxon and a consistent reduction of A to some n-tuple , such that (i) respects α-majority, (ii) for all consistent reductions and (iii) A and only differ in the elements where is in .
Proof. Note that in the above proposition it is easy to show the existence of a taxon that satisfies conditions (i) and (ii). This is because is a trivial solution that satisfies condition (i), and the set of candidates that satisfies is non-empty, hence here exists a taxon that satisfies condition (ii). In order to realize (iii), note that since and has an α-majority, therefore, there exists an annotation in which is at least α fraction of all elements in . Since the reductions are consistent, we can achieve the α majority by simply collapsing the annotations that are descendants of in TNCBI, or specifically, for all annotations where . □
2.2 Algorithm
Since we have outlined a mathematical framework defining α-majority, consistent reductions on the NCBI Tree, and a recursive definition of the entropy of annotations on TNCBI, we can now focus on designing and implementing an algorithm, ComputeLCA*, that calculates an α-majority for a given contig C while minimizing changes to its underlying information entropy.
The input to ComputeLCA* consists of the NCBI taxonomy tree TNCBI, and the n-tuple of ORF annotations A for the ORFs in a contig C, and the threshold α that defines the majority (Algorithm 1). Since we are interested in the taxon that minimizes the change in entropy , our algorithm is designed to exploit the recursive nature of traversal in the TNCBI as well as the recursive delta entropy term (Equation 1).
We use the global hash data-structures and for every node . stores the sum of annotations at node x at its collapsed sub-tree , and similarly stores the sum of entropy terms for each node in the subtree of , i.e. at a given x. ComputeLCA* starts at the root and recursively traverses TNCBI, calculating sums of L and S at all nodes. The algorithm then selects the sum that minimizes the relative entropy and also has sufficient support α.
Algorithm 1.
ComputeLCA*
Require: TNCBI, A, α
Ensure:
1: / * S and L are hashes */
2:
3: call ComputeSL()
4: /* result */
5:
6:
7: /* Subroutine ComputeSL computes the S, L for each taxon */
8: subroutine ComputeSL()
9: if x is a leaf-node in TNCBI
10:
11:
12: else
13:
14: for each c in Children(x)
15: call ComputeSL()
16:
17:
18: return
2.2.1 Implementation
ComputeLCA* for a typical number of annotations on a contig does not take more than a few hundred milliseconds, but the described brute-force method traversing the entire NCBI Tree is computationally inefficient, and for samples with hundreds of thousands of contigs the total computation time could be large. Therefore, in the implementation of the ComputeLCA*, a key optimization step is incorporated that skips the examination of subtrees where no annotations exist.
Consider the set of N ORFs and corresponding set of N annotations originating from contig C. Let M be the total number of taxonomic nodes in TNCBI. Then according to ComputeLCA*, it can take O(MN) steps to compute the LCA* taxonomy for C. Note that at line 14 of ComputeLCA*, it is redundant to visit the sub-tree rooted in the child node stored in loop variable c if there are no annotations in the sub-tree. However, in order to know if annotations are present in a given sub-tree of TNCBI, before running ComputeLCA*, we color all nodes whose subtree contains a non-empty set of annotations. We mark the nodes by considering one annotation at a time, say a, as follows:
we start at the node a in TNCBI and travel upwards towards the root one parent step at a time;
in each step, if the current node p is not marked then mark p, and move to its parent (if present), otherwise we are done with annotation a;
if the parent is already marked we are done with annotation a.
We now describe the relative computational complexity after our optimization. Consider the partially ordered set of annotations on TNCBI, and suppose L is the size of largest subset S of such that any two annotations in S are not comparable via ≼ to each other. Our modified algorithm therefore takes (DL) steps to mark the nodes in the upfront step, where D is the maximum tree-depth in our set of annotations . Since we only visit nodes that have been colored at line 14, our modified algorithm has time complexity O(DL). Although the worst-case time complexity could still be O(MN), where the annotation-breath spans the entire TNCBI, i.e. D = M and L = N, most real-word contig annotations tend to coalesce around a common lineage in the NCBI Tree. This makes L < N and , and hence real-world running time O(DL) is typically much smaller than O(MN).
3 Statistical significance
Although LCA* represents an α-majority taxonomic estimate, this majority might not have statistical confidence, especially for smaller contigs containing only a few ORFs. Here, we apply a multinomial ‘supremacy’ P-value to measure the statistical confidence of the elected taxonomy (Nettleton, 2009). More details on the mapping of LCA* to the hypothesis test and its implementation can be found in Supplementary file 1, Section S1.
4 Methods
Simulated samples, parameter settings, distances and analytical methods used in the performance analysis of LCA* can be found in Section S2 of Supplementary File 1. A RMarkdown document containing the code of this analysis can be found in Supplementary File 2.
5 Results
The performance of LCA* was compared against two other taxonomic estimation methods, LCA2 and Majority. LCA2 is the application of the LCA algorithm to the taxonomic annotations of a contig, while Majority is a simple majority method where taxonomy is ascribed to the most number of annotations (Supplementary File 1, Section S2). We evaluated the relative prediction performance of the three methods against two sets of simulated metagenomic contigs (with and without target genes in the annotation database) and one set of contigs from 201 microbial ‘dark-matter’ (MDM) single-cell Amplified Genomes (SAGs) obtained from the Genomic Encyclopedia of Bacteria and Archaea (GEBA) MDM project (Rinke et al., 2013), a United States Department of Energy Joint Genome Institute (JGI) initiative for sequencing thousands of uncultivated bacterial and archaeal genomes from diverse branches of the Tree of Life (Section 4).
In general, the voting-based measures Majority and LCA* outperformed LCA2 using both the Simple-walk and weighted taxonomic distance (WTD) distances (see methods). In both the Small (10 × 100 randomly sampled contigs from 100 taxa) and Large (10 × 2000 randomly sampled contigs from 2000 taxa) simulations, as well as the GEBA MDM contigs, LCA* and Majority had Simple-walk distances closer to zero when compared with LCA2 (Supplementary Fig. S1). With reference annotations removed from the target database, distances of all three measures were significantly increased in magnitude and variability, with voting based measures offering only a marginal improvement (Supplementary Fig. S2). A similar pattern was observed with the WTD, but here LCA2 is more penalized for predictions widely outside their original taxonomic lineage. Here, LCA2 predictions near the root caused a cluster of negative WTD values in the GEBA MDM contigs (Supplementary Fig. S3). Again, the removal of reference annotations increased WTD distances in terms of magnitude and variability, with the voting-based measures offering relatively minor improvement (Supplementary Fig. S4). From a regression analysis perspective, it is also possible to express these distances as error measurements, and calculate the Root-mean squared error (RMSE) as a measure of accuracy (Fig. 4). Here, the voting-based methods exhibited smaller RMSE values in all cases, but were only significantly different at the 95% confidence level in the Large simulation and GEBA MDM contigs when measured by the WTD. Without reference annotations there was a trend to smaller RMSE values in the voting-based methods, but there was no significant difference between the three methods at the 95% confidence level (Supplementary Fig. S5). In no cases were the RMSE statistics of voting-based methods LCA* and Majority significantly different at the 95% confidence level.
Fig. 4.
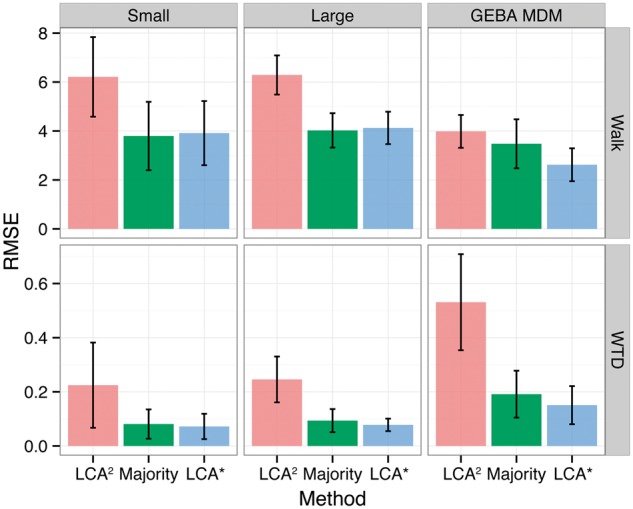
Root-mean-squared error (RMSE) for LCA2, Majority, and LCA*, across experiments and distances. Error bars represent 95% confidence intervals drawn from a Student’s t-distribution
The voting-based LCA* and Majority methods had similar performance in terms of both the simple-walk and WTD with LCA* exhibiting a slightly larger tail with reference annotations present (Supplementary Figs S1 and S3). This difference is not seen without reference annotations (Supplementary Figs S2 and S4). However, the two methods differed significantly in their supremacy P-values with and without the presence of reference annotations, LCA* reporting substantially smaller P-values on average (Supplementary Figs S6 and S7), consistent with LCA* taxonomies providing greater statistical confidence. Moreover, when we compare pairwise P-values, we can see that in the majority of instances, LCA* reported more confident majority taxonomies. However, in many instances the two voting-based methods were convergent, reporting the same P-value when an α-majority is found in the original annotations and no collapsing of annotations was necessary (Supplementary Fig. S8). With reference sequences removed, a similar pattern is observed, although the statistical advantage of LCA* is diminished (Supplementary Fig. S9). The cluster of points where the Majority method’s P-values are 1.0 indicates a definite hazard in interpreting the reported taxonomy. The P-value will equal 1.0 where there is a tie for a majority taxonomy (i.e. Xk = M), and highlights a situation where an arbitrary decision is being made between two or more taxonomies. Interestingly, none of the LCA* estimates in our experiments had a P-value of one, suggesting that because LCA* is compelled to find some majority, occurrence of a such a stalemate election is extremely rare.
6 Discussion
In this work we described, formulated, and implemented LCA*, an entropy-based method to assign taxonomy to contigs assembled from environmental sequence information. By defining a mathematical framework to reason about taxonomy, LCA* identifies a sufficiently strong majority on the NCBI Tree while minimizing entropy changes to the observed taxonomic distribution. This strikes a compromise between the competing goals of obtaining a sufficient majority of at least 50% of annotations as recommended by Condorcet’s Theorem, while minimizing changes to the underlying distribution. A likelihood-ratio test was implemented to test for the supremacy of predicted taxonomies, reporting a P-value that can be used as a measure of confidence and hazard for reported taxonomies. Using simulated and real-world datasets, we empirically demonstrated that voting-based methods, majority vote and LCA*, are consistently more accurate in taxonomically identifying a sequence than the simple LCA2 method, and that LCA* taxonomy results in a balance between specificity and confidence to provide an estimate appropriate to the available information in the data.
While LCA* has a compelling theoretical basis for constructing a majority from a variable taxonomic distribution, it is necessary to consider several assumptions that the statistic makes. Due to the inherent variability in homology-search, annotation databases, or taxonomic summary statistics (e.g. Best-BLAST, LCA, etc.), observed annotations can appear on the tree at various taxonomic levels. This raises a philosophical issue that observed multinomial bins cannot be viewed as completely independent, as internal taxonomic annotations could overlap in this model. One possible remedy is to discard all annotations that do not fall on the leaves of the NCBI Tree. However, annotation of metagenomic contigs can be quite sparse, so discarding internal annotations in the name of independence simply decreases valuable statistical power, and can artificially bias the signal towards an arbitrary taxonomy by removing internal nodes. Alternatively, one could attempt to distribute annotations from internal nodes equally to the observed leaves in the tree. However, this creates its own discretization issue, as in many cases votes cannot be distributed equally, and vote-splitting violates basic assumptions of the multinomial model, i.e. votes are non-negative integers. Moreover, vote-splitting can make the final predicted taxonomy more difficult to interpret, as the distribution in each case could be very different from the observed, and risks electing a more specific taxonomy than supported by the data. In the end, to avoid these issues we opted to leave internal annotations in the election.
Though LCA* will attempt to give reasonable estimates when observed annotations are highly variable, being an alignment-dependent binning method its performance suffers substantially when reference annotations are not present in the database, limiting its capacity to estimate unknown taxonomy. Here, the method could benefit by expanding the current framework to incorporate information from the statistical properties of sequences found in alignment-independent methods and ribosomal or Clusters of Orthogonal Group (COG) marker genes. The election model could also be expanded to incorporate genomic-signature information into a weighted-voting or vote-splitting framework, which perhaps could help improve statistical confidence in cases where observed annotations are extremely sparse. However, expanding our current voting theory model to include continuous variables is not compatible with the current multinomial distribution. Range voting provides an alternative perspective that attempts to accommodate a continuous voting scale, but its theory has numerous impossibility results that challenge all three of the common-sense principles of voting: preserving majority rule, requiring a minimum level of core support, and rewarding sincere voters (Balinski and Laraki, 2007). ‘Taxonomic reconciliation’ between NCBI Tree entries and ribosomal RNA gene or COG alignments (akin to methods recently implemented in PhyloSift (Darling et al., 2014) would allow for an apples-to-apples comparison between taxonomic and functional marker gene binning methods, e.g. ML-TreeMap, MetaPhlan (Segata et al., 2012; Stark et al., 2010), support more powerful integrative alignment-dependent binning models, and facilitate principled placement of taxa from one tree into the other.
7 Conclusions
The LCA* algorithm assigns taxonomy to contigs assembled from environmental sequence information using the NCBI Tree. The algorithm identifies a sufficiently strong majority on the hierarchy while minimizing entropy changes to the observed taxonomic distribution resulting in improved statistical properties. The algorithm and its statistical tests have been implemented as a stand-alone Python library compatible with the MetaPathways pipeline; both of which are available on GitHub with installation instructions and use-cases (http://www.github.com/hallamlab/LCAStar/).
Supplementary Material
Funding
This work was carried out under grants from Genome Canada, Genome British Columbia, Genome Alberta, the Natural Science and Engineering Research Council (NSERC) of Canada, the Canadian Foundation for Innovation (CFI) and the Canadian Institute for Advanced Research (CIFAR). NWH was supported by a UBC four-year doctoral fellowship (4YF). KMK was supported by the Tula Foundation funded Centre for Microbial Diversity and Evolution (CMDE) at UBC.
Conflict of Interest: none declared.
References
- Abubucker S. et al. (2012) Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol., 8, e1002358.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balinski M., Laraki R. (2007) A theory of measuring, electing, and ranking. Proc. Natl. Acad. Sci. U. S. A., 104, 8720–8725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. (2001) Random forests. Mach. Learn., 45, 5–32. [Google Scholar]
- Darling A.E. et al. , (2014) PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ, 2, e243.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estlund D.M. (1994) Opinion leaders, independence, and Condorcet’s Jury Theorem. Theor. Decis., 36, 131–162. [Google Scholar]
- Federhen S. (2012) The NCBI Taxonomy database. Nucleic Acids Res., 40, D136–D143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige U. et al. (1994) Computing with noisy information. SIAM J. Comput., 23, 1001–1018. [Google Scholar]
- Hanson N.W. et al. (2014a) Metabolic pathways for the whole community. BMC Genomics, 15, 619.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson N.W. et al. (2014b). MetaPathways v2.0: a master-worker model for environmental Pathway/Genome Database construction on grids and clouds. In: 2014 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology, pp. 1–7.
- Huson D.H. et al. (2007) MEGAN analysis of metagenomic data. Genome Res., 17, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konwar K.M. et al. (2013) MetaPathways: a modular pipeline for constructing pathway/genome databases from environmental sequence information. BMC Bioinformatics, 14, 202.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konwar K.M. et al. (2015) MetaPathways v2.5: quantitative functional, taxonomic, and usability improvements. Bioinformatics, 31, 3345–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullback S., Leibler R.A. (1951) On Information and Sufficiency. The Annals of Mathematical Statistics, 22, 79–86. [Google Scholar]
- Legendre P., Legendre L. (2012) Numerical Ecology. Elsevier, Amsterdam, Netherlands. [Google Scholar]
- Meyer F. et al. (2008) The metagenomics RAST server – a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics, 9, 386.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nettleton D. (2009) Testing for the Supremacy of a Multinomial Cell Probability. J. Am. Stat. Assoc. 104, 1052–1059. [Google Scholar]
- Prosser J.I. (2010) Replicate or lie. Environ. Microbiol., 12, 1806–1810. [DOI] [PubMed] [Google Scholar]
- Rinke C. et al. (2013) Insights into the phylogeny and coding potential of microbial dark matter. Nature, 499, 431–437. [DOI] [PubMed] [Google Scholar]
- Segata N. et al. (2012) Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods, 9, 811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark M. et al. (2010) MLTreeMap – accurate Maximum Likelihood placement of environmental DNA sequences into taxonomic and functional reference phylogenies. BMC Genomics, 11, 461.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T. et al. (2012) Metagenomics – a guide from sampling to data analysis. Microb. Inf. Exp., 2, 3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade G. (1994) Signal Coding and Processing. Cambridge university press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.