

Abstract
Motivation: Multiple high-throughput approaches have recently been developed and allowed the discovery of enhancers on a genome scale in a single experiment. However, the datasets generated from these approaches are not fully utilized by the research community due to technical challenges such as lack of consensus enhancer annotation and integrative analytic tools.
Results: We developed an interactive database, EnhancerAtlas, which contains an atlas of 2,534,123 enhancers for 105 cell/tissue types. A consensus enhancer annotation was obtained for each cell by summation of independent experimental datasets with the relative weights derived from a cross-validation approach. Moreover, EnhancerAtlas provides a set of useful analytic tools that allow users to query and compare enhancers in a particular genomic region or associated with a gene of interest, and assign enhancers and their target genes from a custom dataset.
Availability and Implementation: The database with analytic tools is available at http://www.enhanceratlas.org/.
Contact: jiang.qian@jhmi.edu or tank1@email.chop.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Enhancers are distal regulatory DNA elements that regulate transcription levels of target genes. They play an important role in development and diseases (Ghavi-Helm et al., 2014; Hnisz et al., 2013). Unlike promoters, enhancers often regulate expression of their target genes independent of their relative location, distance or even the gene orientation (Ong and Corces, 2011). Furthermore, enhancers are often tissue- or cell type-specific (Heinz et al., 2015; Pennacchio et al., 2013). Due to the relative location and tissue specificity, it is challenging to identify enhancers.
Recently, many technologies were developed to map the enhancers on a genome scale. (i) Clusters of transcription factor (TF) binding sites often represent regulatory elements (Spitz and Furlong, 2012), and chromatin immunoprecipitation followed by sequencing (ChIP-seq) or ChIP-chip can be used to identify the binding sites of various TFs or other regulatory factors; (ii) A few enhancer-specific factors have been used to identify enhancers. For example, EP300, a histone acetyltransferase, activates transcription via acetylating the histones. Therefore, the binding sites of EP300 were often used to predict enhancers (Visel et al., 2009); (iii) It was reported that RNA polymerase II (RNAPII) binds to thousands of enhancers (Kim et al., 2010). Therefore, binding sites of POLR2A, the largest subunit of RNAPII, are considered to be the active regulatory regions; (iv) DNase I hypersensitivity sites (DHS) represent the open chromatin regions, many of which cover the enhancers (Thurman et al., 2012); (v) Formaldehyde Assisted Isolation of Regulatory Elements (FAIRE) coupled with sequencing is another method to identify large numbers of active regulatory elements including enhancers (Gaulton et al., 2010); (vi) Some histone modification patterns reflect different chromatin states. For example, the combination of H3K4me1 and H3K27ac is a widely used mark for enhancers (Cotney et al., 2012; Heintzman et al., 2009); (vii) The DNA sequences of enhancers could also be transcribed and these transcribed enhancer RNAs (eRNAs) are an indicator of active enhancers (Andersson et al., 2014); (viii) Approaches to determine the three-dimensional conformation of chromosomes (e.g. 5C and ChIA-PET, Capture-C) are able to provide enhancer-promoter interactions (Fullwood et al., 2009; Jin et al., 2013).
These above approaches have been demonstrated as powerful tools to detect enhancers on a genome-wide scale, and they have been applied to many cell types or tissues. However, the datasets generated by these approaches are not fully utilized by the research community due to several technical challenges. First, these datasets are often disseminated in different databases. A centralized data portal is still lacking that is specially designed for enhancer analysis. Second, enhancer annotations based on different technologies showed large discrepancies. A consensus of enhancers will provide more reliable annotation of enhancers. Third, although several genome annotation databases (e.g. UCSC Genome Browser) are currently available to visualize various datasets, they do not provide useful tools for enhancer analysis, such as comparing enhancers across different cell types, generating network views for enhancer-target interactions, and predict enhancers for a custom dataset.
The goal of our work is of two-fold. First, we combined a large number of available datasets in each cell/tissue type to produce a ‘consensus’ of enhancers that represent the most reliable prediction of enhancers. For this purpose, we develop an unsupervised learning approach to evaluate the quality of each dataset in one cell type and generate a consensus for more reliable enhancer identification. Using this method, we produced enhancer predictions for 76 cell lines and 29 tissues. These enhancer predictions have high quality compared with single dataset. Second, we constructed a user-friendly, interactive online database, EnhancerAtlas.org, to facilitate the analysis of enhancers for different cell types. A series of analytic tools were developed to extract, compare and visualize the enhancers in different cell types.
2 Materials and methods
2.1 Data sources
We used a total of eight types of experimental approaches (i.e. tracks) to identify enhancers. They include DHS, FAIRE, eRNA, P300 binding sites, POL2 binding sites, histone modifications, TF binding sites and CHIA-PET. These tracks represent distinct experimental approaches to detect enhancers. Note that some tracks could contain multiple datasets. For example, replicates sometimes exist for the same experiments that were performed by different labs. The ‘TF-Binding’ track includes all ChIP-Seq datasets for different TFs in a given cell type. The ‘histone’ track includes the datasets for H3K4me1 and H3K27ac. To make a reliable enhancer annotation, we only used cell lines and tissues with at least three tracks. In total, 76 cell lines and 29 tissues were included. We processed and integrated 3785 high throughput datasets (Table 1). All sequencing data were mapped to the hg19 genome assembly. Since some approaches (e.g. DHS) detect all active regulatory regions, promoter and exon regions were removed in our analysis. Here promoters were defined as the region 5 kb upstream of the transcription start sites based on a location analysis of known promoters (Supplementary Figure S1). In addition, matched RNA-seq data were used for predicting enhancer target genes, which were collected from UCSC genome browser, GEO database and Epigenome Roadmap data portal. In total, 48 cell lines and 22 tissues have available RNA-seq data.
Table 1.
Summary of the numbers for collected tracks, datasets and enhancers in 105 cell/tissues
| Sample | Track | Dataset | Enhancer | Sample | Track | Dataset | Enhancer |
|---|---|---|---|---|---|---|---|
| A549 | 7 | 87 | 49760 | Heart | 4 | 8 | 2134 |
| Astrocyte | 5 | 14 | 44489 | HEK293 | 4 | 61 | 13167 |
| BJ | 3 | 16 | 13275 | HEK293T | 5 | 62 | 13426 |
| Bronchia_Epithelial | 4 | 6 | 8776 | Hela | 3 | 47 | 13557 |
| Caco-2 | 3 | 11 | 22358 | Hela-S3 | 6 | 226 | 56247 |
| CD133+ | 3 | 16 | 1812 | HepG2 | 7 | 207 | 50160 |
| CD14+ | 4 | 37 | 45973 | HL-60 | 4 | 15 | 9867 |
| CD19+ | 3 | 16 | 30240 | HMEC | 4 | 25 | 21659 |
| CD20+ | 3 | 8 | 16914 | HSMM | 3 | 23 | 60216 |
| CD34+ | 3 | 51 | 47306 | HUES64 | 3 | 117 | 22368 |
| CD36+ | 4 | 9 | 836 | HUVEC | 5 | 40 | 63977 |
| CD4+ | 6 | 50 | 14465 | IMR90 | 5 | 87 | 85732 |
| CD8+ | 3 | 17 | 35882 | Jurkat | 4 | 16 | 8487 |
| CMK | 3 | 7 | 4731 | K562 | 8 | 282 | 43148 |
| CUTLL1 | 3 | 19 | 18569 | Kasumi-1 | 4 | 15 | 769 |
| ECC-1 | 4 | 52 | 16612 | Left_Ventricle | 3 | 4 | 36128 |
| Esophagus | 3 | 4 | 36217 | liver | 4 | 6 | 2329 |
| Fetal_Brain | 3 | 9 | 37655 | LNCaP | 5 | 87 | 32404 |
| Fetal_heart | 5 | 8 | 2693 | LoVo | 3 | 416 | 15336 |
| Fetal_kidney | 3 | 5 | 8087 | LS174T | 4 | 15 | 3404 |
| Fetal_lung | 3 | 5 | 30805 | lung | 3 | 4 | 45399 |
| Fetal_Muscle_Leg | 3 | 20 | 39372 | Macrophage | 3 | 8 | 34490 |
| Fetal_Placenta | 3 | 6 | 35878 | MCF10A | 3 | 37 | 2528 |
| Fetal_Small_lntestine | 3 | 6 | 39113 | MCF-7 | 8 | 193 | 23858 |
| Fetal_Spinal_Cord | 3 | 5 | 7951 | ME-1 | 3 | 17 | 11334 |
| Fetal_Stomach | 3 | 6 | 36485 | MM1S | 3 | 25 | 2414 |
| Fetal_Thymus | 3 | 6 | 28757 | myotube | 3 | 10 | 59800 |
| Fibroblast_foreskin | 3 | 16 | 40449 | NB4 | 5 | 29 | 24982 |
| Foreskin_Keratinocyte | 3 | 24 | 42297 | NH-A | 3 | 6 | 12609 |
| GM10847 | 3 | 19 | 13935 | NHDF | 3 | 6 | 15730 |
| GM12878 | 7 | 165 | 49672 | NHEK | 6 | 24 | 40361 |
| GM12891 | 5 | 40 | 41435 | NHLF | 3 | 14 | 34281 |
| GM12892 | 5 | 40 | 34086 | NT2-D1 | 3 | 12 | 3875 |
| GM18486 | 3 | 9 | 16988 | OCI-Ly1 | 4 | 26 | 12850 |
| GM18505 | 4 | 22 | 17577 | Osteoblast | 5 | 6 | 23195 |
| GM18507 | 4 | 9 | 5987 | Ovary | 3 | 6 | 14836 |
| GM18508 | 3 | 6 | 5214 | P493-6 | 3 | 24 | 12555 |
| GM18516 | 3 | 6 | 5531 | PANC-1 | 4 | 11 | 5615 |
| GM18522 | 3 | 6 | 7254 | Pancreas | 4 | 7 | 3876 |
| GM18526 | 3 | 18 | 7078 | Pancreatic_Islet | 3 | 4 | 5723 |
| GM18951 | 3 | 20 | 8895 | Raji | 3 | 9 | 3182 |
| GM19099 | 4 | 19 | 9278 | Skeletal_Muscle | 3 | 6 | 24396 |
| GM19141 | 3 | 6 | 5444 | SK-N-MC | 4 | 10 | 4727 |
| GM19193 | 4 | 20 | 15604 | SK-N-SH | 5 | 59 | 38571 |
| GM19238 | 3 | 18 | 30016 | Small_Intestine | 3 | 7 | 29562 |
| GM19239 | 5 | 19 | 37581 | Spleen | 3 | 4 | 39744 |
| GM19240 | 3 | 12 | 21087 | T47D | 3 | 19 | 32456 |
| H1 | 6 | 216 | 58821 | th1 | 3 | 19 | 26137 |
| H128 | 3 | 6 | 11982 | Thymus | 3 | 4 | 31232 |
| H2171 | 3 | 14 | 3828 | U20S | 3 | 30 | 84127 |
| H54 | 4 | 8 | 4711 | U87 | 3 | 15 | 23820 |
| H9 | 4 | 33 | 69481 | VCaP | 3 | 42 | 7683 |
| HCT116 | 5 | 66 | 4418 |
2.2 Track normalization
There are a few steps to normalize the datasets. First, to make datasets comparable, we take fold enrichment of each region as the signal intensity and normalize it in each dataset. For each genomic window of 10 bp, the normalization for each data set is defined as:
Where and are the score and length of peak i (), respectively. Second, many tracks, including DHS, EP300, POL2, FAIRE, may have two or more replicates. We summed the signal intensities in normalized files into one file using bedtools (Quinlan and Hall, 2010) and normalized the merged file again. Furthermore, the ‘TF-binding’ track may contain many ChIP-seq datasets from different TFs, and ‘histone modification’ track contains two different modifications (H3K4me1 and H3K27ac). We also merged different TFs or modifications into one track.
2.3 Generation of consensus tracks
A basic assumption of our approach is that if two datasets are of good quality, they should have a good correlation of the predicted enhancers. On the other hand, if one dataset is of low quality, it will have low correlation with other datasets. By comparing the correlations between different datasets, we obtain the relative quality for each dataset. For two given tracks and , the voting score of on was defined as the Pearson Correlation Coefficient (PCC) between and :
Where and represent the score at genomic position i and the mean of all position scores in track , respectively. Then a PCC matrix can be obtained from a cell line or tissue with m tracks:
Based on the matrix, the weight of track () was set as:
Note that the relative weights are not sensitive to the correlation metrics we used. For example, rank-based Spearman yielded almost identical relative weights as Pearson correlation (Supplementary Figure S2). To combine all tracks, we defined the score of combined track at genomic position i as:
By doing so, the score at each genomic positon in the consensus track came from reasonable weight assignments of all tracks.
2.4 Validation of enhancer predictions using the VISTA enhancer database
To evaluate our consensus track using experimentally validated enhancer documented in the VISTA database (Visel et al., 2007), we first grouped enhancers according to their tissue types. We selected two tissues (i.e. brain and heart) with the largest number of enhancers. The brain and heart have 809 and 96 validated enhancers, respectively. Our evaluation is at base pair level. The Sensitivity and Specificity were defined as below:
2.5 Enhancer-target gene prediction
We used a recent developed algorithm Integrated Method for Predicting Enhancer Targets (IM-PET) to predict enhancer targets (He et al., 2014). IM-PET predicts enhancer-promoter by integrating four features using a Random Forest classifier. Features are derived from transcriptomic, epigenomic and genome sequence data, including enhancer and promoter activity correlation, TF and promoter activity correlation, enhancer and promoter sequence co-evolution and enhancer-to-promoter distance. We showed that IM-PET achieved significant improvement over other state-of-the-art methods. Further, based on our validation experiment using 3C-qPCR we showed that IM-PET has a comparable accuracy to that of the experimental 5C technology. Here, the input data for IM-PET included the genomic positions of predicted enhancers, RNA-Seq data and H3K4me1, H3K4me3, and H3K27Ac ChIP-Seq data for 48 cell lines and 22 tissues. Enhancer targets were predicted using a false discovery rate cutoff of 0.01.
2.6 Implementation of the tools
The visualization tools in EnhancerAtlas were implemented by using html5 canvas element and PHP. The online service of EnhancerAtlas was implemented with Linux-Apache-MySQL-PHP-HTML-JavaScript-Perl and could be run on most PCs (Windows, Mac OSX or Linux). The <canvas> element of HTML was employed to draw the genomic graph and display the datasets for different approaches or cell/tissue types. In the graphic display, a jQuery UI slider widget with two handles was used to zoom in or out on the genome region. We used the plug-in, cytoscape.js, for analysis and visualization of enhancer-gene network in a given cell/tissue type. Links in the graphic presentation or tables were implemented by JavaScript or php functions.
3 Results
3.1 Selection of available datasets
Many approaches have been developed to map enhancers in cells. Although enhancers identified by different approaches have an overall agreement on the enhancer annotation, they showed discrepancies in many genomic regions (Fig. 1A). A consensus of enhancers based on multiple experimental evidences will provide a more reliable enhancer annotation. In this work, we collected eight types of genome-wide experimental datasets (i.e. tracks) to identify the enhancers (Fig. 1B). To obtain reliable enhancer annotation, we only focused on the cell/tissue types with at least three independent experimental tracks for enhancer identification (Fig. 1C). A total of 105 cell/tissue types were collected for the enhancer annotation in the study. Note that some tracks could contain multiple datasets (e.g. replicates, multiple histone modifications for ‘Histone’ track or multiple TF binding datasets for ‘TF-Binding’ track). On the other hand, we kept the tracks for EP300 and POLR2A binding sites separated from the ‘TF-binding’ track, because these two proteins are important indicators for enhancers.
Fig. 1.
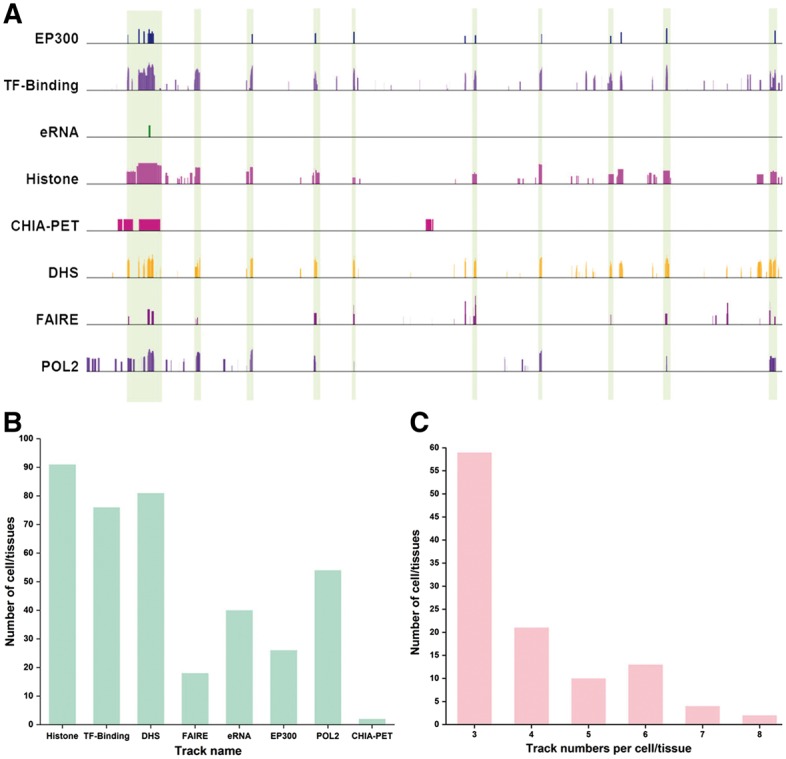
Enhancer annotation and datasets available for enhancer identification. (A) Consistence and discrepancies in enhancer annotation. In the region (chr1:27,515,285-27,848,296 in K562), the enhancers supported by many tracks are highlighted by vertical bars. It is clear that many potential regions are only supported by one or a few tracks. (B) Number of cell/types that contain a certain dataset types. Some technologies were more widely used than others for enhancer identification. (C) Number of cell/tissue types in function of number of independent tracks. Many cell/tissue types include a few tracks (e.g. 3 or 4), while a few cell/tissue types have many tracks (e.g. 7 or 8) (Color version of this figure is available at Bioinformatics online.)
We manually collected and integrated 3785 high throughput experimental datasets (Table 1) mainly from six resources including UCSC genome browser (Kent et al., 2002), NCBI GEO (Barrett et al., 2013), Cistrome database (Qin et al., 2012), ENCODE project data portal (Consortium, 2012), Epigenome Roadmap data portal (Roadmap Epigenomics et al., 2015) and eRNA (Andersson et al., 2014). These datasets will be used to generate consensus enhancers for 105 cell/tissue types.
3.2 Unsupervised learning approach for enhancer identification
To build a consensus of enhancer annotations from multiple datasets, one obvious option is supervised learning approach, which requires a set of validated enhancers as a gold standard. By comparing each dataset to the gold standard, we are able to assess the quality of each dataset and then assign the relative weight to each dataset. An ideal gold standard would be a set of enhancers determined by an in vivo activity-based approach. The VISTA database contains 1746 enhancers identified using comparative genome analysis and validated using mouse transgenic reporter assay (Visel et al., 2007). However, since enhancers are cell type specific, a gold standard is needed for each cell type. In that sense, the dataset in the VISTA is not large enough to serve as a gold standard for most of the cell types.
To overcome this challenge, we employed an unsupervised learning approach to derive the consensus of enhancers from multiple experimental evidences. Our approach evaluates the quality of each dataset by cross-validation. The underlying assumption of our approach is that if two datasets are of good quality, they should have a good correlation among the predicted enhancers. On the other hand, if one dataset is of low quality, it will have low correlations with other datasets. By comparing the correlations between different datasets, we will obtain the relative quality for each dataset.
Here, we used K562 cell line as an example to elucidate our approach (Fig. 2). The K562 has eight tracks available for enhancer annotation, including ‘DHS’, ‘FAIRE’, ‘EP300’, ‘POL2’, ‘Histone’, ‘TF-Binding’, ‘CHIA-PET’ and ‘eRNA’. Among them, 95 TF ChIP-seq datasets in this cell type were available and used to obtain the ‘TF-Binding’ track. First, we normalized the signal intensity in each track to make the tracks comparable (see Section 2 for details). Next, we calculated pairwise Pearson correlation coefficient (PCC) of enhancer signals among the eight tracks. Some tracks have very similar enhancer signals (e.g. locations of EP300 and DHS, with a PCC of 0.490), while other tracks have relatively low similarity (e.g. signals between ChIA-PET and eRNA, with a PCC of 0.019). Next, we summed the correlation coefficients of each track with the other seven tracks. The sum of correlation coefficients for the eight tracks is normalized to obtain the relative weights (Fig. 2A). Finally, we obtained the consensus track based on the weighted sum of the signals from individual tracks (Fig. 2B).
Fig. 2.
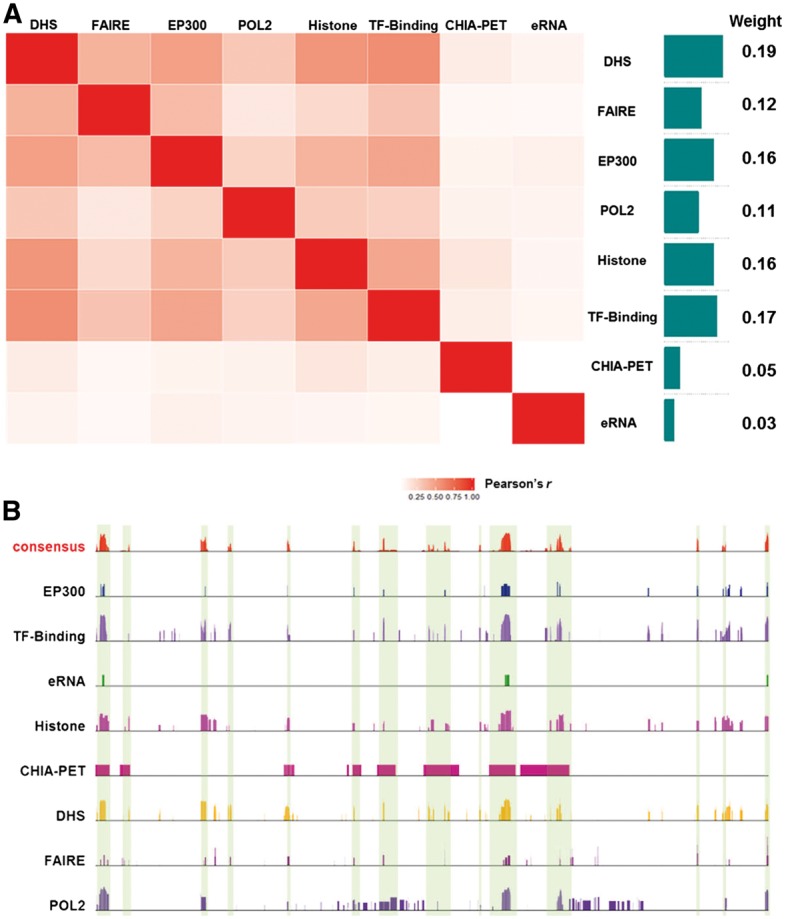
Overall approach to derive the consensus enhancers. (A) Calculation of relative weight for each track. The correlation coefficients of enhancer signals among the tracks were first calculated. The summation of the correlation coefficients for each track was normalized to obtain the relative weight. (B) Based on the weighted sum of the signals from eight individual tracks, the consensus track was created (Color version of this figure is available at Bioinformatics online.)
3.3 Assessment of relative weights for different tracks
As illustrated by the example of K562, we can see that some tracks such as DHS have relatively high weights, while other tracks such as eRNA have relatively low weights. Note that the weights obtained for one particular cell type do not necessarily reflect the quality of the experimental platform in general, because the same experimental approach might be used to generate datasets with different qualities by different laboratories. We sought to assess the quality of each dataset, rather than an experimental approach. Therefore, the relative weights derived by our approach are specific to each cell type (Fig. 3). For instance, the relative weight for the track of ‘TF-Binding’ varies significantly among the cell/tissue types. In MCF-7 cell line the relative weight of ‘TF-Binding’ is 0.252, while the weight for the same track becomes 0.176 in Hela-S3 cell line. Similarly, while eRNA has a small weight (0.03) in K562, it has a relatively high weights in other cell types. For example, the weights for eRNA in adult heart and bronchia epithelial are 0.40 and 0.20, respectively. The dynamic changes of each track among the cell/tissue types reflect the data quality of the specific dataset and the total number of tracks in the cell/tissue type of interest (Fig. 3).
Fig. 3.

Heat map of relative weights for different tracks in 105 cell/tissue types. The tracks with missing data were denoted by white
To confirm that the relative weight indeed reflects the data quality, we performed a simulation on TF binding track in K562 (Fig. 4). Total 95 TF ChIP-seq datasets were used for ‘TF-Binding’ track in K562. We assumed that the quality of track increases with more TF ChIP-seq datasets. While keeping the datasets of other tracks unchanged, we randomly removed certain numbers of TFs in the TF binding track and calculated the relative weight for the TF binding track. We found that the average relative weight decreased with fewer TFs included in the track, suggesting that relative weight of a track indeed correlated with the quality of the track (Fig. 4A). Interestingly, we found that both repressors and activators contributed to the enhancer prediction. If we separated the TFs into activators and repressors based on their known function, we obtained 33 activators and 37 repressors with ChIP-seq data in K562. We found that the weights for both sets increased with increasing number of TFs, although the repressors’ contribution was less significant than activators (Fig. 4B).
Fig. 4.

Relative weight of ‘TF-Binding’ track with varying number of TFs in K562. (A) Simulation results of relative weight of ‘TF-Binding’ track in function of number of TFs included. The weight of track decreased with fewer number of TF ChIP-seq datasets included in the track, suggesting that the relative weight reflects the data quality. (B) Simulation results of relative weight of repressors and activators. In total, 33 activators and 37 repressors with ChIP-seq data in K562 were classified by their known functions. Although the repressors have smaller weight than activators, the weights for both sets increased with increasing number of TFs
3.4 Enhancer calling based on weighted consensus track
To determine enhancers in the consensus track, we first performed simulation to determine the cutoff for enhancer signal intensity. For a given cell/tissue type, we shuffled each track by generating random starting positions within the same chromosome. Using the same relative weights determined as described above, a combined track based on the shuffled tracks was generated and the scores of all positions in this track were sorted. The score at the top 99.5% was selected as the cutoff. Only the peaks in the consensus track with score higher than the cutoff were called as enhancers.
In addition, we had three extra criteria to determine the enhancers in the consensus track. (i) Peaks within 5 kb upstream of the transcription start sites of protein coding genes were removed because they are more likely to be promoters; (ii) We required that more than half of the tracks have non-zero values to call an enhancer. This filtering removed the enhancers that were only supported by one or very few experimental evidence. In other words, this criterion ensured that there were multiple independent lines of experimental evidence to support the annotation of an enhancer. (iii) We removed the enhancers smaller than 50 bp. Using the above approaches, we identified an atlas of 2 534 123 enhancers for 105 cell/tissue types (Table 1).
3.5 Validation using VISTA enhancer database
We next evaluated the performance of our approach. We extracted tissue-specific enhancers from the VISTA database. For example, 809 and 96 validated enhancers were found for fetal brain and heart. The other tissue types have very small number of enhancers available and are not suitable for the validation. The enhancers annotated in VISTA are grouped as a positive set. The union of the peaks in all tracks is the entire prediction space and the regions not overlapping with the positive set are considered as the negative set. We calculated the sensitivity and specificity of the consensus enhancers in the unit of base pair. For heart enhancers, the area under the receiver operating characteristic (AROC) for the consensus is 0.89, while the AROC for individual tracks are all under 0.84, suggesting that the consensus have the better enhancer annotation than any individual track. Because only binary data are available for the tracks of ‘EP300’, ‘eRNA’ and ‘POL2’ in heart (Andersson et al., 2014; Barrett et al., 2013), they do not have an ROC curve. They showed quite good specificity (>0.95) relatively low sensitivity (<0.01) (Fig. 5A). Similar results were observed for brain enhancers (Fig. 5B). We also compared the performance using voting approach in which each track was assigned with uniform weight. The AROC for the fetal brain and heart were 0.70 and 0.86 using uniform weight, respectively, which were lower than the performance using our cross-validation derived weights.
Fig. 5.
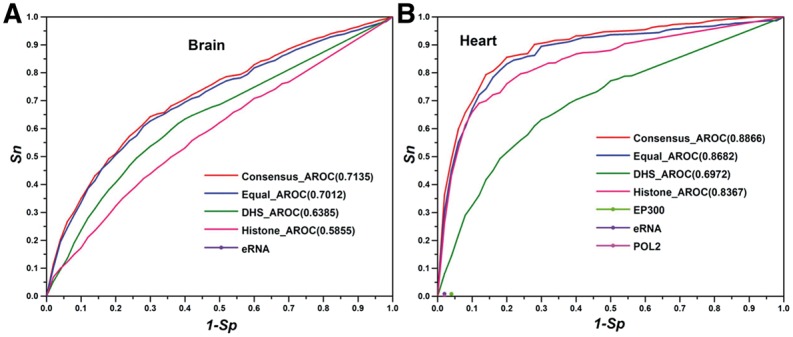
Evaluation of the consensus enhancer annotation. We used experimentally validated enhancers from VISTA database as the gold standard. Sn is the sensitivity, which represents the percentage of enhancers from VISTA recovered by different tracks. Sp is the specificity, which represents the percentage of negative regions in the prediction space (i.e. not in VISTA enhancers) that are correctly predicted as negative by the tracks
3.6 Enhancer-target gene relationships
We predicted the target genes of enhancers using a recent developed algorithm, namely Integrated Method for Predicting Enhancer Targets (IM-PET) (He et al., 2014). IM-PET predicts enhancer-promoter by integrating four features using a Random Forest classifier. Features are derived from transcriptomic, epigenomic and genome sequence data, including enhancer and promoter activity correlation, TF and promoter activity correlation, enhancer and promoter sequence co-evolution and enhancer-to-promoter distance. The input data for enhancer-target prediction included the gene expression dataset (RNA-seq) and genomic positions of predicted enhancers, histone modification data for 48 cell lines and 22 tissues. Overall, we predicted 2 488 394 enhancer-target relationships for the 70 cell/tissue types. On average, one enhancer is associated with 2.4 target genes, and each gene is associated with 4.1 enhancers in one cell/tissue type.
3.7 Web server
EnhancerAtlas is accessible to the research community in the web server (http://www.enhanceratlas.org/). The database has the following features to facilitate the usage of enhancer annotation. First, users are allowed to select a particular genomic region and examine the consensus enhancers within the region. In the meantime, we also show the experimental data (individual tracks) that support the consensus. Second, users can identify the enhancers that associated with a gene of interest. Similarly, both the consensus and all the experimental tracks are shown. Third, users can compare the enhancers across different cell/tissue types. Such comparison will allow users to identify the enhancers that are conserved or specific to cell/tissue types. Fourth, we allow users to upload their own custom data and annotate the potential enhancers for their datasets.
Specifically, we designed six modules to facilitate the enhancer analysis within the EnhancerAtlas (Fig. 6): (A) Search enhancers by region. This module would graphically display enhancers of any genomic region of interest in a selected cell/tissue type. The ‘consensus’ track summarizes all information, while the individual tracks are the experimental datasets from independent high-throughput approaches (Fig. 6A). We also provide a link to the Epigenome browser (Zhou et al., 2011) for an alternative display. (B) Search enhancers by gene name. This module would graphically show the enhancers that are associated with a given gene of interest in one special cell/tissue type. Multiple input options are provided for the gene ID, including Ensembl, EMBL, UCSC, PDB, RefSeq and UniProt (Fig. 6B). (C) Compare enhancers across cell/tissues. Using this module, users could easily compare the enhancers across multiple selected cell/tissue types in a given genomic region and identify possible specific or conserved enhancers. If users click on one cell/tissue type in the display, supporting experimental evidence will be shown for the cell/tissue type (similar to module (A)) (Fig. 6C); (D) Compare enhancers of genes across different cell/tissues. In this module, a common scheme often observed was that one gene is associated with different clusters of enhancers in different cell/tissue types (Fig. 6D). By comparing enhancer-gene interactions across 105 cell lines, conserved enhancer-gene interactions can be identified. When clicking on individual cell/tissue type, users can view the detailed experimental evidence in this cell/tissue type (similar to module (B)); (E) Generate enhancer-gene network. We provide a Cytoscape network presentation between enhancers and associated genes in a given genomic region and a cell type. Interactions that are specific to the cell type or conserved in other cell types are marked with different colors (Fig. 6E); (F) Predict target genes in defined genomic regions. After a user has identified a set of potential cis-regulatory regions (e.g. peaks identified by ChIP-seq for a TF), the user can upload the regions in bed format and select the cell types that are most relevant to the cell type of interest. This module will compare uploaded regions with the enhancers in the selected cell types and obtain the target genes of the enhancers in these cell types. If some of the uploaded regions are in promoters, the module will also provide the flanking target genes of the regulatory regions (Fig. 6F).
Fig. 6.

Analytic tools in EnhancerAtlas. (A) Search enhancers by region. (B) Search enhancers by gene name. (C) Compare enhancers across cells. (D) Compare enhancers of gene across cells. (E) Generate enhancer-gene network. (F) Predict target genes for genomic regions (Color version of this figure is available at Bioinformatics online.)
4 Conclusions and discussions
In this work, we developed an unsupervised learning approach to derive the consensus enhancers by integrating a variety of experimental datasets. Application of the approach to 105 cell/tissue types yielded an atlas of 2 488 394 enhancers. These consensus enhancers were of better quality than individual dataset. To facilitate the usage of the enhancer annotation, we developed EnhancerAtlas, an online database, which is specifically designed for enhancer annotation and analysis. A set of analytic tools was provided in the database.
We noticed that some tracks have relatively small weights such as those for eRNAs and ChIA-PET. The main reasons are that eRNA signal often marks much less enhancers than other tracks and that the resolution of ChIA-PET is often very low. On the other hand, it is not completely surprising that eRNA and ChIA-PET have a much lower correlation with such tracks, since eRNA and ChIA-PET capture a completely different layer of genome regulation. eRNAs typically mark active enhancers, while other tracks might include all types of enhancers including active and poised enhancers. In the future, we plan to classify enhancers so that the eRNA information will be fully appreciated.
While the IM-PET method showed great performance, its effectiveness has not been tested across such a diverse collection of cell types and tissues. Since it is a supervised model, the training cell types may have a significant effect on the performance of the model in different cell types. We used newly reported enhancer-promoter interactions as the gold standard to assess the quality of our IM-PET prediction (He et al., 2014). The performance of predictions in these new cell types (AUC 0.9) are similar the predictions we evaluated in the original IM-PET prediction (Figure S3).
Since enhancers only occupy a tiny fraction of genome, the number of positive is much smaller than the number of negative. The imbalanced data will yield an inflated estimation of the performance. However, we are focusing on the relative performance by comparing the consensus track and each individual track. Therefore, even though the absolute AUC values might be overestimated, the relationship of relative performance should still hold. An alternative measurement would be using precision to estimate the performance. Because only a handful enhancers were validated and vast majority of enhancers were not tested, those untested enhancers are currently included in the negative set. Therefore, many corrected predicted enhancers are called as false positive and thus a deflated precision will be obtained. For example, the precision for the consensus and EP300 tracks in fetal heart are 0.00093 and 0.00013, respectively, which we do not believe reflect the quality of the datasets.
A comprehensive enhancer database is always a moving target. Therefore, the database will be updated routinely as new datasets become available. For example, recently several high-throughput enhancer screens (e.g. STARR-seq) (Arnold et al., 2013) have been reported. Similarly, more hi-C based datasets are also available for different cell types. We plan to include these new data types in the next version of the database.
Supplementary Material
Acknowledgements
We thank Donald Zack and Hongkai Ji for discussion, and the Research Information Services at the Children's Hospital of Philadelphia for providing computing support.
Funding
This work was supported by National Institutes of Health grants [EY024580, GM111514, EY023188 to J.Q; HG006130, GM104369, and GM108716 to K.T].
Conflict of Interest: none declared.
References
- Andersson R. et al. (2014) An atlas of active enhancers across human cell types and tissues. Nature, 507, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold C.D. et al. (2013) Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science, 339, 1074–1077. [DOI] [PubMed] [Google Scholar]
- Barrett T. et al. (2013) Ncbi geo: archive for functional genomics data sets–update. Nucleic Acids Res., 41 (Database issue), D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium,E.P. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotney J. et al. (2012) Chromatin state signatures associated with tissue-specific gene expression and enhancer activity in the embryonic limb. Genome Res., 22, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullwood M.J. et al. (2009) An oestrogen-receptor-alpha-bound human chromatin interactome. Nature, 462, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton K.J. et al. (2010) A map of open chromatin in human pancreatic islets. Nat. Genet., 42, 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavi-Helm Y. et al. (2014) Enhancer loops appear stable during development and are associated with paused polymerase. Nature, 512, 96–100. [DOI] [PubMed] [Google Scholar]
- He B. et al. (2014) Global view of enhancer-promoter interactome in human cells. Proc. Natl. Acad. Sci. USA, 111, E2191–E2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman N.D. et al. (2009) Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature, 459, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S. et al. (2015) The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol., 16, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D. et al. (2013) Super-enhancers in the control of cell identity and disease. Cell, 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F. et al. (2013) A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature, 503, 290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J. et al. (2002) The human genome browser at UCSC. Genome Res., 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.K. et al. (2010) Widespread transcription at neuronal activity-regulated enhancers. Nature, 465, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong C.T., Corces V.G. (2011) Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet., 12, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio L.A. et al. (2013) Enhancers: five essential questions. Nat. Rev. Genet., 14, 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin B. et al. (2012) Cistromemap: a knowledgebase and web server for chip-seq and dnase-seq studies in mouse and human. Bioinformatics, 28, 1411–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. (2010) Bedtools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics,C. et al. (2015) Integrative analysis of 111 reference human epigenomes. Nature, 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F., Furlong E.E. (2012) Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet., 13, 613–626. [DOI] [PubMed] [Google Scholar]
- Thurman R.E. et al. (2012) The accessible chromatin landscape of the human genome. Nature, 489, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A. et al. (2007) Vista enhancer browser–a database of tissue-specific human enhancers. Nucleic Acids Res., 35 (Database issue), D88–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A. et al. (2009) Chip-seq accurately predicts tissue-specific activity of enhancers. Nature, 457, 854–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X. et al. (2011) The human epigenome browser at washington university. Nat. Methods, 8, 989–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.