

Abstract
Summary: Top-down mass spectrometry enables the observation of whole complex proteoforms in biological samples and provides crucial information complementary to bottom-up mass spectrometry. Because of the complexity of top-down mass spectra and proteoforms, it is a challenging problem to efficiently interpret top-down tandem mass spectra in high-throughput proteome-level proteomics studies. We present TopPIC, a tool that efficiently identifies and characterizes complex proteoforms with unknown primary structure alterations, such as amino acid mutations and post-translational modifications, by searching top-down tandem mass spectra against a protein database.
Availability and Implementation: http://proteomics.informatics.iupui.edu/software/toppic/
Contact: xwliu@iupui.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
In the past decade, top-down mass spectrometry (MS) has rapidly developed because of the advances in protein separation and mass spectrometry techniques. It has unique advantages in identifying and characterizing proteoforms with primary structure alterations, such as amino acid mutations and post-translational modifications (PTMs), because top-down tandem mass spectra cover whole proteoform sequences (Catherman et al., 2014). Although several software tools, such as ProSightPC (LeDuc et al., 2004; Zamdborg et al., 2007) and pTop (Sun et al., 2016), have been developed for top-down MS data analysis, efficient interpretation of high throughput top-down MS data is still a challenging computational problem because intact proteoforms often contain unknown primary structure alterations compared with the unmodified forms in the protein database (Smith and Kelleher, 2013).
We developed TopPIC (TOP-down mass spectrometry-based Proteoform Identification and Characterization), a software tool for high-throughput proteome-wide proteoform identification and characterization by top-down MS, which integrates algorithms for protein filtering, spectral alignment, E-value computation and Bayesian models for characterization of unknown amino acid mutations and PTMs. In addition, a user friendly web browser-based interface is provided. TopPIC focuses on the identification and characterization of unknown primary structure alterations and the discovery of novel proteoforms. The performances of TopPIC with various parameter settings were evaluated on a top-down tandem mass spectrometry (MS/MS) data set of Escherichia coli K-12 MG1655.
2 Methods
Top-down MS/MS spectra containing highly charged isotopic peaks are usually preprocessed with two steps: (a) raw profile data files are converted to XML format files with centroided peaks using a file format conversion tool, and (b) a deconvolution method, e.g. Thrash (Horn et al., 2000) and MS-Deconv (Liu et al., 2010), is employed to extract a list of monoisotopic masses from each top-down MS/MS spectrum. Combining TopPIC and the above two steps provides a top-down MS/MS data analysis pipeline (Fig. 1) from raw data to identified proteoform spectrum-matches (PrSMs), some of which contain characterized amino acid mutations and/or PTMs.
Fig. 1.
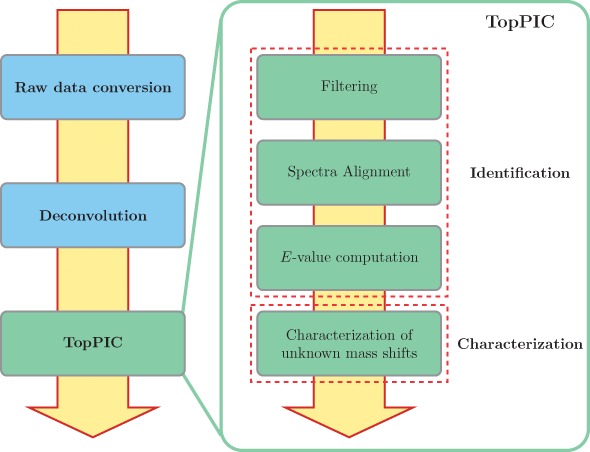
TopPIC is a main step in the software pipeline (left) for the identification and characterization of proteoforms by top-down MS. TopPIC consists of two components: proteoform identification and unknown mass shift characterization (right)
TopPIC consists of two components: the identification of proteoforms and the characterization of unknown mass shifts in PrSMs (Fig. 1). The first component includes algorithms for protein filtering, spectral alignment and the computation of E-values of PrSMs. The second component uses the MIScore method (Kou et al., 2016), which is based on Bayesian models, to find one or two modifications that best explain an unknown mass shift in a reported PrSM. Proteoform identification is the main function of TopPIC, and the MIScore method is an optional feature of TopPIC for automated characterization of unknown mass shifts.
In the first component, TopPIC searches a deconvoluted mass spectrum against a protein sequence database to identify a best scoring PrSM with three steps (Fig. 1): (a) a filtering algorithm based on linked lists (Liu et al., 2012b) is used to reduce the number of candidate proteins from thousands to dozens. (b) A spectral alignment algorithm (Liu et al., 2012a) is employed to find the best alignment between the spectrum and each protein candidate. (c) E values of candidate PrSMs (alignments) are computed by either an extended generation function method (Liu et al., 2014) or a lookup table, and the PrSM with the best E value is reported.
TopPIC has many new features compared with MS-Align+ (Liu et al., 2012a): (a) indexes of protein databases are used to reduce the memory requirement. (b) A lookup table-based method is employed to significantly speed up the computation of E values of PrSMs. (c) Multiple filtering algorithms are added to increase the sensitivity of the filtering step (see the Supplementary material for details). (d) It can combine MS/MS spectral pairs or triplets generated from the alternating fragmentation mode of mass spectrometers to improve the coverage of protein sequences.
The generating function method is time consuming for computing E values of PrSMs. In TopPIC, a lookup table is precomputed for speeding up the computation. A PrSM is mapped a cell in the table to find the E value by using three features: the number of unknown mass shifts, the number of deconvoluted fragment masses and the number of matched fragment masses (each cell is defined by a number of mass shifts and two intervals for the other two features). Each cell stores the average of the logarithms of the E values of 10 randomly generated PrSMs that are mapped to it.
The alternating fragmentation mode generates an MS/MS spectral pair or triplet with different fragmentation methods from the same precursor ion. In the multiple spectra mode of TopPIC, deconvoluted monoisotopic masses in a spectral pair or triplet are combined for protein identification and characterization. Combining these spectra improves protein sequence coverage in identified proteoforms, which is essential for accurate characterization and localization of modifications.
In the second component, the MIScore method (Kou et al., 2016), which is based on Bayesian models, is employed to identify and localize modifications using a list of common modifications provided by the user when identified PrSMs contain unknown mass shifts. For each reported unknown mass shift, the MIScore method evaluates possible explanations with one or two common modifications and reports the best one as well as the confidence score for each characterized modification.
3 Results
TopPIC was tested on a data set of E. coli (EC) K-12 MG1655 with 2027 collision-induced dissociation (CID) and 2027 electron-transfer dissociation (ETD) top-down MS/MS spectra, which were generated by an LTQ Orbitrap Velos mass spectrometer using the alternating fragmentation mode. The details of the MS experiment are given in the Supplementary material. The tests were performed on a desktop computer with an Intel(R) Core i7-3770 3.40 GHz CPU and 16 GB memory.
The maximum number of allowed unknown mass shifts in a proteoform is a main parameter that determines the search space and sensitivity of TopPIC. We compared the performances of TopPIC with three settings for the parameter: 0, 1 and 2. The target-decoy approach was used to filter out low quantity matches; all other parameters were set as the default values (Table 1 in the Supplementary material). Figure 1 in the Supplementary material shows an example of the web pages for the visualization of PrSMs. The running times were 405, 1104 and 31 930 seconds and the numbers of identified PrSMs with a 1% spectrum-level false discovery rate (FDR) were 875 (21.6%), 1860 (45.9%) and 1914 (47.2%) for at most 0, 1 and 2 unknown mass shifts, respectively (Supplementary Tables S1–S3). The 1914 PrSMs were mapped to 301 proteins. The maximum number and the average number of matched spectra for a protein were 102 and 6.36, respectively. When four threads in the CPU were used, the running time was 15 835 s for at most two unknown mass shifts. The maximum memory usage was about 2.5 GB. TopPIC achieved a balance between the speed and the number of identifications when the maximum number of unknown mass shifts was set as 1.
Of the 1914 identified PrSMs (at most two unknown mass shifts were allowed), 997 contain one unknown mass shift and 31 contain two unknown mass shifts (Supplementary Table S3). The MIScore method identified and localized 25 modification sites with an MIScore ≥ 45% from 74 mass shifts by using four common PTMs: acetylation, methylation, oxidation, and phosphorylation (Supplementary Table S4).
The lookup table approach is faster, but less accurate, than the extended generating function approach for computing E values of PrSMs. The two approaches were compared in speed and the number of identifications on the EC data set using the same parameter settings as the previous test. The maximum number of unknown mass shifts was set as 2. The running times for E value computation were 133 and 86 283 s, and the numbers of identifications (with a 1% spectrum-level FDR) were 1914 and 1955 for the lookup table approach and the generating function approach, respectively (Supplementary Tables S3 and S5).
The multiple spectra mode of TopPIC is capable of combining spectral pairs in proteoform identification. We compared the sequence coverage of PrSMs reported by single CID and ETD spectra as well as combined spectral pairs using the default parameter settings except that at most two unknown mass shifts were allowed and the target-decoy approach was used (Supplementary Tables S6–S8). A total of 452 spectral pairs were identified by all three approaches with a 1% spectrum-level FDR. The average peptide bond coverage percentages were 40.4%, 38.1%, and 63.7% for the CID only, ETD only, and combined approaches, respectively. The results demonstrated that the combined approach significantly increased the sequence coverage in identified proteoforms.
MS-Align+ was employed to analyze the same data set and compared with TopPIC. In the two tools, the target-decoy approach was used, at most two unknown mass shifts were allowed in a PrSM, and all other parameters were set as the default values (Tables 1 and 2 in the Supplementary material). MS-Align+ identified 1995 PrSMs with a 1% spectrum-level FDR. Its running time was 89 750 s (about 25 h) and its memory usage was about 11.5 GB. Compared with MS-Align+, TopPIC significantly reduced the memory usage and achieved about 81.3 and 5.7 times speedup for at most 1 and 2 unknown mass shifts, respectively. When at most two unknown mass shifts were allowed, TopPIC identified 104 PrSMs missed by MS-Align+. The main reason is that the filtering algorithms in TopPIC are more sensitive than those in MS-Align+. TopPIC also missed 185 PrSMs identified by MS-Align+. Because many of these PrSMs contained a mass shift larger than 500 Dalton (Da) and the default setting for the maximum absolute value of an unknown mass shift in TopPIC is 500 Da (unlimited for MS-Align+), TopPIC failed to identify the PrSMs. By setting the maximum absolute value of a mass shift as 50 000 Da, the number of PrSMs identified by TopPIC was increased from 1914 to 2040, and the number of PrSMs that were identified by MS-Align+ and missed by TopPIC was decreased to 97. Another reason why some PrSMs were missed by TopPIC is that the lookup table method is less accurate than the generating function method (used in MS-Align+) in the computation of E values. When the generating function method was used, TopPIC identified 2097 PrSM with a 1% spectrum-level FDR (Table S9 in the Supplementary material) and missed only 34 PrSMs identified by MS-Align+ (Table S10 in the Supplementary material), all of which are not highly confident identifications.
4 Discussion
TopPIC focuses on identifying proteoforms with unknown primary structure alterations and characterizing unknown modifications. It includes many useful features, such as proteoform identification by combining spectral pairs or triplets generated by the alternating fragmentation mode of mass spectrometers. It is much faster and more memory efficient compared with MS-Align+. In addition, a web server based graphical user interface is provided.
TopPIC is capable of identifying proteoforms with primary structure alterations, but it still has limitations in the analysis of very complex proteoforms. First, the filtering algorithms in TopPIC are based on finding protein segments without alterations that match to a part of the query spectrum with a large number of matched fragment masses. When a proteoform has many alterations, TopPIC may fail to identify it because the filtering algorithms cannot find such a segment in the target protein. Second, variable PTMs are not incorporated into database search for proteoform identification. A proteoform with several variable PTMs, such as oxidation, may not be identified by TopPIC. Third, a simple mass counting score is used as the scoring function in TopPIC. A scoring function that takes into account peak intensities can further improve the sensitivity of proteoform identification and the accuracy of modification characterization.
Supplementary Material
Acknowledgement
We thank Prof. Si Wu at the University of Oklahoma for providing us with the top-down MS data.
Funding
The research was partially supported by the National Institute of General Medical Sciences, National Institutes of Health (NIH) through Grant R01GM118470.
Conflict of Interest: none declared.
References
- Catherman A.D. et al. (2014) Top down proteomics: facts and perspectives. Biochem. Biophys. Res. Commun., 445, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn D.M. et al. (2000) Automated reduction and interpretation of high resolution electrospray mass spectra of large molecules. J. Am. Soc. Mass Spectrom., 11, 320–332. [DOI] [PubMed] [Google Scholar]
- Kou Q. et al. (2016) Characterization of proteoforms with unknown post-translational modifications using the MIScore. J. Proteome Res., doi: 10.1021/acs.jproteome.5b01098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDuc R.D. et al. (2004) ProSight PTM: an integrated environment for protein identification and characterization by top-down mass spectrometry. Nucl. Acids Res., 32, W340–W345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. et al. (2010) Deconvolution and database search of complex tandem mass spectra of intact proteins a combinatorial approach. Mol. Cell. Proteomics, 9, 2772–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. et al. (2012a) Protein identification using top-down spectra. Mol. Cell. Proteomics, 11, M111–008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. et al. (2012b) Speeding up tandem mass spectral identification using indexes. Bioinformatics, 28, 1692–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. et al. (2014) Spectral probabilities of top-down tandem mass spectra. BMC Genomics, 15, S9.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L.M., Kelleher N.L. (2013) Proteoform: a single term describing protein complexity. Nat. Methods, 10, 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R.X. et al. (2016) pTop 1.0: a high-accuracy and high-efficiency search engine for intact protein identification. Anal. Chem., 88, 3082–3090. [DOI] [PubMed] [Google Scholar]
- Zamdborg L. et al. (2007) ProSight PTM 2.0: improved protein identification and characterization for top down mass spectrometry. Nucl. Acids Res., 35, W701–W706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.