

Abstract
Rett syndrome (RTT; OMIM 312750), a progressive neurological disorder, is caused by mutations in methyl-CpG-binding protein 2 (MECP2; OMIM 300005), a ubiquitously expressed factor. A genetic suppressor screen designed to identify therapeutic targets surprisingly revealed that downregulation of the cholesterol biosynthesis pathway improves neurological phenotypes in Mecp2 mutant mice. Here, we show that MeCP2 plays a direct role in regulating lipid metabolism. Mecp2 deletion in mice results in a host of severe metabolic defects caused by lipid accumulation, including insulin resistance, fatty liver, perturbed energy utilization, and adipose inflammation by macrophage infiltration. We show that MeCP2 regulates lipid homeostasis by anchoring the repressor complex containing NCoR1 and HDAC3 to its lipogenesis targets in hepatocytes. Consistently, we find that liver targeted deletion of Mecp2 causes fatty liver disease and dyslipidemia similar to HDAC3 liver-specific deletion. These findings position MeCP2 as a novel component in metabolic homeostasis. Rett syndrome patients also show signs of peripheral dyslipidemia; thus, together these data suggest that RTT should be classified as a neurological disorder with systemic metabolic components. We previously showed that treatment of Mecp2 mice with statin drugs alleviated motor symptoms and improved health and longevity. Lipid metabolism is a highly treatable target; therefore, our results shed light on new metabolic pathways for treatment of Rett syndrome.
Introduction
Mutations in the X-linked gene encoding methyl-CpG-binding protein 2 (MECP2) are the primary cause of the neurological condition Rett syndrome (1). Females with loss-of-function mutations in MECP2 achieve developmental milestones during the first 6–18 months of life, followed by progressive loss of acquired linguistic and motor skills, stereotypic hand movements, difficulty walking, irregular breathing, and seizures. MECP2 hemizygous males typically die perinatally of encephalopathy. However, Mecp2 hemizygous (Mecp2/Y) male mice survive through birth and develop Rett syndrome (RTT)-like symptoms beginning at 4 weeks of age that progressively worsen until death occurs between 6 and 12 weeks. Mecp2/Y mice are the primary model of RTT; their study has been vital towards understanding the etiology of the disease as Mecp2 heterozygous (Mecp2/+) female mice have a wide range in phenotype severity due to random X-chromosome inactivation (2–5).
Mecp2 encodes a ubiquitously-expressed, intrinsically disordered protein (IDP) that, in murine neurons, binds DNA at near histone-octomer levels (6,7). Characteristic of an IDP, MeCP2 is targeted by a multitude of post-translational modifications, which determine cofactor association and may account for its seemingly endless roles in maintaining cellular homeostasis (8). To this effect, the mechanistic contribution of mutant MeCP2 to RTT pathology is highly complex and remains elusive.
As the culprit gene of a devastating neurological disorder, MECP2 has been largely studied in relation to central nervous system (CNS) development, maturation, and signaling. In neurons, MeCP2 can repress gene transcription by binding the nuclear receptor co-repressor/silencing mediator for retinoid or thyroid-hormone receptors (NCoR/SMRT) complex at MeCP2 amino acids 302–306 (9,10). MeCP2-NCoR/SMRT suppresses transcription by delivering the histone deacetylase HDAC3 to target genes, which removes acetyl marks left by histone acetyltransferases, resulting in a closed chromatin state (11,12). A R306C missense mutation in mouse Mecp2 abolishes interaction with the NCoR/SMRT co-repressor complex and prevents MeCP2-mediated transcriptional repression. Importantly, this mutation is sufficient to cause severe RTT-like phenotypes, including hindlimb clasping, hypoactivity, and reduced brain weight, thus highlighting the importance of the NCoR/SMRT-HDAC3 interaction with MeCP2 in RTT pathology. Furthermore, the amino acid 302–306 region of human MECP2 contains a missense mutation hotspot, causing classical RTT in patients (10,13).
Mouse models have established the vital role of the NCoR corepressor complex in energy metabolism. NCoR-HDAC3 regulates the circadian metabolic cycle of the liver, governing the fluctuation between lipid and glucose utilization in hepatocytes during light/dark cycles (14,15). Loss of Hdac3 from the liver of embryonic or adult mice promotes a constitutively active transcriptional environment, which increases expression of de novo lipogenesis enzymes, resulting in fatty liver disease (16,17). Further, Hdac3 liver deletion increases transcription of several genes in the cholesterol biosynthesis pathway, elevating serum cholesterol. One target of HDAC3 is squalene epoxidase (Sqle), a rate-limiting enzyme of the committed cholesterol biosynthesis pathway, whose transcription is increased ∼170-fold in response to Hdac3 liver deletion (16,18,19). Curiously, a forward genetic suppressor screen found that a nonsense mutation in Sqle improved RTT-like motor symptoms and overall health in Mecp2 null mice. This was intriguing, as both RTT patients and Mecp2 mutant mice can have increased serum cholesterol and triglycerides (20–22). Furthermore, treatment of Mecp2 hemizygous male and heterozygous female mice with HMG-CoA reductase inhibitors (statins) phenocopies symptom improvement by Sqle mutation (20). Notably, RTT-like phenotypes are improved following treatment with fluvastatin, a hydrophilic statin that does not readily cross the blood-brain barrier suggesting that MeCP2 plays a role in regulating systemic cholesterol metabolism (23,24). As such, we hypothesized that genetic loss of Mecp2 would cause metabolic perturbations in mice. Here, we report that Mecp2 deletion causes fatty liver disease, metabolic syndrome, insulin resistance, and alters overall energy homeostasis. We show that hepatic MeCP2 works in conjunction with the NCoR-HDAC3 corepressor complex to orchestrate transcription of enzymes in the triglyceride and cholesterol synthesis pathways. In support of this, liver-specific loss of Mecp2 is sufficiently deleterious to increase lipogenic enzyme transcription leading to fatty liver; however, these mice are insulin sensitive, suggesting that MeCP2 plays a role in insulin signaling outside of the liver.
Results
Mecp2 deletion increases lipogenic enzyme expression, resulting in non-alcoholic fatty liver
Surprisingly, we observed a consistent pale coloration of 129.Mecp2/Y livers (Fig. 1A). Oil Red O staining suggested an accumulation of lipid vesicles and subsequent quantification of lipids in Mecp2/Y null liver showed a ∼2-fold increase in liver triacylglycerol (TAG) over wild-type (Fig. 1B). We examined hepatic expression of genes in the fatty acid and TAG synthesis pathways at three (pre-symptomatic) and eight (symptomatic) weeks of age. Expression of four enzymes involved in fatty acid synthesis (acetyl-CoA carboxylase A and B [Acca, Accb], fatty acid synthase [Fasn] and stearoyl-CoA desaturase 1 [Scd-1]) was increased in pre- and post-symptomatic Mecp2/Y mice liver. Expression of fatty acid translocase (Cd36), a transporter of fatty acids, and perilipin 2 and 5 (Plin2 and Plin5), proteins that coat TAG-dense lipid droplets, was also increased. However, levels of sterol-regulatory element-binding protein 1c (Srebp-1c), the transcription factor responsible for regulating the fatty acid synthesis pathway, was not altered in Mecp2/Y liver (Fig. 1C–E) (25,26).
Figure 1.

Mecp2 null mice develop fatty liver and aberrant hepatic lipogenic enzyme expression. (A) Representative livers from eight-week old mice. Liver sections were subjected to Oil Red O staining. Scale bar represents 50 μM. (B) Levels of TAGs and total cholesterol in liver of eight-week old male mice (n = 6–7 mice per genotype). (C) mRNA expression in liver dissected from three-week old male mice (n = 3–4 mice per genotype) quantified by real-time PCR. (D) mRNA expression in liver dissected from eight-week old male mice (n = 6–7 mice per genotype) quantified by real-time PCR. (E) A simplified schematic of the enzymes and transcription factors in the hepatic cholesterol and TAG synthesis pathways. (F) Western blot of immature SREBP2 and mature, nuclear SREBP2 (nSREBP2) in liver lysates from eight-week old male Mecp2/Y mice (n = 4–5 mice per genotype). (G) Quantification of nSREBP2/SREBP2 western blot normalized to GAPDH. B–D, G. Values are mean ± SEM. ns, no significance; *P < 0.05, **P ≤ 0.01, ***P < 0.001 versus wild-type littermates. Statistical analyses were performed using Student's t-test.
Transcription of HMG-CoA reductase (Hmgcr), squalene epoxidase (Sqle) and lanosterol synthase (Lss), enzymes of the cholesterol biosynthesis pathway, were all increased ∼3-fold in 129.Mecp2/Y liver by 8 weeks of age (Fig. 1C–E). Concurrently, 129.Mecp2/Y null mice have increased serum cholesterol by six weeks of age (20). Therefore, we analyzed the expression and maturation of SREBP2, the transcription factor responsible for expression of enzymes in the cholesterol biosynthesis pathway (27). However, there were no differences in levels of Srebp2 transcript (Fig. 1C and D) nor levels of unspliced or mature nuclear nSREBP2 between 129.Mecp2/Y null and wild-type livers (Fig. 1F and G). These data suggest that a SREBP-independent pathway is responsible for aberrant hepatic lipogenesis in Mecp2/Y null mice. Notably, Mecp2/+ heterozygous female mice also accumulate fat in their livers by 6 months of age, indicating that mosaic loss of Mecp2 is sufficient to cause fatty liver (Supplementary Material, Figure S1A and B).
Impaired glucose and insulin homeostasis in Mecp2 mice
Non-alcoholic fatty liver disease is closely linked to glucose intolerance and insulin resistance. Mecp2/Y mice display hyperglycemia in the fed state at 8-weeks of age, but glucose levels are similar to wild-type when fasted (Supplementary Material, Figure S2A and B). Even so, plasma insulin levels of fasted and non-fasted Mecp2/Y mice are significantly greater than wild-type littermates at both pre- and post-symptomatic ages (4 and 8 weeks, respectively) (Fig. 2A and B). Concurrently, 129.Mecp2/Y null mice are glucose intolerant at both four and eight-weeks of age and insulin resistance is apparent by 9-weeks (Supplementary Material, Figure S2C–E). Glucose intolerance also arises in 129.Mecp2/+ heterozygous females by 20-weeks of age and insulin resistance is evident at 24-weeks of age (Supplementary Material, Figure S3A–C).
Figure 2.

Mecp2 null mice are insulin resistant, resulting in perturbed glucose uptake. (A) Serum insulin concentration in Mecp2/Y and wild-type (+/Y) littermates at four-weeks of age in the fed state (n = 5–7 mice per genotype) or following a 6-h fast (n = 10–12 mice per genotype). (B) Serum insulin concentration at eight-weeks of age in the fed state or following a 6-h fast (n = 10–12 mice per genotype). (C) A schematic showing the hyperinsulinemic-euglycemia clamps protocol (n = 4–5 mice per genotype) performed at eight-weeks of age. (D) The GIR needed to maintain euglycemia (100–140 mg/dl serum glucose) in response to insulin clamps. (E) HGP in the basal and clamped state. (F) Rate of insulin-stimulated glucose disposal (Rd). (G) The rate of glucose uptake in WAT, muscle, cerebral cortex, hypothalamus and thalamus, cerebellum, and hippocampus. (H) In vivo lipolysis assays in 4-h fasted male mice at eight-weeks of age. Serum glycerol and NEFAs levels are shown at basal metabolic state (basal) and 15-min after lipolysis induction by injection with CL316243 (CL), a β3-adrenoceptor agonist (n = 7–9 mice per genotype). (I) mRNA expression of inflammatory markers TNFa, CD68, IL6, and IL10 in epididymal WAT from eight-week male mice (n = 6–8 mice per genotype) quantified by real-time PCR. (J) Representative epididymal WAT sections stained for F4/F80+ cells at eight-weeks. Scale bar represents 100 μM. (K) Number of crown-like structures in epididymal WAT sections (n = 3 mice per genotype) at 9-weeks. A,B, D–I, K. Values are mean ± SEM. ns, no significance, *P < 0.05, **P ≤ 0.01, ***P ≤ 0.001 versus wild-type littermates. Statistical analyses were performed using Student's t-test.
To critically assess global insulin sensitivity, Mecp2/Y null mice were subjected to hyperinsulinemic-euglycemic clamp studies (Fig. 2C). In this gold standard assay for assessing insulin resistance, [3-3H]-glucose and insulin are infused in fasted, unrestrained mice to maintain a euglycemic ‘clamped’ state (100–140 mg/dl serum glucose). The radioisotope allows for measurement of basal and clamped hepatic glucose production (HGP) and glucose disappearance or disposal (Rd). These studies revealed that Mecp2/Y null mice require a lower rate of exogenous glucose infusion (GIR) to maintain euglycemia (Fig. 2D). When clamped, insulin infusion did not decrease HGP in mutant mice (Fig. 2E). Further, Mecp2/Y mice have a lower rate of whole-body insulin-stimulated glucose disposal (Rd) (Fig. 2F). Together, these data reflect a state of profound insulin resistance in Mecp2 null liver, skeletal muscle, and white adipose tissues (WATs). To this effect, glucose uptake by Mecp2 null WAT and muscle is markedly suppressed (Fig. 2G). However, glucose uptake is increased in the cerebral cortex and hypothalamus/thalamus regions of the brain, possibly due to the insulin-independent nature of the major glucose transporters (GLUTs) of the brain.
Mecp2 deletion induces macrophage infiltration, adipose inflammation and increased lipolysis
Lipolysis is precisely regulated by hormones such as insulin. We asked whether Mecp2 loss and subsequent insulin resistance would affect adipose lipolysis. Basal levels of circulating glycerol and non-esterified fatty acids (NEFA) were significantly increased at eight-weeks in Mecp2/Y null mice and at 24-weeks in Mecp2/+ heterozygous female mice compared with wild-type littermates (Fig. 2H and Supplementary Material, Figure S3D), suggesting an increased rate of lipolysis. A selective β3-adrenergic receptor agonist CL316243 (CL), was used to measure response to lipolysis induction. Lipolysis stimulation resulted in an approximately 2-fold increase in circulating glycerol and fatty acids in both male and female Mecp2 mutant and wild-type mice (Fig. 2H and Supplementary Material, Figure S3D). No increase in basal circulating NEFA or glycerol was observed when lipolysis was examined at earlier time points (4-weeks of age in Mecp2/Y males and 12-weeks of age in Mecp2/+ females), indicating that glucose intolerance precedes these events (Supplementary Material, Figures S2F and S3E).
Adipose expansion and increased lipolysis are often accompanied by WAT inflammation. As such, we investigated inflammatory markers in serum and in epididymal (gonadal) WAT using gene expression analysis. Serum leptin, a pro-inflammatory factor, was highly elevated in Mecp2/Y serum while there was no observable difference in adiponectin (Supplementary Materials, Figure S2G and H). Mecp2/Y null WAT has increased expression of macrophage-derived interleukin-6 (IL-6) and TNFα, cytokines implicated in dramatically increasing adipocyte lipolysis, and CD68, a macrophage marker, but exhibit no change in the anti-inflammatory marker IL10 (28) (Fig. 2I). These data indicate macrophage infiltration in Mecp2/Y WAT, which was confirmed by increased F4/F80 immunohistochemistcal staining in gonadal WAT (Fig. 2J). Adipose macrophages in Mecp2/Y null WAT formed a significantly larger number of crown-like structures, which envelope necrotic adipocytes (Fig 2K).
Mecp2 deletion modifies whole body substrate metabolism
To further characterize metabolic changes in Mecp2/Y null mice, we assessed energy expenditure (EE) and substrate metabolism by indirect calorimetry over a 20-h period (11-h dark period, cumulative 9-h day period), following a 4-h acclimation period. 129.Mecp2/Y null male mice weigh significantly more than wild-type (+/Y) littermates by seven weeks of age, due to an expansion of white and brown adipose tissue, despite a decrease in lean mass (Fig. 3A and B and Supplementary Material, Figure S4A and B). Because Mecp2/Y mice are heavier at eight weeks of age, we employed analysis of covariance (ANCOVA) to control for both the weight factor and genotype (29) (Fig. 3C). Mecp2/Y null mice display a slight, but significant decrease (P = 0.03) in EE during the day, but have no difference in EE at night (Fig. 3D). This is somewhat surprising considering severe hypoactivity is a hallmark of symptomatic (eight-week old) Mecp2/Y null mice (3). As expected, both wild-type and Mecp2 null mice displayed diurnal rhythms in their whole-body EE and respiratory exchange ratio (RER). RER, an indicator of energy derived from oxidizing glucose (RER = 1.0) or fatty acids (RER = 0.7), increases for both genotypes at night, reflective of food intake and carbohydrate utilization (Fig. 3E). However, Mecp2 null mice have a markedly decreased RER versus wild-type littermates during both day and night cycles, consistent with preferential lipid oxidation over carbohydrate metabolism (Fig. 3E and F).
Figure 3.

Mecp2 deletion shifts whole body substrate metabolism. (A) Total weight of Mecp2/Y and wild-type (+/Y) littermates (n = 10 mice per genotype). (B) Lean and fat mass (n = 7–8 mice per genotype). (C) Linear regression for EE as a function of genotype and body weight, separated by day (+/Y: y = 0.0228 * x – 0.1398; r2=0.4247; Mecp2/Y: y = 0.0102 * x + 0.1159; r2 = 0.6417) and night (+/Y: y = 0.0209 * x – 0.0245; r2=0.4716; Mecp2/Y: y = 0.0125 * x + 0.1709; r2 = 0.7990) periods. (D) EE adjusted for weight (average weight = 24.35g) using ANCOVA for wild-type control and Mecp2/Y mice (n = 7–9 mice per genotype) at eight-weeks. (E) Respiratory rate (VCO2/VO2) across a 20-h period. (F) Average respiratory rate, separated by day and night periods. Values are mean ± SEM. ns, no significance, *P < 0.05, ** P ≤ 0.01, *** P ≤ 0.001 versus WT. EE averages were adjusted for weight using ANCOVA and statistical analyses were performed using Student's t-test.
MeCP2 regulates expression of liver lipogenic enzymes through interaction with the NCoR/HDAC3 corepressor complex
In neurons, MeCP2 interacts with the nuclear receptor co-repressor 1 (NCoR1) complex, which includes the proteins histone deactylase 3 (HDAC3) and transducin-(beta) like 1 X-linked receptor 1 (TBLR1) to facilitate gene repression (9,10). In hepatocytes, the NCoR-HDAC3-TBLR1 corepressor complex is a potent regulator of deactylase-dependent gene transcription. As such, we hypothesized that this corepressor complex contributes to the metabolic perturbations in Mecp2 null mice. To address this question, we first assessed native MeCP2 ability to bind the NCoR complex in vivo, in the liver. Co-immunoprecipitation of HDAC3 indeed revealed association with MeCP2, as well as its corepressor complex partners NCoR1 and TBLR1 (Fig. 4A). This binding partnership was validated using three different HDAC3 immunoprecipitation (IP) antibodies, all of which pulled down MeCP2 in complex (not shown).
Figure 4.

MeCP2 interacts in murine hepatocytes with the NCoR-HDAC3 complex and loss of MeCP2 prevents lipogenic enzyme repression at the locus. (A) IP of NCoR1, MeCP2, and TBLR1 from murine liver nuclear extracts using an antibody to HDAC3. i, input. (B) Anti-HDAC3 and anti-H3K27ac ChIP-qPCR of sites surrounding the Sqle TSS (n = 3 mice per genotype). (C) Anti-HDAC3 and anti-H3K27ac ChIP-qPCR of sites surrounding the Fasn TSS (n = 3 mice per genotype). (D) Anti-HDAC3 and anti-H3K27ac ChIP-qPCR of sites surrounding the Cd36 TSS (n = 3 mice per genotype). Values are mean ± SEM. ns, no significance, *P < 0.05, ** P ≤ 0.01, *** P ≤ 0.001 versus WT. Statistical analyses were performed using Student's t-test.
Next, we investigated the effect of MeCP2 loss on HDAC3 binding and activity. Specifically, we performed ChIP-qPCR to assess the binding of HDAC3 to regulatory regions surrounding the transcription start sites (TSS) of several rate-limiting enzymes that are upregulated in Mecp2 null liver. We found that loss of MeCP2 significantly hinders the capability of HDAC3 to bind regulatory regions surrounding the TSS of Sqle (Fig. 4B), Fasn (Fig. 4C), and Cd36 (Fig. 4D) in the liver. HDAC3 binding at the promoter region of the attachment region binding protein (Arbp) gene was used as a negative control (Fig. 4B). Deletion of HDAC3 from the liver or genetic abolishment of its deacetylase activity results in increased local histone acetylation in liver, specifically of the transcriptional activating marker Histone 3 lysine 27 acetylation (H3K27ac) (15,16,30). Therefore, we hypothesized that a reduction in chromatin-bound HDAC3 in Mecp2 null mice would result in increased H3K27ac at the same locations. Remarkably, we saw increased accumulation of H3K27ac at all three genes in Mecp2 null liver lysates, correlating with decreased HDAC3 binding (Fig. 4B–D).
A conditional deletion of Mecp2 from mouse liver dissociates fatty liver and insulin resistance
In light of these findings, we hypothesized that Mecp2 loss from the liver alone would be sufficient to cause fatty liver disease through aberrant expression of genes such as Sqle. To test this hypothesis, we generated a B6.Mecp2 liver-specific deleted mouse line. We removed Mecp2 from Albumin-expressing cells by breeding B6.Mecp2tm1Bird mice (Mecp2flox/+), in which exons 3 and 4 are flanked by loxP sites, to B6.Alb-Cre21Mgn BAC transgenic mice. Alb regulatory elements drive Cre expression in transgenic mice during embryonic development (as early as E15.5) and after birth in hepatocytes, with some restricted expression in bile ducts. Recombination of the floxed Mecp2 allele is robust but not complete in the Mecp2flox/Y;Alb-Cre liver, resulting in a ∼90% decrease in Mecp2 transcription, as evidenced by qRT-PCR (Fig. 5A). However, recombination does not occur in the brain of Mecp2flox/Y;Alb-Cre mice and they did not develop neurological symptoms, as expected (Supplementary Material, Figure S5A and B). It is important to note that the Mecp2floxallele (Mecp2tm1Bird) utilized in these studies is hypomorphic, resulting in an approximate 20% decrease in Mecp2 mRNA in littermate control males lacking Cre (Fig. 5B) (31). Therefore, we anticipated that male mice carrying this allele would display mild phenotypes due to a decrease in MeCP2 dosage.
Figure 5.

Loss of Mecp2 from the liver dissociates fatty liver and insulin resistance. (A) PCR amplification of the Mecp2 locus in mouse liver shows presence of a hemizygous Mecp2flox allele in mice lacking the Alb-Cre transgene. Offspring with the genotype Mecp2flox/Y; Alb-Cre have recombination of the Mecp2 floxed locus to a null allele. This recombination does not occur in the brain of Mecp2flox/Y; Alb-Cre mice. (B) Mecp2 mRNA expression in liver (n = 5 per genotype). (C) mRNA expression in liver dissected from 6-month old male mice (n = 5 per genotype). (D) Livers from 6-month-old mice subjected to Oil Red O staining. Scale bar represents 100 μM. (E) Levels of liver lipids of from 6-month-old male mice (n = 6–7 per genotype). (F) Glucose tolerance tests (GTT). Glucose was administered intraperitoneally after a 5-h fast, and blood glucose was measured at the indicated times. Male mice were tested at 6 months of age (n = 6–8 mice per genotype). (G) Insulin tolerance tests (ITT). Insulin was administered intraperitoneally after a 4-h fast, and blood glucose was measured at the indicated times. Male mice were tested at 6 months of age (n = 4–5 mice per genotype). B–C, E–G. Values are mean ± SEM. ns, no significance, * P < 0.05, ** P ≤ 0.01, *** P ≤ 0.001. Statistical analyses were performed using one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc analysis.
Conditional deletion of Mecp2 from hepatocytes increases transcription of genes in the lipogenesis pathways, including the rate-limiting enzymes of cholesterol biosynthesis Hmgcr and Sqle, as well as rate-limiting enzymes of the triglyceride synthesis pathway Scd1 and Fasn (Fig. 5C). Similarly to a liver-specific deletion of Hdac3, B6.Mecp2flox/Y;Alb-Cre mice develop fatty liver, as evidenced by Oil Red O staining and a quantitative assessment of liver lipids (Fig. 5D and E). Perturbation of these pathways translates to an increase in serum cholesterol in B6.Mecp2flox/Y;Alb-Cre, but not in wild-type, Alb-Cre, or B6.Mecp2flox/Y littermate controls (Table 1). B6.Mecp2flox/Y;Alb-Cre mice gain weight through an expansion of fat mass; however, B6.Mecp2flox/Y littermates display the same phenotype. Therefore, we cannot attribute weight gain to a direct MeCP2 mechanism in the liver.
Table 1.
Metabolic and serum parameters in liver-specific Mecp2 null mice
| Parameter | 6 months | |||
|---|---|---|---|---|
| B6.+/Y | B6.Alb-Cre | B6.Mecp2flox/Y | B6.Mecp2flox/Y; Alb-Cre | |
| Weight (g) | 33.8 0.8 | 35.6 0.8ns | 41.7 1.9** | 41.8 2.0** |
| Fat mass (g) | 6.4 0.6 | 7.8 0.6ns | 13.2 1.5*** | 12.1 1.3*** |
| Lean mass (g) | 22.2 0.3 | 22.7 0.4ns | 23.1 0.6ns | 23.2 0.8ns |
| Cholesterol (mg/dl) | 113.5 7.5 | 102.9 14.4ns | 135.2 10.6ns | 166.5 14.2** |
| Triglycerides (mg/dl) | 46.9 6.8 | 48.7 4.1ns | 49.0 5.9ns | 42.1 2.4ns |
| NEFA (mE/l) | 0.6 0.1 | 0.8 0.1ns | 0.7 0.1ns | 0.7 0.1ns |
| Glycerol (mg/dl) | 24.2 1.7 | 33.2 3.8ns | 30.5 2.5ns | 32.9 1.2ns |
| Leptin (ng/ml) | 15.2 2.9 | 15.9 3.1ns | 42.6 5.5** | 42.2 4.9** |
n = 6–7 mice per genotype. Values represent mean ± SEM. Statistics were performed using one-way ANOVA followed by Tukey’s post-hoc analysis. Ns, no significance,
P < 0.05,
P ≤ 0.01,
P ≤ 0.001 compared with B6.+/Y Male (WT).
Fatty liver is closely associated with metabolic syndrome; therefore, we performed glucose tolerance and insulin resistance tests in tissue-specific deleted B6.Mecp2 null animals and control littermates. Mecp2flox/Y;Alb-Cre mice are not glucose intolerant nor insulin resistant (Fig. 5F and G). Furthermore, Mecp2flox/Y;Alb-Cre display no significant increase in circulating fatty acids or glycerol (Table 1). As such, Mecp2 loss from the liver dissociates fatty liver and insulin sensitivity.
Discussion
Thought to be entirely a neurological disorder, RTT research has focused on the role of Mecp2 in the CNS; however, the wide variety of phenotypes in Mecp2 null mice and RTT patients points to vital roles for Mecp2 outside the nervous system (32–36). Here we show that Mecp2 null male mice develop severe dyslipidemia due to aberrant transcription of rate-limiting de novo lipogenesis and cholesterol synthesizing enzymes in the liver. Strikingly, expression of neither Srebp1c nor nSREBP2, the transcriptional regulators of fatty acid and cholesterol synthesis, respectively, changed in Mecp2/Y liver (27,37). As such, we hypothesized that increased transcription of cholesterol and de novo lipogenesis enzymes results from a direct loss of MeCP2 transcriptional repression at targeted loci, rather than increased SREBP activity, similar to a liver-specific deletion of Hdac3.
MeCP2 represses gene transcription through association with chromatin-remodeling complexes containing Type I histone deactylases (38,39), which cannot readily bind promoter regions in the absence of MeCP2, resulting in aberrant transcription of targets (40). In neurons, NCoR1-HDAC3 associates with MeCP2 to regulate gene transcription in an activity-dependent manner (9,10). The NCoR1-HDAC3 complex regulates a wide range of vital cellular processes: its disruption is implicated in metabolism, inflammatory pathways, and perturbed CNS development (reviewed in 41). Whole-body deletion of Hdac3 results in embryonic death, limiting its mechanistic study to individual tissues and organ systems (42). Hepatic HDAC3 regulates the diurnal balance between gluconeogenesis and lipid synthesis thereby balancing energy oxidation versus storage during the fasted and fed states, respectively. Liver-specific deletion of Hdac3 increases activating acetylation marks at the promoters of enzymes in the cholesterol and triglyceride synthesizing pathways, including Sqle. This results in aberrant transcription of the de novo lipogenesis pathway in a Srebp-independent manner, subsequently leading to fatty liver disease and increased serum cholesterol (15–17). We hypothesized that MeCP2 interacts with the NCoR-HDAC3-TBLR1 complex in the liver to repress enzyme transcription (20). Indeed, hepatic MeCP2 interacts in vivo with endogenous HDAC3, NCoR1, and TBLR1 in the liver. Loss of MeCP2 decreases HDAC3 binding, resulting in an open chromatin state, as defined by increased H3K27ac marks at the Sqle, Fasn, and Cd36 loci (Fig. 6). This histone modification profile in the liver is highly consistent with loss of enzymatically active HDAC3 (30).
Figure 6.
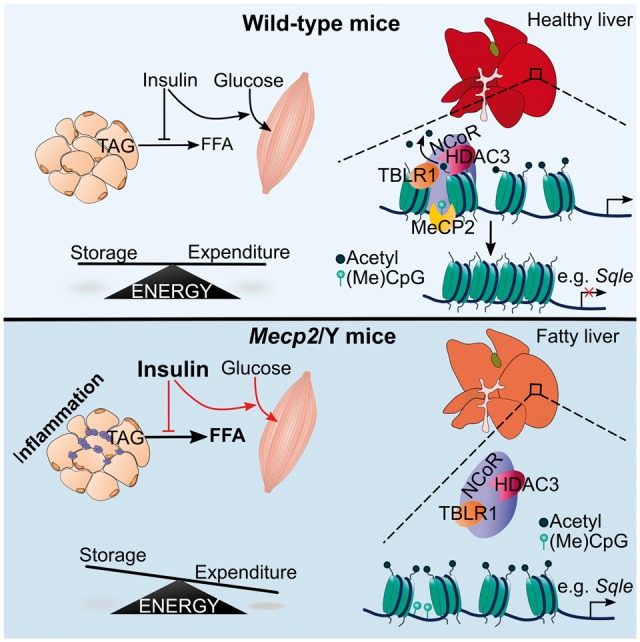
A schematic model of metabolic phenotypes in Mecp2 null mice. MeCP2 co-ordinates hepatic lipid metabolism in vivo through binding HDAC3, which deactylates histone tails leading to reduced transcription at lipogenic loci. In the absence of MeCP2, HDAC3 fails to bind promoter regions, which prevents the removal of histone aceylation marks. This leads to downstream aberrant transcription of enzymes such as Sqle, which subsequently increases hepatic triglyceride and cholesterol production. Mecp2 null mice then develop severe metabolic syndrome, including reduced insulin signaling and glucose uptake (indicated by red lines), resulting in increased lipolysis, and shifting the balance between energy storage and expenditure.
In addition to fatty liver, Mecp2 null mice develop glucose intolerance and hyperinsulinemia as early as four weeks of age, before onset of neurological phenotypes. Insulin resistance in these animals is global, as evidenced by poor glucose uptake in muscle and WAT when under a euglycemic, insulin clamped state. Lipolysis is significantly increased in Mecp2 null mice at eight-weeks of age, but not as early as four-weeks. This timing suggests that increased lipolysis and subsequent WAT inflammation is a downstream effect of hyperinsulinemia and glucose intolerance. Consistent with increased lipolysis, a decreased RER in Mecp2/Y mice during both day and night cycles indicates a preferential shift in energy utilization from carbohydrate to lipids. This may be due to the inability of skeletal muscle to utilize glucose properly in its insulin resistant state, in which case the tissue will switch to fatty acid oxidation for energy.
Neither transport nor usage of glucose in the CNS is effected by insulin action (43,44). Glucose is transported into the brain through a gradient sink, such that a faster rate of glucose metabolism (glycolysis) in the brain allows for faster uptake of glucose from the blood via brain-specific GLUTs. As such, we hypothesize that increased glucose uptake by the Mecp2/Y null brain may be due to an upregulation in insulin-insensitive GLUT or abnormal neuronal glycolysis in response to synaptic hyper-excitability reported in Mecp2 mutant neurons (45–48). Further investigation of this phenotype will shed light on this mechanism and may inform on a root cause of poor neuronal communication in the Mecp2 mutant brain.
Here we show, for the first time, that MeCP2 directly controls expression of rate-limiting metabolic enzymes, such as Sqle, in the liver. As such, loss of MeCP2 from the liver causes aberrant expression of lipogenesis pathways leading to dyslipidemia and non-alcoholic fatty liver disease. Although the brain regulates energy metabolism through neuroendocrine feedback, we reasoned that MeCP2 deletion specifically from the liver would also result in fatty liver disease. Surprisingly, this is only the second time a conditional deletion of Mecp2 outside of the CNS has been published (49). Consistent with our hypothesis, liver-specific deletion of Mecp2 recapitulates most of the phenotypes of a whole-body deletion: the mice have increased de novo lipogenesis, fatty liver and increased serum cholesterol. This genetic experiment further supports a hepatocyte-autonomous role for Mecp2 in repression of enzymes of the cholesterol and triglyceride biosynthesis pathways. Dissociation of fatty liver and insulin resistance is reminiscent of an Alb-Cre induced liver deletion of Hdac3 (17). Mecp2 deleted from Sim-1 or Pomc hypothalamic cells results in obesity, reduced Pomc expression, and subsequent leptin resistance (50,51). It is unknown if these hypothalamic-specific Mecp2 deletion models develop insulin resistance. However, we would predict that dysfunctional systemic feedback between the hypothalamus, adipose, and liver in the whole-body Mecp2 null animals account for insulin resistance and excessive lipolysis, which are absent in a liver-specific deletion of Mecp2.
Importantly, we found that Mecp2 heterozygous (Mecp2/+) female mice develop similar metabolic phenotypes to the male null mice, indicating that a mosaic dose of MeC2 is not sufficient to maintain metabolic homeostasis in mice. Fatty liver disease has not been reported in RTT patients in the literature. However, we and others have reported abnormal cholesterol and triglycerides levels in RTT patients (21,22,52). Furthermore, glucose intolerance has been reported in pediatric RTT patients (53). Certainly, our data open the possibility that RTT should be reclassified as a neurological disorder with systemic metabolic components, and suggest that RTT patients should be examined further for metabolic anomalies. Before labelled as a monogenic neurological disorder, RTT was popularly thought to be a metabolic disease of mitochondria. Abnormal carbohydrate metabolism, increased reactive oxygen species load and poor mitochondrial function have all been reported in RTT patients or Mecp2 mouse models (54–57). These and other metabolic anomalies can be utilized as biomarkers to alert physicians of impending complications associated with Rett syndrome.
In addition to a severe systemic dyslipidemia, we have previously reported perturbed cholesterol homeostasis in the brains of Mecp2/Y null mice (20). Cholesterol is a vital molecule for brain development and health as it is heavily incorporated in myelin sheaths and lipid rafts and is required for axonal guidance, synaptogenesis, and dendrite formation (58). Genetic perturbations in brain cholesterol homeostasis cause a host of severe, life-threatening neurological disorders (reviewed in 59). We showed that cholesterol-lowering statins successfully improved motor phenotypes, overall health and increased lifespan in Mecp2 mutant mice (20). Our data here suggest that statin treatment primarily influenced improvement of phenotypes through altered brain lipids. Strikingly, statins account for only one family of numerous metabolic modulators, many of which are being developed to treat lipid accumulation in Type II diabetes, and many of which cross the blood brain barrier. Further study will illuminate new therapeutic options in highly targetable metabolic pathways that may prevent the progressing morbidities of Rett syndrome.
Materials and Methods
Animals
All animal experiments were conducted under protocols approved by local Animal Care and Use Committees in the AALAC or CCAC-accredited animal facilities at Baylor College of Medicine or the Toronto Center for Phenogenomics, respectively. Congenic 129.Mecp2tm1.1Bird/Y (Mecp2/Y null) feature a deletion of the last two exons (exons 3–4) of the Mecp2 transcript. Mecp2/Y and wild-type male offspring were obtained by backcrossing Mecp2tm1.1Bird/+ females to males of the 129SvEvS6/Tac strain. Mecp2/Y liver-specific deletion lines (Mecp2flox/Y; Alb-Cre), conditional-ready control (Mecp2flox/Y) lines (exons 3 and 4 flanked by loxP sites), and wild-type littermates were obtained by crossing B6J.Mecp2tm1Bird/+ (Mecp2flox/+) heterozygous female mice to male mice heterozygous for the Albumin-Cre transgene on a C57BL/6J background. Mice were fed a standard diet (Harlan Teklad 2918), ad libitum, consisting of 18% protein, 6% fat, and 44% carbohydrates. Mice were housed in a 12-h light/dark cycle facility and were euthanized between the hours of 3 P.M. and 5 P.M. (ZT 8–10) to control for circadian rhythm fluctuations. Lean mass and fat mass was determined in live mice using a Bruker Minispec NMR body composition analyzer.
Histology
A small section of liver (∼5 mm3) was extracted from Mecp2/Y (eight-weeks of age) and age-matched wild-type littermates. Liver was placed immediately in 10% neutral buffered formalin and was fixed for 48 h at room temperature. The liver was washed with exchanges of PBS and was incubated in increasing sucrose gradients (15% then 30%) in PBS overnight at 4°C. Tissue was frozen in OCT and 7 μm slices were prepared. Sections were then stained in 0.3% Oil Red O in isopropanol and then counterstained with hematoxylin for 5 s.
Quantitative PCR
Tissue (liver, WAT) was snap-frozen and stored at −80 until processed for RNA extraction. Details are described in Supplementary Materials. Primers are listed in Supplementary Material, Table S1.
Hyperinsulinemic-euglycemic clamps
Mice were anesthetized and a midline neck incision was made to expose the jugular vein. A microcannula was inserted into the jugular vein, threaded into the right atrium, and anchored at the venotomy site. Mice were allowed to recover for 4 days with ad libitum access to water and food. Following an overnight fast, the conscious mice received a primary infusion (10 uCi) and then a constant rate intravenous infusion (0.1 uCi/min) of chromatography-purified [3-3H]-glucose using a syringe infusion pump. For determination of basal glucose production, blood samples were collected from the tail vein after 50, 55, and 60 min of labeled glucose infusion. After 60 min, mice received a priming bolus of insulin (40 mU/kg body weight) followed by a 2-h insulin infusion (4 mU/kg/min). Simultaneously, 10% glucose was infused using another infusion pump at a rate adjusted to maintain the blood glucose level at 100–140 mg/dl (euglycemia). Blood glucose concentration was measured every 10 min by a glucometer. At 100, 110, and 120 min, blood was collected to measure HGP under clamped conditions. To estimate insulin-stimulated GLUT activity in individual tissues 2-[14C]deoxyglucose was administered as a bolus (10 uCi) at 45 min before the end of the clamps. After 120 min, mice were euthanized and tissues were extracted. Brain regions were rapidly dissected from both hemispheres of the brain. Thalamic and hypothalamic tissue was pooled. Glucose uptake in different tissues was calculated from plasma by tissue enrichment of 2-[14C]deoxyglucose by gas chromatography-mass spectrometry.
Tolerance tests and lipolysis analysis
In vivo lipolysis analyses were performed by the Diabetes and Endocrinology Research Center at BCM. Details are described in Supplementary Materials.
Nuclear extraction
Freshly dissected liver (50 mg) was washed with 1X volume of ice-cold PBS and centrifuged at 500 g for 5 min at 4°C. Tissue was then transferred cleanly to a dounce homogenizer on ice containing 500 μl of cold NP40 lysis buffer (10 mM Hepes pH7.9, 3 mM MgCl2, 10 mM KCl, 0.5% NP40, 0.5mM DTT, and Complete EDTA-free protease inhibitor cocktail (Roche, CA, USA)). Tissue was incubated in buffer on ice for 45 min and then tissue was disrupted with 10 strokes of the tight pestle in the dounce tissue homogenizer. Homogenized tissue was transferred into a new, cold microfuge tube and centrifuged at 2000 g for 10 min at 4°C. The supernatant was removed and the pellet was resuspended in 250 μl of Benzonase extraction buffer (10 mM Hepes pH7.9, 3 mM MgCl2, 280 mM NaCl, 0.2 mM EDTA pH8.0, 0.5% NP40, 0.5 mM DTT, and Complete EDTA-free protease inhibitor cocktail) with 250 U of Benzonase. Nuclei were incubated in extraction buffer for 1 h on at rotator at 4°C. Following incubation, lysates were centrifuged at 17 000g for 20 min at 4°C. Lysates were snap frozen and stored at −80.
Co-immunoprecipitation
IP was carried out using the Millipore Catch and Release kit (Millipore, MA, USA). Following the instructions, 500 μg of nuclear lysates was incubated with 12 μg of anti-HDAC3 (ab7030), 10 μg of anti-HDAC3 (Upstate, 06-890), or 10 μg of anti-HDAC3 (SantaCruz, H-303) at 4°C for 3 h. Following elution, half of the immunoprecipitated sample was loaded onto an SDS-PAGE gel for downstream western blotting. Input samples represent 25 μg of total nuclear extracted protein.
Western blotting
The following antibodies were used for western blotting: anti-MeCP2 (Millipore 07-013) at a concentration of 1:700 in 3% Milk-PBS; anti-SREBP2 (ab30682) at a concentration of 1:400 in 5% Milk-TBST; anti-GAPDH (Cell Signaling 5174) at a concentration of 1:8000 in 5% Milk-TBST; anti-NCoR1 (sc-1609) at a concentration of 1:450 in 1% casein-TBST; anti-TBLR1 at a concentration of 1:1200 in 1% casein-TBST. For co-IP, a light-chain specific secondary antibody (Jackson ImmunoResearch) was used to avoid heavy chain detection at 50kDa. Quantification of band intensity was achieved using ImageJ (NIH) and normalized to GAPDH.
Chromatin-IP
ChIP was performed as described in (60). Details are provided in Supplementary Materials.
Indirect calorimetry
Respiratory metabolic function was assessed using the Oxymax Deluxe System indirect calorimeter equal flow eight chamber system (Columbus Instruments, Columbus, Ohio) and analyzed using Oxymax Windows V3.32 Software. Details are described in the Supplementary Materials.
Subjective health assessments
Mice were assayed for general health once per week from 5 weeks of age to 24-weeks of age. Scoring was blinded by genotype. Mice were scored using the assessment published in (61), where a score of 0 is not different from wildtype, 1 is more severe, and 2 is very severe. Mice were assessed for limbclasping, tremors, home-cage activity, and grooming for a combined possible score of 0–8.
Rotarod
At 5-months of age, male mice were placed on a grooved rotating rod at a speed of 4 r.p.m. Over the course of 5 min, the revolution rate increased steadily to a maximum of 40 r.p.m. The time each mouse stayed on the rod was recorded for four trials spread across two consecutive days, totaling eight trials for each mouse, with a 30-min break between trials. A trial ended when the mouse fell from the rotating rod, spun with the rod for two consecutive revolutions, or successfully stayed on the rod for 5 min.
Statistics
Values are expressed at mean ± SEM. The significance of the differences in mean values among different groups was evaluated by two-tailed Student t-tests or, for comparing means of more than two groups, one-way ANOVA followed by Tukey’s post-hoc analysis. P-values < 0.05 were considered statistically significant (P0.05 = *, P ≤ 0.01 = **, P ≤ 0.001 = ***). Linear regression, Student t-test, and ANOVA was analyzed using GraphPad Prism 6.
Supplementary Material
Supplementary Material is available at HMG online.
Acknowledgements
We thank Misty Hill, Jennifer Borkey, Julie Ruston, Stephen McDonald, and Natalie Tully of the Justice lab for technical assistance. We thank Dr. Michael Wilson and Lina Antounians for valuable guidance in ChIP experiments. We also thank the Centre for Modeling Human Disease (CMHD) for assistance in IHC sample processing.
Conflict of Interest statement. None declared.
Funding
This work was supported by the Rett Syndrome Research Trust (RSRT) [M.J.J.]; and the National Institutes of Health [T32 GM008307 to S.M.K., P30 DK079638 to L.C.C.]. Funding to pay the Open Access publication charges for this article was provided by The Hospital for Sick Children Operating Funds [M.J.J.].
References
- 1.Amir R.E., Van den Veyver I.B., Wan M., Tran C.Q., Francke U., Zoghbi H.Y. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet., 23, 185–188. [DOI] [PubMed] [Google Scholar]
- 2.Braunschweig D., Simcox T., Samaco R.C., LaSalle J.M. (2004) X-Chromosome inactivation ratios affect wild-type MeCP2 expression within mosaic Rett syndrome and Mecp2−/+ mouse brain. Hum. Mol. Genet., 13, 1275–1286. [DOI] [PubMed] [Google Scholar]
- 3.Guy J., Hendrich B., Holmes M., Martin J.E., Bird A. (2001) A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet., 27, 322–326. [DOI] [PubMed] [Google Scholar]
- 4.Katz D.M., Berger-Sweeney J.E., Eubanks J.H., Justice M.J., Neul J.L., Pozzo-Miller L., Blue M.E., Christian D., Crawley J.N., Giustetto M. et al. (2012) Preclinical research in Rett syndrome: setting the foundation for translational success. Dis. Model. Mech., 5, 733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young J.I., Zoghbi H.Y. (2004) X-Chromosome Inactivation Patterns Are Unbalanced and Affect the Phenotypic Outcome in a Mouse Model of Rett Syndrome. Am. J. Hum. Genet., 74, 511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adams V.H., McBryant S.J., Wade P.A., Woodcock C.L., Hansen J.C. (2007) Intrinsic Disorder and Autonomous Domain Function in the Multifunctional Nuclear Protein, MeCP2. J. Biol. Chem., 282, 15057–15064. [DOI] [PubMed] [Google Scholar]
- 7.Skene P.J., Illingworth R.S., Webb S., Kerr A.R.W., James K.D., Turner D.J., Andrews R., Bird A.P. (2010) Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell, 37, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ausió J., Paz AM. de, Esteller M. (2014) MeCP2: the long trip from a chromatin protein to neurological disorders. Trends Mol. Med., 20, 487–498. [DOI] [PubMed] [Google Scholar]
- 9.Ebert D.H., Gabel H.W., Robinson N.D., Kastan N.R., Hu L.S., Cohen S., Navarro A.J., Lyst M.J., Ekiert R., Bird A.P. et al. (2013) Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature, 499, 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lyst M.J., Ekiert R., Ebert D.H., Merusi C., Nowak J., Selfridge J., Guy J., Kastan N.R., Robinson N.D., de Lima Alves F. et al. (2013) Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci., 16, 898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guenther M.G., Barak O., Lazar M.A. (2001) The SMRT and N-CoR Corepressors Are Activating Cofactors for Histone Deacetylase 3. Mol. Cell. Biol., 21, 6091–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J., Wang J., Wang J., Nawaz Z., Liu J.M., Qin J., Wong J. (2000) Both corepressor proteins SMRT and N‐CoR exist in large protein complexes containing HDAC3. Embo J., 19, 4342–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heckman L.D., Chahrour M.H., Zoghbi H.Y. (2014) Rett-causing mutations reveal two domains critical for MeCP2 function and for toxicity in MECP2 duplication syndrome mice. eLife, 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alenghat T., Meyers K., Mullican S.E., Leitner K., Adeniji-Adele A., Avila J., Bućan M., Ahima R.S., Kaestner K.H., Lazar M.A. (2008) Nuclear receptor corepressor and histone deacetylase 3 govern circadian metabolic physiology. Nature, 456, 997–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng D., Liu T., Sun Z., Bugge A., Mullican S.E., Alenghat T., Liu X.S., Lazar M.A. (2011) A Circadian Rhythm Orchestrated by Histone Deacetylase 3 Controls Hepatic Lipid Metabolism. Science, 331, 1315–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knutson S.K., Chyla B.J., Amann J.M., Bhaskara S., Huppert S.S., Hiebert S.W. (2008) Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. Embo J, 27, 1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun Z., Miller R.A., Patel R.T., Chen J., Dhir R., Wang H., Zhang D., Graham M.J., Unterman T.G., Shulman G.I. et al. (2012) Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat. Med., 18, 934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gill S., Stevenson J., Kristiana I., Brown A.J. (2011) Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab., 13, 260–273. [DOI] [PubMed] [Google Scholar]
- 19.Hidaka Y., Satoh T., Kamei T. (1990) Regulation of squalene epoxidase in HepG2 cells. J. Lipid Res., 31, 2087–2094. [PubMed] [Google Scholar]
- 20.Buchovecky C.M., Turley S.D., Brown H.M., Kyle S.M., McDonald J.G., Liu B., Pieper A.A., Huang W., Katz D.M., Russell D.W. et al. (2013) A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat. Genet., 45, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Justice M.J., Buchovecky C.M., Kyle S.M., Djukic A. (2013) A role for metabolism in Rett syndrome pathogenesis: New clinical findings and potential treatment targets. Rare Dis. Austin Tex., 1, e27265.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Segatto M., Trapani L., Di Tunno I., Sticozzi C., Valacchi G., Hayek J., Pallottini V. (2014) Cholesterol metabolism is altered in rett syndrome: a study on plasma and primary cultured fibroblasts derived from patients. PLoS One, 9, e104834.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guillot F., Misslin P., Lemaire M. (1993) Comparison of fluvastatin and lovastatin blood-brain barrier transfer using in vitro and in vivo methods. J. Cardiovasc. Pharmacol., 21, 339–346. [DOI] [PubMed] [Google Scholar]
- 24.Saheki A., Terasaki T., Tamai I., Tsuji A. (1994) In vivo and in vitro blood-brain barrier transport of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors. Pharm. Res., 11, 305–311. [DOI] [PubMed] [Google Scholar]
- 25.Matsumoto M., Ogawa W., Akimoto K., Inoue H., Miyake K., Furukawa K., Hayashi Y., Iguchi H., Matsuki Y., Hiramatsu R. et al. (2003) PKClambda in liver mediates insulin-induced SREBP-1c expression and determines both hepatic lipid content and overall insulin sensitivity. J. Clin. Invest, 112, 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimomura I., Matsuda M., Hammer R.E., Bashmakov Y., Brown M.S., Goldstein J.L. (2000) Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol. Cell, 6, 77–86. [PubMed] [Google Scholar]
- 27.Horton J.D., Goldstein J.L., Brown M.S. (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest., 109, 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guilherme A., Virbasius J.V., Puri V., Czech M.P. (2008) Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol., 9, 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tschöp M.H., Speakman J.R., Arch J.R.S., Auwerx J., Brüning J.C., Chan L., Eckel R.H., Farese R.V., Galgani J.E., Hambly C. et al. (2012) A guide to analysis of mouse energy metabolism. Nat. Methods, 9, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.You S.H., Lim H.W., Sun Z., Broache M., Won K.J., Lazar M.A. (2013) Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat. Struct. Mol. Biol., 20, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samaco R.C., Fryer J.D., Ren J., Fyffe S., Chao H.T., Sun Y., Greer J.J., Zoghbi H.Y., Neul J.L. (2008) A partial loss of function allele of Methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum. Mol. Genet, 17, 1718–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chahrour M., Zoghbi H.Y. (2007) The story of Rett syndrome: from clinic to neurobiology. Neuron, 56, 422–437. [DOI] [PubMed] [Google Scholar]
- 33.Gong M., Liu J., Sakurai R., Corre A., Anthony S., Rehan V.K. (2015) Perinatal nicotine exposure suppresses PPARγ epigenetically in lung alveolar interstitial fibroblasts. Mol. Genet. Metab., 114, 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hara M., Takahashi T., Mitsumasu C., Igata S., Takano M., Minami T., Yasukawa H., Okayama S., Nakamura K., Okabe Y. et al. (2015) Disturbance of cardiac gene expression and cardiomyocyte structure predisposes Mecp2-null mice to arrhythmias. Sci. Rep, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu B., Gharaee-Kermani M., Wu Z., Phan S.H. (2011) Essential role of MeCP2 in the regulation of myofibroblast differentiation during pulmonary fibrosis. Am. J. Pathol., 178, 1500–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li C., Jiang S., Liu S.Q., Lykken E., Zhao L., Sevilla J., Zhu B., Li Q.J. (2014) MeCP2 enforces Foxp3 expression to promote regulatory T cells’ resilience to inflammation. Proc. Natl. Acad. Sci. USA, 111, E2807–E2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murphy C., Ledmyr H., Ehrenborg E., Gåfvels M. (2006) Promoter analysis of the murine squalene epoxidase gene. Identification of a 205 bp homing region regulated by both SREBP’S and NF-Y. Biochim. Biophys. Acta, 1761, 1213–1227. [DOI] [PubMed] [Google Scholar]
- 38.Bienvenu T., Chelly J. (2006) Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat. Rev. Genet., 7, 415–426. [DOI] [PubMed] [Google Scholar]
- 39.Stancheva I., Collins A.L., Van den Veyver I.B., Zoghbi H., Meehan R.R. (2003) A mutant form of MeCP2 protein associated with human Rett syndrome cannot be displaced from methylated DNA by notch in Xenopus embryos. Mol. Cell, 12, 425–435. [DOI] [PubMed] [Google Scholar]
- 40.Martinowich K., Hattori D., Wu H., Fouse S., He F., Hu Y., Fan G., Sun Y.E. (2003) DNA Methylation-Related Chromatin Remodeling in Activity-Dependent Bdnf Gene Regulation. Science, 302, 890–893. [DOI] [PubMed] [Google Scholar]
- 41.Karagianni P., Wong J. (2007) HDAC3: taking the SMRT-N-CoRrect road to repression. Oncogene, 26, 5439–5449. [DOI] [PubMed] [Google Scholar]
- 42.Bhaskara S., Chyla B.J., Amann J.M., Knutson S.K., Cortez D., Sun Z.W., Hiebert S.W. (2008) Deletion of Histone Deacetylase 3 Reveals Critical Roles in S Phase Progression and DNA Damage Control. Mol. Cell, 30, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mergenthaler P., Lindauer U., Dienel G.A., Meisel A. (2013) Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci., 36, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banks W.A., Owen J.B., Erickson M.A. (2012) Insulin in the brain: there and back again. Pharmacol. Ther, 136, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Calfa G., Hablitz J.J., Pozzo-Miller L. (2011) Network hyperexcitability in hippocampal slices from Mecp2 mutant mice revealed by voltage-sensitive dye imaging. J. Neurophysiol., 105, 1768–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang W., Peterson M., Beyer B., Frankel W.N., Zhang Z. (2014) Loss of MeCP2 from forebrain excitatory neurons leads to cortical hyperexcitation and seizures. J. Neurosci. Off. J. Soc. Neurosci., 34, 2754–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kron M., Howell C.J., Adams I.T., Ransbottom M., Christian D., Ogier M., Katz D.M. (2012) Brain Activity Mapping in Mecp2 Mutant Mice Reveals Functional Deficits in Forebrain Circuits, Including Key Nodes in the Default Mode Network, that are Reversed with Ketamine Treatment. J. Neurosci. Off. J. Soc. Neurosci, 32, 13860–13872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lundgaard I., Li B., Xie L., Kang H., Sanggaard S., Haswell J.D.R., Sun W., Goldman S., Blekot S., Nielsen M. et al. (2015) Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nat. Commun., 6, 6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conti V., Gandaglia A., Galli F., Tirone M., Bellini E., Campana L., Kilstrup-Nielsen C., Rovere-Querini P., Brunelli S., Landsberger N. (2015) MeCP2 affects skeletal muscle growth and morphology through non cell-autonomous mechanisms. PLoS One, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fyffe S.L., Neul J.L., Samaco R.C., Chao H.T., Ben-Shachar S., Moretti P., McGill B.E., Goulding E.H., Sullivan E., Tecott L.H. et al. (2008) Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron, 59, 947–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X., Lacza Z., Sun Y.E., Han W. (2014) Leptin resistance and obesity in mice with deletion of methyl-CpG-binding protein 2 (MeCP2) in hypothalamic pro-opiomelanocortin (POMC) neurons. Diabetologia, 57, 236–245. [DOI] [PubMed] [Google Scholar]
- 52.Sticozzi C., Belmonte G., Pecorelli A., Cervellati F., Leoncini S., Signorini C., Ciccoli L., De Felice C., Hayek J., Valacchi G., (2013) Scavenger receptor B1 post-translational modifications in Rett syndrome. FEBS Lett., 10.1016/j.febslet.2013.05.042. [DOI] [PubMed] [Google Scholar]
- 53.Cooke D.W., Naidu S., Plotnick L., Berkovitz G.D. (1995) Abnormalities of thyroid function and glucose control in subjects with Rett syndrome. Horm. Res., 43, 273–278. [DOI] [PubMed] [Google Scholar]
- 54.Cronk J.C., Derecki N.C., Ji E., Xu Y., Lampano A.E., Smirnov I., Baker W., Norris G.T., Marin I., Coddington N. et al. (2015) Methyl-CpG binding protein 2 regulates microglia and macrophage gene expression in response to inflammatory stimuli. Immunity, 42, 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Felice C., Della Ragione F., Signorini C., Leoncini S., Pecorelli A., Ciccoli L., Scalabrì F., Marracino F., Madonna M., Belmonte G. et al. (2014) Oxidative brain damage in Mecp2-mutant murine models of Rett syndrome. Neurobiol. Dis., 68, 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsuishi T., Urabe F., Percy A.K., Komori H., Yamashita Y., Schultz R.S., Ohtani Y., Kuriya N., Kato H. (1994) Abnormal carbohydrate metabolism in cerebrospinal fluid in rett syndrome. J. Child Neurol., 9, 26–30. [DOI] [PubMed] [Google Scholar]
- 57.Müller M., Can K. (2014) Aberrant redox homoeostasis and mitochondrial dysfunction in Rett syndrome. Biochem. Soc. Trans., 42, 959–964. [DOI] [PubMed] [Google Scholar]
- 58.Pfrieger F.W., Ungerer N. (2011) Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res., 50, 357–371. [DOI] [PubMed] [Google Scholar]
- 59.Kanungo S., Soares N., He M., Steiner R.D. (2013) Sterol metabolism disorders and neurodevelopment—an update. Dev. Disabil. Res. Rev., 17, 197–210. [DOI] [PubMed] [Google Scholar]
- 60.Schmidt D., Wilson M.D., Spyrou C., Brown G.D., Hadfield J., Odom D.T. (2009) ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods San Diego Calif, 48, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guy J., Gan J., Selfridge J., Cobb S., Bird A. (2007) Reversal of neurological defects in a mouse model of Rett syndrome. Science, 315, 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.