

Abstract
In contrast to white adipose tissue (WAT) that stores energy in the form of triglycerides, brown adipose tissue (BAT) dissipates energy by producing heat to maintain body temperature by burning glucose and fatty acids in a process called adaptive thermogenesis. The presence of an inducible thermogenic adipose tissue, and its beneficial effects for maintaining body weight and glucose and lipid homeostasis, have raised intense interest in understanding the regulation of thermogenesis. Elucidating the regulatory mechanisms underlying the thermogenic adipose program may provide excellent targets for therapeutics against obesity and diabetes. Here, we review recent research on the role of epigenetics in the thermogenic gene program, focusing on DNA methylation and histone modifications.
Keywords: adipose, thermogenesis, browning, epigenetic, DNA methylation, Histone modifications
Brown and beige adipocytes, and adaptive thermogenesis
Brown adipose tissue (BAT) (see glossary) is an adipose depot with the capacity to regulate body temperature via adaptive thermogenesis. BAT is enriched in mitochondria whose inner membrane harbors Uncoupling protein 1 (UCP1) that uncouples oxidative respiration from ATP synthesis to dissipate energy in the form of heat. However, in addition to BAT, upon cold exposure or activation of β-adrenergic receptor (β-AR), clusters of adipocytes that express UCP1 can be detected in white adipose tissue (WAT) [1], so called beige or brite (hereafter beige) adipocytes [2, 3]. While human infants are known to have BAT to maintain body temperature, it was thought to undergo rapid involution in early childhood. Recently, however, substantial depots of thermogenic adipose tissue have been detected in adult humans upon cold exposure or activation of β-AR [4–6]. Moreover, increased mass of these depots has been associated with lower body weight and improved glucose [7] and lipid [8, 9] homeostasis.
The reversible process by which beige cells in WAT are induced to express UCP1 and other BAT-enriched genes to develop thermogenic capacity is referred to as browning of WAT [10]. Beige adipocytes can express UCP1 at levels comparable to those found in classic brown adipocytes, and become thermogenic. However, they do not appear to arise from the same cell lineage [11] and, to date, it is still debated whether beige adipocytes derive from de novo differentiation of precursors [12, 13], or arise from transdifferentiation of mature white adipocytes or reactivation of “masked” beige cells [14]. Despite the controversy in their distinct developmental origin, accumulating evidence suggests that brown and beige adipocytes share common transcriptional regulators involved in the activation of thermogenic genes [15]. Considering the potential beneficial metabolic effects of enhancing energy expenditure and glucose homeostasis, efforts are directed toward elucidating the signaling pathways and transcriptional mechanisms that regulate the thermogenic adipose program.
Transcriptional activation and signaling in thermogenic gene induction
Identifying effectors involved in the commitment and differentiation of precursors to brown or beige lineage, and the induction of the thermogenic program in mature cells is a compelling but challenging area of research. The co-regulator PR domain containing 16 (PRDM16), which is enriched in BAT compared to WAT, was the first effector reported to be critical for thermogenic gene program. PRDM16 has been shown to be recruited to the promoter regions of BAT-enriched genes, such as UCP1, Deionidase 2 (Dio2) or Cell death-inducing DNA fragmentation factor alpha-like effector A (Cidea), by various transcription factors, including Peroxisome Proliferator Activated Receptor Gamma (PPARγ) [16], PPARγ coactivator 1-alpha (PGC1α) [17], CAAT-enhancer-binding protein β (C/EBPβ) [18] and Zinc finger protein 516 (Zfp516) [28], to potentiate transcriptional activation. In addition, PRDM16 has been reported to enhance transcription through recruitment of the MED1/Mediator complex to BAT-enriched genes [19, 20]. Transgenic mice overexpressing PRDM16 in all adipose depots displayed beige cells in their WAT and had increased energy expenditure [21]. Conversely, whereas deletion of PRDM16 in all adipose depots impaired beige adipocyte function [22], deletion in brown adipose precursor cells using Myf5-Cre demonstrated its role in BAT maintenance in adults, but not in BAT development [23]. Although PPARγ is critical for both brown and white adipogenesis, genome-wide chromatin immunoprecipitation combined with sequencing (ChIP-seq) in BAT and WAT revealed that approximately 10% of PPARγ binding sites are depot-specific [24]. BAT-specific targets included UCP1, PPARα and PRDM16. Treatment with PPARγ agonists in mice induces UCP1 and other thermogenic genes by stabilization of PRDM16, thus promoting browning of WAT [25]. In addition, Early B-cell factor 2 (EBF2), a transcription factor which is expressed at higher level in BAT and beige adipocytes compared to white adipocytes, has been shown to bind at/or near the PPARγ sites of the BAT-enriched genes to potentiate PPARγ binding [24] and to activate the thermogenic gene program [24, 26]. The critical role of EBF2 was evidenced by the impaired BAT development in EBF2 knockout (KO) embryos [24], whereas development of beige adipocytes in WAT was enhanced in EBF2 transgenic mice [24, 26]. Interferon regulatory factor-4 (IRF4), a transcription factor which is induced by cold exposure in BAT and WAT and by cAMP treatment in primary brown adipocytes [27], increases expression of, and interacts with, PGC1α to activate thermogenic genes in brown adipocytes [27]. IRF4 overexpression in all adipose depots enhanced thermogenesis, hence causing resistance to diet induced obesity. Deletion of IRF4 using UCP1-Cre blocked browning of WAT and decreased thermogenesis and energy expenditure [27]. Moreover, Zfp516, a BAT-enriched_zinc-finger transcription factor, has been identified by screening known and putative transcription factors at a global level for activation of UCP1 promoter. Zfp516 is induced upon cold exposure or β-adrenergic stimulation and activates numerous BAT genes, including UCP1 and PGC1α, to induce thermogenic gene program during BAT development and also browning of WAT [28]. Interestingly, Zfp516, not only directly interacts with PRDM16, but also with Lysine-Specific histone Demethylase 1 (LSD1), which contributes to thermogenic gene activation by removing repressive marks on H3K9 residues [28, 29].
It is well established that thermogenesis is induced by cold exposure through activation of β-ARs via sympathetic stimulation [10]. The p38 mitogen-activated protein Kinase (MAPK) and ATF2/CREB pathways are known to be downstream effectors of β-AR stimulation in brown adipocytes [28, 30]. Many of the transcription factors and coregulators that are involved in activation of thermogenic genes, such as PGC1α [31], IRF4 [27], Zfp516 [28] and LSD1 [29, 32], are all induced in response to cold or β-AR stimulation also. However, PRDM16 is not cold inducible [17].
While sympathetic stimulation of thermogenesis involves β-AR, growth factors, such as bone morphogenic factor 7 (BMP7) [33] and fibroblast growth factor 21 (FGF21) [34] induce thermogenesis through β-AR-independent mechanisms. BMP7 induces brown adipogenesis by activating the BAT gene program. BMP7 is the first factor reported to promote the commitment of mesenchymal progenitors into brown adipocytes in vitro [33]. BMP7 deletion in mice showed its requirement for BAT development and thermogenesis in vivo [33]. Notably, BMP7 is secreted also by brown adipocytes and thus can act through an autocrine loop [35]. FGF21, secreted mainly by the liver, is released also by BAT [34] and by WAT [36] upon cold exposure in mice. FGF21 activates thermogenic genes and thus FGF21 KO mice exhibit impaired cold tolerance and reduced browning of WAT [34, 36].
Epigenetic modifications for activation of thermogenic genes and biogenesis of thermogenic adipocytes
Epigenetic regulation is a heritable mechanism affecting gene transcription without changes in DNA sequence. Epigenetic regulation includes DNA modifications, mainly DNA methylation (see Box 1), and histone modifications (see Box 2), as well as regulation by small and long non-coding RNAs. While exploring new regulators of thermogenic adipocytes, the role of epigenetic regulation on the BAT gene program and thermogenesis has been uncovered. The study of epigenetics in thermogenic tissue represents an emerging field that will help unveil the molecular mechanisms underlying activation of the thermogenic adipose program and also adipose tissue plasticity. This review focuses on the contribution of DNA methylation and histone modifications in the regulation of thermogenic adipocytes.
Text box 1. DNA methylation.
DNA methylation is a covalent modification occurring on position 5 of cytosine (C) within CpG dinucleotides of the DNA sequence [89]. In mammals, 80% of CpG sites is estimated to be methylated. DNA methylation patterns are established by de novo methyltransferases 3 (DNMT3) and maintained by methyltransferase DNMT1. DNA demethylation can occur by two modes. First, DNA methylation can be lost by DNA replication. Second, ten eleven translocation (TET) family of iron-dependent dioxygenases [89] is involved in oxidative demethylation by converting 5-methylcytosine to 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxylcytosine, which then can be excised out by the thymine-DNA glycosylase (TDG), coupled with the base excision repair (BER) for reversion to cytosine [89]. Moreover, readers of these oxidized DNA demethylation intermediates may have a function independent of demethylation.
CpG methylation is generally associated with gene silencing, whereas demethylation is thought to be permissive for transcription. Methylation and demethylation of CpG sites on DNA have been proposed as regulatory mechanism for tissue-specific gene expression and cell fate determination and differentiation [90]. Methylated DNA provides binding sites for Methylated CpG binding proteins (MBDs and MeCP2) and BTB/POZ family of proteins that are presumed to mediate the effects of DNA methylation, and these proteins are often associated with chromatin remodeling or transcription repressor complexes. However, certain DNA-binding transcription factors have been identified to bind methylated DNA for transcriptional activation. Interestingly, there is a complex interplay between DNA methylation and histone modifications for the regulation of chromatin remodeling and gene regulation [50, 89]. This crosstalk involves interactions between epigenetic readers, histone modifying enzymes and DNA methylation marks.
Text box 2. Histone modifications.
The most studied epigenetic mechanisms are histone modifications that affect chromatin organization. The first level of chromatin organization involves nucleosomal particles containing histone octamers that consist of two copies of each core histones, H2A, H2B, H3B, and H4, around which 145–147 base pairs (bp) of DNA is wrapped. Nucleosomal particles are maintained by linker histone H1 that stabilizes the chromatin structure thereby forming a nucleosome, the fundamental repeated unit of chromatin. The N-terminal tail region of histone H3 and H4 is flexible and protrudes from the nucleosome, thus is more accessible and, as such, undergoes posttranslational modifications that impact transcription through two mechanisms. First, directly, histone tail modifications affect interactions between nucleosomes and DNA and thus alter chromatin condensation and structure. Decompacted chromatin is transcriptionally active, whereas compacted chromatin is less accessible and thus transcriptionally inactive. In addition, histone modifications have been shown to affect binding of various chromatin-associated effectors.
Acetylation, the transfer of an acetyl group to ɛ-amino groups of lysine (K) side chains of histones, decreases the positive charge thus weakens electrostatic interactions between histones and DNA, and reduces chromatin compaction. For example, H3K9 and H3K27 acetylation are activation marks [91]. Furthermore, acetylated lysines bind bromodomains that are often found in chromatin remodeling enzymes, thus directing their recruitment. Histone methylation mainly occurs on ɛ-amino groups of specific lysine (K) and at the terminal guanidinyl group of arginine (R) residues. Lysines may be mono-, di- or tri-methylated, whereas arginines may be mono-, symmetrically or asymmetrically di-methylated. Unlike histone acetylation, histone methylation does not alter the electronic charge of histone proteins but rather affects DNA accessibility to the binding of chromatin modifiers, transcription factors or epigenetic readers. For instance, effector proteins bearing a chromodomain or a plant homeodomain (PHD) are differentially recruited to the chromatin to either activate or repress transcription, depending on the number of methyl groups added. Thus, H3K4me2/3 and H3K9me2/3 at the tail region of histone H3 are best known as activation and repressive marks for transcription, respectively [92]. However, the combined effects of histone modifications are context-dependent, and are also determined by chromatin effectors that specifically bind these modifications. While much effort has been devoted to histone tail modifications, histone core regions, including those on the lateral surface of the nucleosome have been reported to be modified, as well. These modifications appear to affect transcription and nucleosome stability (reviewed in [93]).
DNA methylation
Previous in vitro studies suggested a role for DNA methylation (see Box 1) in the commitment of precursor cells into the adipocyte lineage [37, 38]. Treatment of mouse fibroblasts with an inhibitor of DNA methylation was shown to induce differentiation of cells into the adipose lineage. More recently, a genome-wide DNA methylation profiling showed noticeable changes during adipocyte differentiation of 3T3-L1 cells, suggesting a dynamic regulatory mechanism for adipogenesis [39]. In addition, global methylation profiles of both white and brown adipocyte precursors showed a distinguishable methylation pattern [40]. However, few studies have demonstrated a clear relationship between DNA methylation and gene expression in the context of adipogenesis. During differentiation, demethylation at the promoter regions of leptin and glucose transporter type 4 (Glut4), two late markers of both WAT and BAT differentiation, has been shown to correlate with gene activation [41, 42]. Moreover, in rodents under high fat diet, DNA methylation at the leptin promoter region has been associated with lower circulating leptin levels and higher body weight [43]. In humans, DNA methylation at specific loci of markers for low or high response to caloric restriction was correlated with weight changes [44]. However, genome-wide studies comparing DNA methylation profiles between white and brown (or beige) adipocytes are yet to be reported. Nevertheless, several studies demonstrated the potential role of DNA methylation for regulation of BAT-enriched and thermogenic genes. An early study exploring the silencing of UCP1 involving the corepressor receptor-interacting protein 140 (RIP140) [45] suggested that DNA methylation, along with histone H3K4 modification described in the later section of this review, are involved in transcriptional repression in white adipocytes [46]. Indeed, the methylation status of the UCP1 promoter region has been shown to be important for its tissue-specific and regulated expression in BAT in comparison to visceral WAT. In addition, cold exposure has been reported to induce chromatin remodeling at the UCP1 promoter region toward a more active state, consistent with the increased UCP1 expression [47]. Furthermore, upon ablation of Jak kinase Tyk2 in mice, DNA hypermethylation was observed in addition to decreased histone H3K4 trimethylation (H3K4me3) at the UCP1 and Cidea promoter regions, correlating with reduced gene expression and defective thermogenesis [48]. More recently, studies on global epigenetic marks showed an increased global DNA methylation in addition to decreased histone acetylation (H3K23Ac) correlating with the repression of thermogenic genes in BAT of hibernating rodents, limiting energy expenditure [49]. Although these studies show evidence that variations in DNA methylation are associated with thermogenic gene expression, further investigation is needed to understand how fate of thermogenic cells is controlled through DNA methylation, and to identify specific epigenetic effectors that are involved. It would also be important to study interplay between gene expression and environmental conditions that may affect DNA methylation in brown and beige adipocytes in vivo.
Histone modifications
In addition to DNA methylation, histone modifications (see Box 2) represent an important mode of epigenetic regulation of transcription in directing cell lineage specification and establishment of tissue-specific gene expression [50]. Here, we focus our discussion on histone acetylation and methylation. Specific epigenetic writers and erasers for histone modifications and readers that recognize these modifications govern transcription, and as such, contribute to regulation of the thermogenic adipose program (See key Figure).
Key Figure. Epigenetic regulation of thermogenic adipose program.
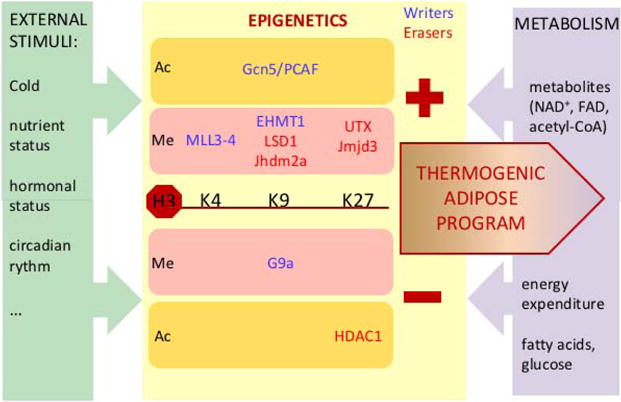
Both the expression and activity of epigenetic effectors are modulated by environmental signals and external stimuli such as cold temperature and nutritional or hormonal status. Chromatin-modifying enzymes add (writers) or remove (erasers) epigenetic marks on lysine (K) residues of histone H3, such as histone methylation (Me) or acetylation (Ac), thereby affecting transcription of the thermogenic genes and also metabolic enzymes in adipocytes. Thus, epigenetics allows the integration of various external stimuli leading to activation (+) or repression (−) of the thermogenic adipose program that contributes to energy homeostasis. Intracellular metabolic pathways regulate, in a feedback loop, epigenetics at the level of metabolites such as FAD, NAD+ and acetyl-CoA but also thermogenesis by impacting on fatty acids and glucose availability. However, more effort is required to further elucidate epigenetic molecular mechanisms and their specific targets and validate their physiological relevance for thermogenic adipose program.
Histone acetylation
Histone acetylation is dynamically regulated by histone acetyltransferases (HATs) and deacetylases (HDACs) (see Box 3). Histone acetyltransferases, Gcn5 and its homolog PCAF, were found to be critical for brown adipocyte differentiation in culture by promoting elongation of PPARγ transcript and by facilitating recruitment of RNA polymerase II to PRDM16 promoter region [51]. Thus, downregulating HDACs has shown to stimulate adipocyte differentiation in culture [52]. Treating genetically obese mice with class I HDAC inhibitors was shown to induce PGC1α and mitochondrial biogenesis, enhancing oxidative metabolism and increasing BAT mass and browning of WAT, likely through inhibition of HDAC3 [53]. However, others reported promotion of the thermogenic program by class I HDAC inhibitors, via inhibition of HDAC1 which was found at lower level in BAT compared to WAT, and was suppressed upon cold exposure or β3-adrenergic stimulation [54]. Moreover, β-adrenergic stimulation of brown adipocytes in culture induced dissociation of HDAC1 from BAT-specific genes, resulting in increased H3K27 acetylation, followed by H3K27me3 demethylation thus allowing transcription [54]. Similarly, ablation of a class II deacetylase, HDAC9, in mice increased energy expenditure and adaptive thermogenesis by inducing beige adipocyte formation in subcutaneous WAT [55]. Collectively, accumulating evidence demonstrated a role of histone acetylation in the transcriptional control of BAT gene program and thermogenesis. However, recruitment of specific HATs or HDACs and regulation of the thermogenic gene program, and the molecular consequences of the acetylation of specific lysine residues need to be defined to better understand the underlying molecular details.
Text box 3. Histone modifying enzymes.
Histones modifications are catalyzed by histone modifying enzymes that are recruited to the chromatin and are often part of larger protein complexes.
Histone acetylation is a dynamically regulated process involving HATs and HDACs [91]. HATs catalyze the transfer of an acetyl group onto lysine residues, neutralizing their positive charge, to affect electrostatic interactions leading to the destabilization of chromatin structure. Decompacted chromatin structure allows transcription and, as such, HATs are generally associated with transcriptional activation. Conversely, HDACs catalyze removal of acetylation marks restoring the compacted structure of chromatin and predominantly repressing transcription. HATs are diverse and are classified into subgroups including HAT1, Gcn5/PCAF, MYST and CBP/p300 families. Notably, the HAT1 group acetylates free histones for their organization into chromatin, however, thereafter, these marks are removed. There are four classes of HDAC: class I (HDAC1-3,8), class II (HDAC4-7,9-10), class IV containing only HDAC 11 and lastly class III also referred to as Sirtuins (Sirt1-7) that, unlike the other classes, uses NAD+ as a cofactor.
Histone methylation mainly occurs on specific lysine and arginine residues and is catalyzed by HMTs, while the reverse reaction is catalyzed by histone demethylases (reviewed in [94]). HMTs are organized in two families; lysine-specific and arginine-specific methyltransferases that both utilize S-adenosyl methionine (SAM) as a cofactor. EHMT1, G9a (also known as EHMT2) and MLL3-4, mentioned in the review, belong to the lysine-specific methyltransferase family. HMTs exhibit relative specificity towards the residue they target (H3K4 or H3K9 for instance) and the degree of methylation they catalyze (mono, di or tri-methylation). Histone demethylases are also divided into two families. The first family referred to as KDM1 comprises only two members LSD1 (or KDM1A) and LSD2 (or KDM1B), which are FAD-dependent amino oxidase enzymes. Using FAD as a cofactor, these lysine-specific histone demethylase enzymes target mono and di -methylated lysine K4 and K9 on histone H3, where LSD1 acts on both H3K4 and H3K9, whereas LSD2 targets only H3K4. The second, larger, family, called Jumonji-type histone demethylases, is characterized by a conserved Jumonji C (JmjC) catalytic domain and comprised of the KDM2 to KDM7 subgroups. These enzymes are 2-oxoglutarate oxygenases and depend on iron (Fe) as cosubstrate. Jmjd3, Jmjd1a and UTX mentioned in this review belong to this family. Both histone methyltransferases and demethylases exhibit specificity due to intrinsic enzyme properties and specific interaction with regulatory complexes.
Unlike the class I and II HDACs the class III deacetylases Sirtuin 1 (Sirt1) and Sirt3, may affect thermogenic function, not by histone deacetylation, but by deacetylating PGC1α. Thus, in contrast to the activated thermogenic function by inhibition of HDAC activity described above, mice overexpressing Sirt1 exhibited enhanced BAT activity and thermogenesis, resulting in increased energy expenditure and improved glucose homeostasis [56]. Notably, this effect did not arise from enhanced brown adipogenesis, but from higher transcriptional response of mature cells to β3-adrenergic stimulation. In this regard, the cAMP-PKA pathway induced both PGC1α expression and Sirt1 deacetylase activity independent of NAD+, leading to deacetylation of PGC1α that, in turn, activated genes associated with fatty acid oxidation and energy expenditure [57]. In addition, Sirt3 overexpression in brown adipocytes in culture increased mitochondrial respiration and expression of UCP1 and PGC1α [58]. However, the sirtuin family of deacetylases has been shown to have specific HDAC activity. For example, H3K57 is a target of Sirt1/Sirt2 [59]. Therefore, the potential function of Sirtuin family in epigenetic regulation of thermogenic program via histone deacetylation needs to be further examined.
Histone methylation
Several histone methyltransferases (HMTs) and demethylases that govern histone methylation status (see Box 3) have been implicated in BAT development and the thermogenic gene program. The histone methyltransferase, MLL3 (KMT2C), and its paralog MLL4 (KMT2D) may function at the enhancer regions for cell type specific gene expression by catalyzing methylation of H3K4. MLL3 ablation in mice caused changes in gene expression, in both WAT and BAT. In BAT, more than 500 genes were altered, 46% of them being involved in metabolism, suggesting a potential role of H3K4 methylation status and MLL3 in the regulation of BAT function [60]. Notably, these mice exhibited lower body weight, improved glucose tolerance and insulin sensitivity, and increased energy expenditure. More recently, MLL4 was identified to be a methyltransferase promoting H3K4 mono and di -methylation (H3K4me1/2), having partial functional redundancy with MLL3 and required for adipogenesis. MLL4 ablation in cells markedly decreased H3K4me1/2, as well as H3K27 acetylation, at the enhancer regions for tissue specific gene expression [61].
The H3K9 methylation status has also been shown to affect thermogenesis in various studies on H3K9 demethylases. Firstly, two independent studies have reported a role for the histone lysine demethylase Jhdm2a (also known as Jmjd1a or Kdm3a) in regulating metabolic genes and energy homeostasis. Mice carrying a disruption of the Jhdm2a gene exhibited obesity and hyperlipidemia due to impaired fatty acid metabolism [62] and reduced energy expenditure [63]. Their BAT function was altered with decreased expression of mitochondrial genes, causing reduction in oxygen consumption. The authors reported that induction and binding of Jhdm2a at PPAR Responsive Elements (PPREs) upon β-adrenergic stimulation not only decreases H3K9 demethylation but also facilitates the recruitment of PPARγ and PGC1α at the UCP1 and fatty acid oxidation gene promoters. In addition, upon β-adrenergic stimulation, Jhdm2a is phosphorylated at serine 265 (S265) by PKA that increases its interaction with the SWI/SNF chromatin-remodeling complex and DNA-bound PPARγ, providing an additional role of Jhdm2a besides H3K9 demethylation [64]. Furthermore, via direct interaction with Zfp516, the histone demethylase LSD1 is recruited to activate thermogenic genes and to promote thermogenic adipose program in vivo [29]. LSD1 ablation in mice using UCP1-Cre impaired BAT development and BAT function, resulting in the “whitening” of BAT. Besides, compromising the thermogenic function, LSD1 ablation in all adipose tissues using adiponectin-Cre prevented browning of WAT. While LSD1 is known to be involved in both repression and activation of transcription, LSD1 specifically induces demethylation of H3K9me1/2 for transcriptional activation of the thermogenic adipose program in adipose tissue through its interaction with Zfp516 [29].
Euchromatic histone N-lysine methyltransferase 1 (EHMT1), that catalyzes H3K9 di or tri-methylation (H3K9me2/3), has also been reported to be part of PRDM16 complex to control BAT cell fate. Ablation of EHMT1 in brown adipocytes caused loss of brown adipocyte characteristics, whereas EHMT1 expression promoted BAT selective thermogenic program via stabilization of PRDM16 protein [65]. Notably, adipose-specific deletion of EHMT1 driven by adiponectin-Cre in mice led to a marked reduction of BAT-mediated adaptive thermogenesis, leading to obesity and systemic insulin resistance. This study suggests EHMT1 as an essential switch that may control brown adipose cell fate and energy homeostasis. Notably, expression levels of BAT-enriched genes in human brown adipocytes were correlated with the formation of PRDM16-EHMT1 complex [66]. Additionally, the repressive mark H3K9me2 induced by methyltransferase G9a was selectively enriched on the entire PPARγ locus. Interestingly, both G9a expression and H3K9me2 mark have been reported to decrease during adipogenesis, and deletion of G9a or inhibition of its methyltransferase activity promoted brown adipogenesis [67].
Tetratricopeptide repeat on chromosome X (UTX), a histone demethylase for H3K27me2/3, is induced during brown adipocyte differentiation and by cold exposure in both BAT and WAT [68]. Upon β-adrenergic stimulation of brown adipocytes, UTX was recruited to UCP1 and PGC1α promoters to decrease H3K27me3 and to interact with the HAT CBP, that increased H3K27 acetylation, thereby inducing a switch from a repressive to an active state for thermogenic gene activation. Additionally, mice overexpressing Jmjd3, a demethylase targeting H3K27me3, were shown to have reduced body weight, whereas overexpression of dominant negative Jmjd3 reduced cold tolerance [69]. H3K27me3 was reported to distinguish a subset of BAT-enriched genes from common adipose genes in both brown and white preadipocytes. Moreover, recruitment of Jmjd3 for H3K27me3 demethylation was required for BAT gene expression and browning of WAT. Thus, the authors proposed that H3K27me3 may be an epigenetic mark discriminating BAT from WAT lineage, and Jmjd3 may regulate fate between BAT and WAT [69].
Altogether, a growing number of studies show a role for histone modifications in regulating the thermogenic adipose program and plasticity of WAT (See key Figure). However, it will require not only better understanding of the molecular mechanisms but also validation of their physiological relevance for brown and beige adipocytes in vivo. Epigenetic effectors may represent relevant targets in brown and beige adipocytes to increase energy expenditure and prevent obesity and development of related metabolic diseases, including diabetes and dyslipidemia.
Regulation of epigenetic modifications
Histone modifications are controlled at various levels, such as expression levels of chromatin modifiers, their posttranslational modifications that may affect their activities or interactions with specific proteins for recruitment, as well as by the availability of intracellular metabolites that function as co-substrates or cofactors.
Expression, posttranslational modifications and recruitment of chromatin modifiers
Several reports documented that, in adipose tissue, particularly in BAT, the expression of enzymes involved in posttranslational modifications of histones is altered during differentiation. More specifically, the demethylase Jhdm2a is induced by β-AR signaling in brown adipocytes [62]. LSD1 is also induced by cold exposure in both WAT [32] and BAT [29], as well as during brown adipocyte differentiation [29]. Sirt3 expression has been reported to be higher in BAT compared to WAT, to be induced by cold-exposure and caloric restriction, but to be downregulated in BAT of genetically obese mice [58].
The histone deacetylase Sirt1 is phosphorylated and activated in BAT after acute cold exposure through the cAMP/PKA pathway. Phosphorylation of serine 434 (S434) at the catalytic domain increases Sirt1 deacetylase activity, independent of NAD+, leading to deacetylation of PGC1α [57]. Casein kinase 2 (CK2) phosphorylates and activates class I HDACs in white adipocytes where it inhibits thermogenesis [70].
Interestingly, transient hyperglycemia has been reported to promote the recruitment of LSD1 to the NFκB p65 promoter in vitro, inversely correlating with H3K9 methylation at the promoter region [71]. In brown adipocytes, by direct interaction with cold-inducible Zfp516, LSD1 is recruited to the UCP1 promoter where it functions as a co-activator by decreasing H3K9 methylation. Therefore, Zfp516 knockdown prevents the binding of LSD1 at the UCP1 promoter region, which is required for transcriptional activation [29].
Metabolite availability
The availability of metabolites, such as acetyl-CoA, FAD and NAD+ that serve as co-substrates or cofactors for histone modifying enzymes, offers an additional level of control of epigenetic modifications. For example, HATs use as co-substrate, acetyl-CoA, whose level is known to be sensitive to glucose availability, to affect histone acetylation during white adipocyte differentiation [72]. Thus, physiological changes in nutrient levels may regulate histone acetylation that in turn, affects metabolic and thermogenic gene expression. In this regard, a recent study established that N-acetyl aspartate (NAA), generated by acetylation of aspartate using acetyl-CoA, regulates thermogenic gene expression and lipolysis. NAA affects acetyl-CoA levels and thus histone acetylation in brown adipocytes [73]. Interestingly, in BAT, an increase in NAD+ levels affects Sirt1 activity to activate transcription of thermogenic genes, including PGC1α, and enhances oxidative metabolism [74]. Besides, cellular NAD+ levels can be affected by caloric restriction or by high fat diet [75, 76]. More recently, nicotinamide N-methyltransferase (Nnmt) has been identified as a regulator of adiposity and energy expenditure in WAT, decreasing S-adenosine-methionine (SAM) and NAD+ levels, that affect histone methylation and Sirt1 activity, respectively [77]. Furthermore, during white adipogenesis, the cellular content of FAD was reported to increase markedly to affect LSD1-dependent transcription [78]. These studies indicate that cellular metabolite levels, that reflect the metabolic status of the cell affect the epigenome, hence altering gene regulation to contribute to adaptations to the environment. Thus, acetyl-CoA and FAD are examples of metabolites whose intracellular concentrations are affected by glucose and fatty acid metabolism, and that also control epigenetic regulation. Moreover, recent investigations have uncovered other types of histone modification [reviewed in [79]], which require as co-substrates metabolites generated during fatty acid and amino acid metabolism such as butyrate, propionate, glutarate or hydroxybutyrate, and predicting to also participate in epigenetic regulation. By serving as co-substrates or cofactors for epigenetic effectors, metabolites could provide a feed-back loop in regulating the expression of metabolic enzymes and other proteins to adapt to physiological changes [80]. In adipocytes, such interplay may represent a finely-controlled mechanism integrating information from different environmental signals to regulate gene expression (See key Figure).
Epigenetics of thermogenic adipose tissue in humans
Genome-scale DNA methylation analysis from human DNA samples indicated that variably methylated regions exhibited covariation with body mass index (BMI), and that those regions were located near or in genes known to regulate body weight [81]. Few human studies have investigated changes in DNA methylation in adipose tissue samples as marker of epigenetic adaptation. Significant differences in DNA methylation were found in WAT of obese subjects between low and high responders to calorie restriction, suggesting that the epigenetic profile could predict weight loss upon diet [82]. Furthermore, changes in DNA methylation have been reported in subcutaneous WAT between monozygotic twins discordant for type 2 diabetes [83]. In line with these results, a study of the global methylation of H3K4 in obese subjects with or without type 2 diabetes showed that H3K4me3 was higher in diabetic compared to non-diabetic subjects, whether obese or lean [84], suggesting that the H3K4 methylation status might affect insulin sensitivity in humans. Interestingly, a large-scale genome-wide analysis showed association between increased BMI and methylation of hypoxia inducible transcription factor 3A (HIF3A) gene in adipose tissue [85].
As mentioned above, metabolically active BAT or BAT-like tissues can be detected in humans after cold exposure, and the level of activation is inversely correlated with adiposity and BMI [86]. Seasonal temperature-dependent variation in BAT thermogenesis was also reported in this study, suggesting plasticity of the BAT program. In addition, a recent study confirmed that repeated intermittent cold exposure induces BAT activation and is associated with increased adaptive thermogenesis [87]. Moreover, cold exposure increases BAT activity and energy expenditure even in subjects exhibiting a low basal BAT activity [88]. To date, however, there is no study establishing an association between epigenetic changes and BAT program in humans. From early life to adulthood, humans are exposed to various environmental factors such as varied nutritional conditions, hormonal signals or temperature stress that may trigger chromatin remodeling. Thus, epigenetic mechanisms, inherited or acquired, could contribute significantly to the regulation of the thermogenic adipose program to maintain energy homeostasis. Further studies in humans are required to establish causal link between epigenetic modifications and thermogenic fat activation. Finally, although the therapeutic potential of WAT browning in humans requires more investigation, epigenetic effectors may represent attractive targets to induce thermogenic reprogramming in WAT.
Concluding Remarks and Future Perspectives
Understanding how environmental conditions can be relayed through epigenetics to the transcriptional machinery to regulate the thermogenic adipose program and the enzymes involved in the epigenetic regulation may be promising therapeutic targets. However, important questions remain in integrating recently characterized epigenetic effectors into a cohesive network in the regulation of thermogenic program. For example, studies suggest the existence of a cell autonomous epigenetic memory in brown and beige cells in humans and rodents, but how histone modifiers build up an epigenetic memory in beige cells allowing for improved responsiveness to reiterated stimulation, or how an epigenetic profile contributes to the heterogeneity and plasticity of brown, beige, and white adipocytes, is unclear (see Outstanding Questions Box). Future research is needed to answer these questions and to dissect pathways involving histone modifiers and readers regulating thermogenic adipose program and browning of WAT. Accessibility to defined human adipose samples will facilitate the examination of epigenetic profiles in association with activation of the thermogenic program and browning of WAT during changing environmental conditions. Furthermore, characterizing the epigenetic signature of beige cells using developing new techniques (see Box 4) will contribute to a better understanding of their origin, and how browning of WAT could specifically be induced in a cell fate reprogramming strategy. Interestingly, the epigenetic reprogramming of adult stem cells located in WAT towards a thermogenic lineage, could offer relevant therapeutic prospects. Finally, exploring epigenetic mechanisms allowing transition from a white adipocyte phenotype to a thermogenic phenotype may provide therapeutic prospects for obesity and diabetes.
OUTSTANDING QUESTIONS BOX.
How do environmental and external stimuli affect the availability and activity of chromatin modifiers modulating the chromatin landscape in thermogenic adipose tissue? What are the signaling pathways that mediate chromatin remodeling in brown and beige cells?
Do epigenetic effectors represent metabolic sensors integrating external stimuli into thermogenic response at the transcriptional level? In humans, as in rodents, activation of brown or beige adipocytes has been associated with improved glucose and lipid clearance. Hence, at the interface between nutrient status and gene regulation, does epigenetics contribute to the effects of brown/beige adipocytes on systemic glucose and lipid homeostasis?
Do brown, beige, and white adipocytes display a distinct epigenetic profile contributing to their heterogeneity and plasticity? The study of epigenetic marks and corresponding chromatin writers and readers in white, brown and beige adipocytes could be crucial in better understanding cell fate determination and may also address the open question on the origin of beige adipocytes.
Do histone modifiers build up an epigenetic memory in beige cells allowing for improved responsiveness to reiterated stimulation? Induced by prolonged or intermittent cold stress, and likely other environmental factors, specific epigenetic profiles could explain how cells are more efficiently recruited or activated and, globally more responsive in inducing thermogenic genes. Such an adaptive mechanism relying on epigenetic effectors in BAT and WAT raises new perspectives to induce cells whose function is protective against hyperglycemia and dyslipidemia.
Text box 4. Developing techniques in epigenomics.
Progress in high-throughput DNA sequencing and, recently, development of next-generation sequencing methodologies provided larger scale genome-wide profiling of chromatin landscape enabling to better understand the role of epigenetic modifications in regulating and maintaining cell identity. With bisulfite-based sequencing, detecting changes in CpG island methylation at a single nucleotide resolution [95], or chromatin immunoprecipitation (ChIP) identifying posttranslational histone modifications [96], it is possible to characterize tissue-specific or lineage-specific changes in epigenomics at a global level (ChIP-Seq). Mikkelsen et al., [97] compared histone methylation and acetylation of lysines K4, K27 and K36 on histone H3 (H3K4me3/me2/me1, H3K27me3/ac, and H3K36me3) of two models of adipogenesis, murine 3T3-L1 cells and human adipose stromal cells. Dynamic changes in epigenetic marks were significantly correlated with the expression pattern of genes directly related to the cell phenotype. By comparing adipose precursors and mature brown and beige adipocytes at different stages of differentiation, such approach would allow the mapping of histone modifications crucial for thermogenic adipocyte biogenesis. However, the identification of epigenetic determinants with thermogenic potential in beige cells within WAT requires examination of dynamic changes during cell-fate transition at a single-cell resolution. A new fluorescent protein-based reporter system may allow visualization of endogenous DNA methylation dynamics at a single-cell level [98]. The system is based on a gene promoter sequence that exhibits an inherent sensitivity to the methylation state of adjacent genomic regions and drives the expression of green fluorescent protein (GFP). Combining this technique with other conventional strategies, such as conditional knockout or a gene reporter system, could allow epigenetic lineage-tracing of beige adipocytes and elucidation of epigenetic reprogramming of thermogenic adipocytes in WAT. Moreover, methods using CRISPR-Cas9 system and epigenome editing may be employed to target histone modifiers to specific gene loci to test the significance of specific histone modifications [99]. Genome wide screening using CRISPR-Cas9 system may also allow identification of modifiers critical for thermogenic gene program. Advances in proteomic techniques may enable us to identify other, low frequency, histone modifications such as propionylation, butyrylation or crotonylation. Moreover, using locus specific ChIP-mass spectrometry (MS) methods, it would be possible to identify single gene locus associated proteins, including specific histone modifiers, as well as chromatin effector and possibly proteins that provide co-substrates [100].
Table 1.
epigenetic modifications in thermogenic adipocytes
| Epigenetic modification | Effector | Pathway affected/target | Epigenetic mark | Effect | REF | |
|---|---|---|---|---|---|---|
| DNA methylation | N/A | adipogenesis-commitment | N/A | N/A | [40] | |
| N/A | thermogenic adipose program-UCP1 | N/A | repression | [46] | ||
| N/A | thermogenic adipose program-UCP1 | N/A | repression | [47] | ||
| N/A | thermogenic adipose program | N/A | repression | [48] | ||
| N/A | thermogenic adipose program | N/A | repression | [49] | ||
| histone acetylation | HAT | Gcn5/PCAF | Brown differentiation | H3K9 | activation | [51] |
| HDAC | HDAC class I | thermogenic adipose program-browning | N/A | repression | [53] | |
| HDAC9 | thermogenic adipose program -beige differentiation | N/A | repression | [55] | ||
| HDAC1 | thermogenic adipose program -beige differentiation | H3K27 | repression | [54] | ||
| Sirt1 | thermogenic adipose program –BAT function | PGC1a, N/A | activation | [56,57] | ||
| Sirt3 | thermogenic adipose program –BAT function | N/A | activation | [58] | ||
| histone methylation | methyltransferase | MLL3-4 | Differentiation- brown and white adipocytes | H3K4 | activation | [60,61] |
| G9a | Differentiation- brown and white adipocytes | H3K9 | repression | [67] | ||
| EHMT1 | thermogenic adipose program – differentiation | H3K9 | activation | [65,66] | ||
| demethylase | Jhdm2a | Thermogenic adipose program- BAT-WAT | H3K9 | activation | [62–64] | |
| LSD1 | thermogenic adipose program -browning | H3K9 | activation | [29] | ||
| UTX | thermogenic adipose program/brown adipocytes | H3K27 | activation | [68] | ||
| Jmjd3 | Differentiation/thermogenic adipose program/BAT-WAT | H3K27 | activation | [69] | ||
TRENDS BOX.
Recent progress in our understanding of thermogenic adipose development and function has suggested a role for epigenetic mechanisms in the regulation of thermogenic adipose program and browning of WAT.
Characterizing the establishment of differential epigenetic signatures between adipose cell types may allow to better understand thermogenic cell fate determination and differentiation.
Environmental stimuli that are known to affect brown and beige adipocyte development and function have been shown to also alter epigenetic effector activity and thus chromatin landscape and gene regulation.
A bi-directional interplay exists between metabolism and chromatin remodeling through metabolites, with epigenetic effectors playing a role in the integrative control of gene regulation for energy homeostasis.
Acknowledgments
We apologize for not citing other relevant publications due to space limitations. This work was supported by DK95338 to HSS.
GLOSSARY BOX
- Adaptive thermogenesis
also called non-shivering thermogenesis, refers to the dissipation of chemical energy to produce heat that occurs in BAT or beige cells upon external stimuli, such as cold or stress, to ensure the maintenance of body temperature.
- Brown adipose tissue (BAT)
is an adipose depot in which glucose and fatty acids are oxidized to produce heat and ensure thermoregulation. BAT expresses UCP1 and contributes to energy expenditure. In rodents and newborn humans, BAT is mainly located in the interscapular region, while in adults BAT or BAT-like depots are spread in supraclavicular, cervical and paravertebral regions. Brown adipocytes have high mitochondrial content and multilocular lipid droplets. BAT is highly vascularized and innervated by the sympathetic nervous system.
- Beige (or brite) adipocytes
initially described as brown-like adipocytes emerging in WAT, beige adipocytes are highly inducible under cold (or other conditions), express UCP1 at a level similar to that in brown adipocytes upon stimulation, exhibit a multilocular morphology and high mitochondrial content as well, but may have distinct developmental origin.
- Epigenetics
refers to the study of molecular mechanisms controlling stable changes in chromatin organization, without alterations in DNA sequence, and playing an important role in transcriptional regulation and cell fate determination and differentiation.
- Epigenetic Erasers
enzymes that catalyze removal of epigenetic marks to regulate gene transcription.
- Epigenetic Readers
effector proteins that bind epigenetic marks for their recruitment to the chromatin and that are involved in the remodeling of the chromatin landscape and/or gene regulation.
- Epigenetic Writers
enzymes that catalyze addition of epigenetic marks to regulate gene transcription
- Histone modifications
enzymatic reactions catalyzed by histone modifying enzymes that are recruited to the chromatin and are often part of larger protein complexes. The most studied covalent modifications of histones to date are histone acetylation, methylation and phosphorylation. However, other modifications have been described including deimination, glycosylation, ADP ribosylation, ubiquitylation, sumoylation, as well as, propionylation, butyrylation, crotonylation, succinylation, malonylation, glutarylation and hydroxyisobutyrylation.
- Thermogenic adipocytes
adipocytes that display, constitutively or upon induction, a thermogenic function, burning chemical energy that results in generation of heat, comprises brown and beige adipocytes.
- Thermogenic adipose program
refers to the expression of BAT-enriched genes, including UCP1, in brown adipocytes and beige adipocytes upon browning of WAT.
- Uncoupling protein 1 (UCP1)
is a proton channel located in the inner mitochondrial membrane. UCP1 catalyzed proton transport dissipates proton gradient generated by the respiratory chain, preventing ATP production thereby generating heat. UCP1 is thus critical for the thermogenic function of brown and beige adipocytes.
- White adipose tissue (WAT)
the prominent form of adipose tissue in mammals. Its main function is to store energy in the form of lipids when energy intake exceeds energy expenditure. There are two types of WAT depending on their location; visceral WAT and subcutaneous WAT that can be induced to express thermogenic genes upon cold exposure, so called browning. White adipocytes are characterized by unilocular lipid droplets and contain only few mitochondria.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kajimura S, et al. Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell metabolism. 2015;22:546–559. doi: 10.1016/j.cmet.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishibashi J, Seale P. Medicine. Beige can be slimming. Science (New York, NY) 2010;328:1113–1114. doi: 10.1126/science.1190816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petrovic N, et al. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. The Journal of biological chemistry. 2010;285:7153–7164. doi: 10.1074/jbc.M109.053942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cypess AM, et al. Identification and importance of brown adipose tissue in adult humans. The New England journal of medicine. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Marken Lichtenbelt WD, et al. Cold-activated brown adipose tissue in healthy men. The New England journal of medicine. 2009;360:1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 6.Virtanen KA, et al. Functional brown adipose tissue in healthy adults. The New England journal of medicine. 2009;360:1518–1525. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 7.Chondronikola M, et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes. 2014;63:4089–4099. doi: 10.2337/db14-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ouellet V, et al. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. The Journal of clinical investigation. 2012;122:545–552. doi: 10.1172/JCI60433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chondronikola M, et al. Brown Adipose Tissue Activation Is Linked to Distinct Systemic Effects on Lipid Metabolism in Humans. Cell metabolism. 2016;23:1200–1206. doi: 10.1016/j.cmet.2016.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nedergaard J, Cannon B. The browning of white adipose tissue: some burning issues. Cell metabolism. 2014;20:396–407. doi: 10.1016/j.cmet.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 11.Wu J, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150:366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang QA, et al. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nature medicine. 2013;19:1338–1344. doi: 10.1038/nm.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee MW, et al. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell. 2015;160:74–87. doi: 10.1016/j.cell.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenwald M, et al. Bi-directional interconversion of brite and white adipocytes. Nature cell biology. 2013;15:659–667. doi: 10.1038/ncb2740. [DOI] [PubMed] [Google Scholar]
- 15.Wu J, et al. Formation and activation of thermogenic fat. Trends in genetics : TIG. 2015;31:232–238. doi: 10.1016/j.tig.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seale P, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seale P, et al. Transcriptional control of brown fat determination by PRDM16. Cell metabolism. 2007;6:38–54. doi: 10.1016/j.cmet.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kajimura S, et al. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature. 2009;460:1154–1158. doi: 10.1038/nature08262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harms MJ, et al. PRDM16 binds MED1 and controls chromatin architecture to determine a brown fat transcriptional program. Genes & development. 2015;29:298–307. doi: 10.1101/gad.252734.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iida S, et al. PRDM16 enhances nuclear receptor-dependent transcription of the brown fat-specific Ucp1 gene through interactions with Mediator subunit MED1. Genes & development. 2015;29:308–321. doi: 10.1101/gad.252809.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seale P, et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. The Journal of clinical investigation. 2011;121:96–105. doi: 10.1172/JCI44271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen P, et al. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell. 2014;156:304–316. doi: 10.1016/j.cell.2013.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harms MJ, et al. Prdm16 is required for the maintenance of brown adipocyte identity and function in adult mice. Cell metabolism. 2014;19:593–604. doi: 10.1016/j.cmet.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rajakumari S, et al. EBF2 determines and maintains brown adipocyte identity. Cell metabolism. 2013;17:562–574. doi: 10.1016/j.cmet.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohno H, et al. PPARgamma agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell metabolism. 2012;15:395–404. doi: 10.1016/j.cmet.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stine RR, et al. EBF2 promotes the recruitment of beige adipocytes in white adipose tissue. Molecular metabolism. 2016;5:57–65. doi: 10.1016/j.molmet.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kong X, et al. IRF4 is a key thermogenic transcriptional partner of PGC-1alpha. Cell. 2014;158:69–83. doi: 10.1016/j.cell.2014.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dempersmier J, et al. Cold-inducible Zfp516 activates UCP1 transcription to promote browning of white fat and development of brown fat. Molecular cell. 2015;57:235–246. doi: 10.1016/j.molcel.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sambeat A, et al. LSD1 Interacts with Zfp516 to Promote UCP1 Transcription and Brown Fat Program. Cell reports. 2016;15:2536–2549. doi: 10.1016/j.celrep.2016.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao W, et al. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Molecular and cellular biology. 2004;24:3057–3067. doi: 10.1128/MCB.24.7.3057-3067.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Puigserver P, et al. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 32.Duteil D, et al. LSD1 promotes oxidative metabolism of white adipose tissue. Nature communications. 2014;5:4093. doi: 10.1038/ncomms5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tseng YH, et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature. 2008;454:1000–1004. doi: 10.1038/nature07221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hondares E, et al. Hepatic FGF21 expression is induced at birth via PPARalpha in response to milk intake and contributes to thermogenic activation of neonatal brown fat. Cell metabolism. 2010;11:206–212. doi: 10.1016/j.cmet.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park JH, et al. A multifunctional protein, EWS, is essential for early brown fat lineage determination. Developmental cell. 2013;26:393–404. doi: 10.1016/j.devcel.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fisher FM, et al. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes & development. 2012;26:271–281. doi: 10.1101/gad.177857.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor SM, Jones PA. Multiple new phenotypes induced in 10T1/2 and 3T3 cells treated with 5-azacytidine. Cell. 1979;17:771–779. doi: 10.1016/0092-8674(79)90317-9. [DOI] [PubMed] [Google Scholar]
- 38.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 39.Sakamoto H, et al. Sequential changes in genome-wide DNA methylation status during adipocyte differentiation. Biochemical and biophysical research communications. 2008;366:360–366. doi: 10.1016/j.bbrc.2007.11.137. [DOI] [PubMed] [Google Scholar]
- 40.Sakamoto H, et al. Cell type-specific methylation profiles occurring disproportionately in CpG-less regions that delineate developmental similarity. Genes to cells : devoted to molecular & cellular mechanisms. 2007;12:1123–1132. doi: 10.1111/j.1365-2443.2007.01120.x. [DOI] [PubMed] [Google Scholar]
- 41.Melzner I, et al. Leptin gene expression in human preadipocytes is switched on by maturation-induced demethylation of distinct CpGs in its proximal promoter. The Journal of biological chemistry. 2002;277:45420–45427. doi: 10.1074/jbc.M208511200. [DOI] [PubMed] [Google Scholar]
- 42.Yokomori N, et al. DNA demethylation during the differentiation of 3T3-L1 cells affects the expression of the mouse GLUT4 gene. Diabetes. 1999;48:685–690. doi: 10.2337/diabetes.48.4.685. [DOI] [PubMed] [Google Scholar]
- 43.Milagro FI, et al. High fat diet-induced obesity modifies the methylation pattern of leptin promoter in rats. Journal of physiology and biochemistry. 2009;65:1–9. doi: 10.1007/BF03165964. [DOI] [PubMed] [Google Scholar]
- 44.Milagro FI, et al. A dual epigenomic approach for the search of obesity biomarkers: DNA methylation in relation to diet-induced weight loss. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2011;25:1378–1389. doi: 10.1096/fj.10-170365. [DOI] [PubMed] [Google Scholar]
- 45.Christian M, et al. RIP140-targeted repression of gene expression in adipocytes. Molecular and cellular biology. 2005;25:9383–9391. doi: 10.1128/MCB.25.21.9383-9391.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kiskinis E, et al. RIP140 directs histone and DNA methylation to silence Ucp1 expression in white adipocytes. The EMBO journal. 2007;26:4831–4840. doi: 10.1038/sj.emboj.7601908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shore A, et al. Role of Ucp1 enhancer methylation and chromatin remodelling in the control of Ucp1 expression in murine adipose tissue. Diabetologia. 2010;53:1164–1173. doi: 10.1007/s00125-010-1701-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Derecka M, et al. Tyk2 and Stat3 regulate brown adipose tissue differentiation and obesity. Cell metabolism. 2012;16:814–824. doi: 10.1016/j.cmet.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Biggar Y, Storey KB. Global DNA modifications suppress transcription in brown adipose tissue during hibernation. Cryobiology. 2014;69:333–338. doi: 10.1016/j.cryobiol.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 50.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nature reviews. Genetics. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 51.Jin Q, et al. Gcn5 and PCAF regulate PPARgamma and Prdm16 expression to facilitate brown adipogenesis. Molecular and cellular biology. 2014;34:3746–3753. doi: 10.1128/MCB.00622-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoo EJ, et al. Down-regulation of histone deacetylases stimulates adipocyte differentiation. The Journal of biological chemistry. 2006;281:6608–6615. doi: 10.1074/jbc.M508982200. [DOI] [PubMed] [Google Scholar]
- 53.Galmozzi A, et al. Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes. 2013;62:732–742. doi: 10.2337/db12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li F, et al. Histone Deacetylase 1 (HDAC1) Negatively Regulates Thermogenic Program in Brown Adipocytes via Coordinated Regulation of Histone H3 Lysine 27 (H3K27) Deacetylation and Methylation. The Journal of biological chemistry. 2016;291:4523–4536. doi: 10.1074/jbc.M115.677930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chatterjee TK, et al. HDAC9 knockout mice are protected from adipose tissue dysfunction and systemic metabolic disease during high-fat feeding. Diabetes. 2014;63:176–187. doi: 10.2337/db13-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boutant M, et al. SIRT1 enhances glucose tolerance by potentiating brown adipose tissue function. Molecular metabolism. 2015;4:118–131. doi: 10.1016/j.molmet.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gerhart-Hines Z, et al. The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+) Molecular cell. 2011;44:851–863. doi: 10.1016/j.molcel.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shi T, et al. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. The Journal of biological chemistry. 2005;280:13560–13567. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- 59.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annual review of pathology. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee J, et al. Targeted inactivation of MLL3 histone H3-Lys-4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:19229–19234. doi: 10.1073/pnas.0810100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee JE, et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife. 2013;2:e01503. doi: 10.7554/eLife.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tateishi K, et al. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature. 2009;458:757–761. doi: 10.1038/nature07777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Inagaki T, et al. Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes to cells : devoted to molecular & cellular mechanisms. 2009;14:991–1001. doi: 10.1111/j.1365-2443.2009.01326.x. [DOI] [PubMed] [Google Scholar]
- 64.Abe Y, et al. JMJD1A is a signal-sensing scaffold that regulates acute chromatin dynamics via SWI/SNF association for thermogenesis. Nature communications. 2015;6:7052. doi: 10.1038/ncomms8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ohno H, et al. EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature. 2013;504:163–167. doi: 10.1038/nature12652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nagano G, et al. Activation of classical brown adipocytes in the adult human perirenal depot is highly correlated with PRDM16-EHMT1 complex expression. PloS one. 2015;10:e0122584. doi: 10.1371/journal.pone.0122584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang L, et al. Histone H3K9 methyltransferase G9a represses PPARgamma expression and adipogenesis. The EMBO journal. 2013;32:45–59. doi: 10.1038/emboj.2012.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zha L, et al. The Histone Demethylase UTX Promotes Brown Adipocyte Thermogenic Program Via Coordinated Regulation of H3K27 Demethylation and Acetylation. The Journal of biological chemistry. 2015;290:25151–25163. doi: 10.1074/jbc.M115.662650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pan D, et al. Jmjd3-Mediated H3K27me3 Dynamics Orchestrate Brown Fat Development and Regulate White Fat Plasticity. Developmental cell. 2015;35:568–583. doi: 10.1016/j.devcel.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shinoda K, et al. Phosphoproteomics Identifies CK2 as a Negative Regulator of Beige Adipocyte Thermogenesis and Energy Expenditure. Cell metabolism. 2015;22:997–1008. doi: 10.1016/j.cmet.2015.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brasacchio D, et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes. 2009;58:1229–1236. doi: 10.2337/db08-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wellen KE, et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science (New York, NY) 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Prokesch A, et al. N-acetylaspartate catabolism determines cytosolic acetyl-CoA levels and histone acetylation in brown adipocytes. Scientific reports. 2016;6:23723. doi: 10.1038/srep23723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Canto C, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell metabolism. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cohen DE, et al. Neuronal SIRT1 regulates endocrine and behavioral responses to calorie restriction. Genes & development. 2009;23:2812–2817. doi: 10.1101/gad.1839209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chalkiadaki A, Guarente L. High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell metabolism. 2012;16:180–188. doi: 10.1016/j.cmet.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kraus D, et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature. 2014;508:258–262. doi: 10.1038/nature13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hino S, et al. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nature communications. 2012;3:758. doi: 10.1038/ncomms1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kebede AF, et al. Novel types and sites of histone modifications emerge as players in the transcriptional regulation contest. The FEBS journal. 2015;282:1658–1674. doi: 10.1111/febs.13047. [DOI] [PubMed] [Google Scholar]
- 80.Ost A, Pospisilik JA. Epigenetic modulation of metabolic decisions. Current opinion in cell biology. 2015;33:88–94. doi: 10.1016/j.ceb.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 81.Feinberg AP, et al. Personalized epigenomic signatures that are stable over time and covary with body mass index. Science translational medicine. 2010;2:49ra67. doi: 10.1126/scitranslmed.3001262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bouchard L, et al. Differential epigenomic and transcriptomic responses in subcutaneous adipose tissue between low and high responders to caloric restriction. The American journal of clinical nutrition. 2010;91:309–320. doi: 10.3945/ajcn.2009.28085. [DOI] [PubMed] [Google Scholar]
- 83.Ribel-Madsen R, et al. Genome-wide analysis of DNA methylation differences in muscle and fat from monozygotic twins discordant for type 2 diabetes. PloS one. 2012;7:e51302. doi: 10.1371/journal.pone.0051302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jufvas A, et al. Global differences in specific histone H3 methylation are associated with overweight and type 2 diabetes. Clinical epigenetics. 2013;5:15. doi: 10.1186/1868-7083-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dick KJ, et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet (London, England) 2014;383:1990–1998. doi: 10.1016/S0140-6736(13)62674-4. [DOI] [PubMed] [Google Scholar]
- 86.Saito M, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009;58:1526–1531. doi: 10.2337/db09-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.van der Lans AA, et al. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. The Journal of clinical investigation. 2013;123:3395–3403. doi: 10.1172/JCI68993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yoneshiro T, et al. Recruited brown adipose tissue as an antiobesity agent in humans. The Journal of clinical investigation. 2013;123:3404–3408. doi: 10.1172/JCI67803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harbor perspectives in biology. 2014;6:a019133. doi: 10.1101/cshperspect.a019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shiota K, et al. Epigenetic marks by DNA methylation specific to stem, germ and somatic cells in mice. Genes to cells : devoted to molecular & cellular mechanisms. 2002;7:961–969. doi: 10.1046/j.1365-2443.2002.00574.x. [DOI] [PubMed] [Google Scholar]
- 91.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell research. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rice JC, et al. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Molecular cell. 2003;12:1591–1598. doi: 10.1016/s1097-2765(03)00479-9. [DOI] [PubMed] [Google Scholar]
- 93.Lawrence M, et al. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends in genetics : TIG. 2016;32:42–56. doi: 10.1016/j.tig.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 94.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nature reviews. Genetics. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 97.Mikkelsen TS, et al. Comparative epigenomic analysis of murine and human adipogenesis. Cell. 2010;143:156–169. doi: 10.1016/j.cell.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stelzer Y, et al. Tracing dynamic changes of DNA methylation at single-cell resolution. Cell. 2015;163:218–229. doi: 10.1016/j.cell.2015.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thakore PI, et al. Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nature methods. 2016;13:127–137. doi: 10.1038/nmeth.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wierer M, Mann M. Proteomics to study DNA-bound and chromatin-associated gene regulatory complexes. Human molecular genetics. 2016 doi: 10.1093/hmg/ddw208. [DOI] [PMC free article] [PubMed] [Google Scholar]