

Abstract
Rationale
Pulmonary hypertension (PH) is characterized by pulmonary vascular remodeling that leads to pulmonary congestion, uncompensated right-ventricle (RV) failure, and premature death. Preclinical studies have demonstrated that the G protein-coupled estrogen receptor (GPER) is cardioprotective in male rats and that its activation elicits vascular relaxation in rats of either sex.
Objectives
To study the effects of GPER on the cardiopulmonary system by the administration of its selective agonist G1 in male rats with monocrotaline (MCT)-induced PH.
Methods
Rats received a single intraperitoneal injection of MCT (60 mg/kg) for PH induction. Experimental groups were as follows: control, MCT + vehicle, and MCT + G1 (400 μg/kg/day subcutaneous). Animals (n = 5 per group) were treated with vehicle or G1 for 14 days after disease onset.
Measurements and main results
Activation of GPER attenuated exercise intolerance and reduced RV overload in PH rats. Rats with PH exhibited echocardiographic alterations, such as reduced pulmonary flow, RV hypertrophy, and left-ventricle dysfunction, by the end of protocol. G1 treatment reversed these PH-related abnormalities of cardiopulmonary function and structure, in part by promoting pulmonary endothelial nitric oxide synthesis, Ca2+ handling regulation and reduction of inflammation in cardiomyocytes, and a decrease of collagen deposition by acting in pulmonary and cardiac fibroblasts.
Conclusions
G1 was effective to reverse PH-induced RV dysfunction and exercise intolerance in male rats, a finding that have important implications for ongoing clinical evaluation of new cardioprotective and vasodilator drugs for the treatment of the disease.
Keywords: GPER, monocrotaline, vascular remodeling, right ventricular dysfunction
Graphical abstract
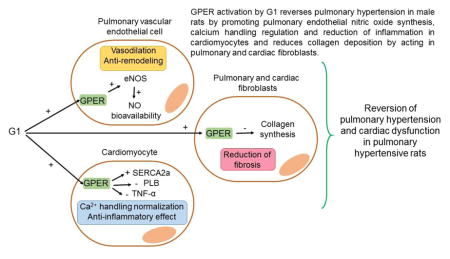
1. Introduction
Pulmonary hypertension (PH) is a multifactorial condition that has high rates of morbidity and mortality (Moreira et al. 2015, Peacock et al., 2007). PH is a socially and scientifically relevant disease, with the cardiopulmonary research field proving its epidemiological importance and identifying new complex pathobiological routes (Humbert and Ghofrani, 2016; Schermuly et al., 2011). The three main characteristics of PH are (1) dysfunctional pulmonary hemodynamics (i.e., exacerbated vasoconstriction and reduced vasodilation), (2) structural changes in the pulmonary vasculature (i.e., wall remodeling and hypertrophy), leading to elevated pulmonary arterial pressure, and (3) long-term right-ventricle (RV) pressure overload and subsequent RV failure (Lourenco et al., 2012; Schermuly et al., 2011). Moreover, the function and size of the RV are not only indicators of the severity and chronicity of PH, but are also the most important determinants of prognosis (Voelkel et al., 2006).
The approved medical treatments for PH include drugs that target impaired pathways in the pulmonary circulation, showing positive results in clinical trials when used as monotherapies or in combination (Badiani and Messori, 2016; Humbert and Ghofrani, 2016). Nevertheless, despite recent medical advances, treatment strategies are frequently refractory in PH subjects, and overall survival is poor, with 5-year survival estimates of less than 60% in registries (McLaughlin and McGoon, 2006). As such, there is a need for new therapies focused not only on the anti-remodeling and vasodilation effects of the pulmonary vasculature, but also myocardial protection.
Estrogens are sex hormones and important mediators of normal cardiovascular function (Babiker et al., 2002; Dubey et al., 2002; Yang and Reckelhoff, 2011). The G protein-coupled estrogen receptor (GPER) has been extensively characterized as a protective target in animal models of left heart and systemic circulation remodeling and dysfunction. Nongenomic mechanisms are triggered, such as rapid estrogen-mediated activation of related protein-serine/threonine kinases (ERK1/2) and cyclic AMP (cAMP) generation, with subsequent induction of beneficial effects on the heart and arterial wall, including vasodilation, inhibition of smooth muscle cell proliferation, inhibition of inflammation, antioxidant effects, and endothelial/cardiac cell survival following injury (Alencar et al., 2016; Deschamps and Murphy, 2009; Haas et al., 2009; Jessup et al., 2010; Lindsey et al., 2009; Liu et al., 2016; Wang et al., 2012; Wang et al., 2015; Weil et al., 2010). Despite classical estrogen receptors (ERα and ERβ) signaling has been well characterized in different animal models of PH (Frump et al., 2015; Mair et al., 2014; Wright et al., 2015; Xu et al., 2013), there is a lack of robust exploration of GPER specific roles during experimental PH and in RV dysfunction. Accordingly, we investigated the effects of the activation of GPER by the administration of its highly selective agonist, G1, in male rats with monocrotaline (MCT)-induced PH. While PH is more common among women, we chose to study the effects of activating GPER in male rats only in order to examine its therapeutic potential independent of sex. Indeed, future studies are needed to examine its role in females with and without endogenous oestradiol (E2). We hypothesized that G1 treatment would reverse the pulmonary vascular and cardiac dysfunction in PH experimental model.
2. Methods
2.1 Animals and experimental design
All experiments were conducted in accordance with the Animal Care and Use Committee at Universidade Federal do Rio de Janeiro (UFRJ). Fifteen male Wistar rats (220–300 g) were housed at 20 ± 3 °C under a 12-h light/12-h dark cycle with free access to food and water. Rats were randomly divided into three groups, five rats per group: control, MCT + vehicle, and MCT + G1. Rats in the MCT groups were given a single intraperitoneal (i.p.) injection of MCT at 60 mg/kg body weight to induce PH as published elsewhere (Alencar et al., 2014; Alencar et al., 2013; Zapata-Sudo et al., 2012). Rats in the control group were injected with the same volume of sterile saline. Two weeks after MCT or saline administration, rats were dosed once daily for 2 weeks with 0.1 mL of peanut oil (control and MCT + vehicle groups) or with G1 at 400 μg/kg body weight, administered subcutaneously (MCT + G1 group). Rats were weighed daily, and volumes of G1 solution were adjusted appropriately.
2.2 Echocardiography
Cardiac function was assessed by an echocardiographic system equipped with a 10-MHz mechanical transducer (Esaote model, CarisPlus, Florence, Italy), following the procedure previously described (Alencar et al., 2014; Lang et al., 2006). In order to confirm PH development induced by MCT, parameters were evaluated before, 14 days and 28 days after injection.
2.3 Exercise test protocol, hemodynamic and RV hypertrophy measurements
Animals performed a graded treadmill (EP–131, Insight, São Paulo, Brazil) run to exhaustion, in different times (before, 14 days after, and 28 days after MCT injection) in order to assess the exercise performance as published elsewhere (Alencar et al., 2014). At the endpoint of the protocol, rats were anesthetized with ketamine (80 mg/kg, i.p.) and xylazine (15 mg/kg, i.p.) and mean arterial blood pressure (MAP), right-ventricle systolic pressure (RVSP) and RV hypertrophy were measured as previously described (Nishida et al., 2009)
2.4 Membrane preparations and Western blot analysis
The preparation of rat lung and RV subcellular fractions and the subsequent western blot analysis for sarco/endoplasmic reticulum Ca2+-ATPase 2a (SERCA2a), phospholamban (PLB), tumor necrosis factor-alpha (TNF-α), endothelial nitric oxide synthase (eNOS), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, used as loading control) proteins were performed as the already published work (Alencar et al., 2014).
2.5 Histomorphometric analysis
For the Histomorphometric analysis lungs were collected, immersed in 10% neutral buffered formalin, and embedded in paraffin (Alencar et al., 2014). RVs were treated in a similar way, except that the processing times were different (20 min in sequential baths with alcohol, xylene, and paraffin). Tissues were sectioned at 4 μm, stained, and analyzed in a blinded manner following the already published methods (Alencar et al., 2014; Alencar et al., 2013).
2.6 Data analysis
Data analysis was performed for all endpoints, and one-way ANOVA was used to determine the significance of differences among groups. Significance of interactions between groups was determined by Tukey post-hoc tests. Pearson correlation was used to test for a relationship between time to exhaustion, RVSP, and PAT, and between TNF-α, SERCA2a, and PLB levels. Differences for all tests were considered significant when the P value was less than 0.05. Analyses were performed using GraphPad Prism, version 6 (GraphPad, San Diego, CA, USA). An expanded methods section is available in the online supplement.
3. Results
3.1 Activation of GPER inhibits MCT-induced heart and lung hypertrophy 4 weeks after MCT treatment
Body, heart, and lung weights are shown in Figure 1 and Table 1. Body weight did not change between the groups throughout the protocol period (Table 1). MCT injection significantly increased the ratios of the heart weight and RV weight to the final body weight compared to each control animal (Table 1). However, the ratio of the LV + S weight to the final body weight was not different between MCT-treated and control animals. MCT-induced RV hypertrophy was further evaluated by measuring the ratio of the RV weight to the LV + S weight. This ratio was significantly larger in MCT + vehicle rats (0.78 ± 0.10) compared to saline-treated control rats (0.30 ± 0.03, P < 0.05, Table 1). Changes in the heart and RV weights observed in MCT-treated rats were suppressed by a daily subcutaneous administration of G1 (P < 0.05, Table 1). The ratio of the lung weight to the final body weight was significantly higher in the MCT-treated groups compared to the control group. GPER activation with G1 reduced this lung index of pulmonary edema and fibrosis (Table 1).
Figure 1.

Effects of the subcutaneous treatment with vehicle or G1 (400 μg/kg/day) on the heart and lung weights of MCT-injected rats. a heart weight, b right ventricle weight, c left ventricle plus septal weight, and d lung weight 29 days after monocrotaline injection. Each column and bar represent the mean ± S.E.M. (n = 5 rats per group). *P < 0.05 compared with control rats; †P < 0.05 compared with MCT + vehicle rats. HW, heart weight; RV, right ventricle; LV, left ventricle; S, septum; MCT, monocrotaline.
Table 1.
Comparative data on body, heart, and lung weights
| Control | MCT + V | MCT + G1 | |
|---|---|---|---|
| FBW, g | 289.0 ± 19.3 | 307.0 ± 14.1 | 274.0 ± 20.4 |
| HW/FBW, mg/g | 2.9 ± 0.1 | 4.4 ± 0.3* | 3.2 ± 0.3† |
| RVW/FBW, mg/g | 0.7 ± 0.05 | 1.9 ± 0.1* | 0.7 ± 0.1† |
| LV + S/FBW, mg/g | 2.3 ± 0.1 | 2.6 ± 0.2 | 2.4 ± 0.2 |
| RVW/LV + S | 0.3 ± 0.03 | 0.8 ± 0.02* | 0.3 ± 0.01† |
| Lung W/FBW, mg/g | 8.2 ± 0.6 | 13.1 ± 0.8* | 9.8 ± 0.9† |
Each value represents the mean ± S.E.M (n = 5 rats per group).
P < 0.05 compared with control rats, and
P < 0.05 compared with MCT + vehicle rats.
FBW, final body weight; HW, heart weight; RVW, right ventricle weight; LV+S, left ventricle plus septum; Lung W, lung weight; MCT, monocrotaline; V, vehicle.
3.2 GPER activation improves PA blood flow in MCT-treated rats
Doppler imaging echocardiographic analysis was used to visualize PA outflow (Figure 2). PH development could be confirmed by a change in the shape of the PA waveform from 14 days to 28 days after MCT injection (Figure 2B–C), in accordance with previous reports (Jones et al., 2002; Koskenvuo et al., 2010). Activation of GPER with G1 partially corrected this MCT-induced triangular flow profile (Figure 2C) and significantly reversed the PA flow impairment by the end of the protocol, as depicted by the increased pulmonary acceleration time (PAT) (Figure 2D) and pulmonary artery velocity time integral (PAVTI) (Figure 2E) after G1 administration.
Figure 2.

Effects of MCT injection on the pulmonary artery outflow profile over 28 days of protocol and subcutaneous treatment with vehicle or G1 (400 μg/kg/day) during 14 days. Representative images of pulmonary artery outflow profile are shown before (panel A), 14 days (panel B) and 28 days (panel C) after MCT injection. Pulmonary artery acceleration time and velocity time integral are shown in D and E, respectively. Data represent the mean ± S.E.M. (n = 5 rats per group). *P < 0.05 compared with control rats; †P < 0.05 compared with MCT + vehicle rats. PAT, pulmonary artery acceleration time; PAVTI, pulmonary artery velocity time; MCT, monocrotaline; V, vehicle
3.3 G1 decreases the progressive RV dilatation and improves LV function in rats with PH
Figure 3A shows the parasternal short-axis views obtained by B-mode echocardiography (all end-diastolic) for all animal groups. MCT treatment induced progressive RV remodeling and dilatation, with a concomitant decrease of the LV area (Figure 3A). At 28 days after MCT injection, the RV wall thickness (MCT + vehicle vs. control, P < 0.05; Table 2) and RV area (MCT + vehicle, 39.3 ± 2.3 mm2 vs. control, 22.3 ± 3.8 mm2, P < 0.05, Figure 3B) were elevated compared to values in the control group. RV hypertrophy was responsible for the reduction of the LV area (MCT + vehicle, 26.4 ± 3.5 mm2 vs. control, 41.2 ± 2.8 mm2, P < 0.05, Figure 3C) and for the impairment of LV function (reduced LV stroke volume and ejection fraction, and increased LV end-systolic volume) in MCT + vehicle rats compared to the control group (P < 0.05, Table 2).
Figure 3.

Effects of the subcutaneous treatment with vehicle or G1 (400 μg/kg/day) on heart structure and function of MCT-injected rats. a representative images of parasternal short-axis views obtained by B-mode echocardiography (all end-diastolic), b right ventricle area, c left ventricle area, and d left ventricular cardiac output 28 days after monocrotaline injection. Each column and bar represent the mean ± S.E.M. (n = 5 rats per group). *P < 0.05 compared with control rats; †P < 0.05 compared with MCT + vehicle rats. RV, right ventricle; LV, left ventricle; LVCO, left ventricular cardiac output; MCT, monocrotaline; V, vehicle.
Table 2.
Echocardiographic parameters
| Control | MCT + V | MCT + G1 | |
|---|---|---|---|
| Heart rate, bpm | 261 ± 19.6 | 300 ± 4.7 | 284 ± 27.0 |
| RV wall thickness, cm | 0.38 ± 0.04 | 0.90 ± 0.07* | 0.53 ± 0.04† |
| LV stroke volume, μL | 261 ± 20.7 | 114 ± 17.0* | 235 ± 31.6† |
| LVESV, μL | 136 ± 11.7 | 362 ± 26.4* | 176 ± 17.9† |
| LVEDV, μL | 400 ± 32.2 | 379 ± 23.5 | 402 ± 36.2 |
| LVEF, % | 65 ± 4.0 | 38 ± 3.9* | 56 ± 4.9† |
Each value represents the mean ± S.E.M (n = 5 rats per group).
P < 0.05 compared with control rats, and
P < 0.05, compared with MCT + vehicle rats.
RV, right ventricle; LV, left ventricle; LVESV, left ventricular end-systolic volume; LVEDV, left ventricular end-diastolic volume; LVEF; left ventricular ejection fraction; MCT, monocrotaline; V, vehicle.
The LV cardiac output, calculated by combining the heart rate and stroke volume, was significantly lower in MCT-treated rats (34.6 ± 5.7 mL/min) compared to the control group (69.2 ± 9.1 mL/min, P < 0.05, Figure 3D). By 28 days after MCT administration, rats receiving G1 treatment showed significantly lower RV wall thickness and RV area, and higher LV area results, compared to MCT-induced rats. In PH rats treated subcutaneously with G1, cardiac output increased to values that were similar to those of the control group, due to the enhanced stroke volume. The heart rate was not altered.
3.4 G1 treatment improves systemic and pulmonary hemodynamics in MCT-induced PH rats
On day 29 of the protocol, rats in the MCT + vehicle group exhibited elevated RVSP values (41.1 ± 1.4 mmHg) compared to control rats (24.6 ± 0.6 mmHg, P < 0.05, Figure 4B). Treatment with G1 at 400 μg/kg/day significantly reduced the RVSP value to 27.5 ± 1.7 mmHg (P < 0.05 compared to MCT + vehicle, Figure 4B). By the end of the protocol, the PH rats had lower MAP values compared to the control group (P < 0.05, Figure 4C). Treatment with G1 increased the MAP values.
Figure 4.

Effects of the subcutaneous treatment with vehicle or G1 (400 μg/kg/day) on hemodynamic parameters of MCT-injected rats. a representative tracing of right ventricular systolic pressure and mean arterial pressure, b right ventricular systolic pressure, and c mean arterial pressure 29 days after monocrotaline injection. Each column and bar represent the mean ± S.E.M. (n = 5 rats per group). *P < 0.05 compared with control rats; †P < 0.05 compared with MCT + vehicle rats. RVSP, right ventricular systolic pressure; MAP, mean arterial pressure; MCT, monocrotaline; V, vehicle.
3.5 Long-term GPER activation reduces exercise intolerance in MCT-treated PH rats
Animals were submitted to a treadmill test before, 14 days after, and 29 days after MCT injection. The time to exhaustion was similar between the groups before MCT administration. After 14 days, the time to exhaustion was significantly reduced in MCT-injected rats compared to the saline-treated control rats (data not shown). By 29 days after PH induction, the time to exhaustion was further reduced from 1042.0 ± 66.5 seconds (control) to 200 ± 63.3 seconds (MCT + vehicle group, P < 0.05, Figure 5A). By the end of the protocol, MCT + G1 rats showed a longer time to exhaustion of 909.4 ± 45.6 seconds compared to the MCT + vehicle group (P < 0.05, Figure 5A). Linear regression analyses revealed that exercise intolerance in PH rats was significantly correlated with the increased RVSP (Figure 5B), reduced PA flow (Figure 5C), and reduced LV cardiac output (Figure 5D).
Figure 5.

Effects of the subcutaneous treatment with vehicle or G1 (400 μg/kg/day) on exercise test protocol of MCT-injected rats. a time to exhaustion, b linear regression between RVSP and time to exhaustion, c linear regression between PAT and time to exhaustion, and d linear regression between LVCO and time to exhaustion 29 days after monocrotaline injection. Each column and bar represent the mean ± S.E.M. (n = 5 rats per group). *P < 0.05 compared with control rats; †P < 0.05 compared with MCT + vehicle rats. RVSP, right ventricular systolic pressure; PAT, pulmonary artery acceleration time, LVCO, left ventricular cardiac output, MCT, monocrotaline; V, vehicle.
3.6 Effects of G1 on SERCA2a, PLB, and TNF-α expression levels in RV tissues from PH rats
Western blot analysis of RV tissues showed that PH increased the relative expression ratio of PLB to SERCA2a (MCT + vehicle vs. control, P < 0.05, Figure 6B). This ratio was normalized after GPER activation with G1 (MCT + G1 vs. MCT + vehicle, P < 0.05, Figure 6B). TNF-α was overexpressed in hearts from PH rats (P < 0.05 vs. control, Figure 6C). Downregulation of SERCA2a and overexpression of PLB were significantly correlated with alterations in TNF-α protein expression (Figure 6D and 6E, respectively). RV tissues from G1-treated rats (MCT + G1) showed lower levels of TNF-α expression compared to MCT- injected rats (Figure 6C).
Figure 6.

Western blot analyses of a SERCA2a, PLB and TNF-α expression in right ventricle from control, MCT+ vehicle, and MCT+ G1 groups, respectively. GAPDH was used for normalization. b relative expression ratio of PLB to SERCA2a, c quantification of TNF-α expression, d linear regression between TNF-α and SERCA2a expression levels, and e linear regression between TNF-α and PLB expression levels. Each column and bar represent the mean ± S.E.M. (n = 3 rats per group). *P < 0.05 compared with control rats; †P < 0.05 compared with MCT + vehicle rats. SERCA2a, sarco/endoplasmic reticulum Ca2+-ATPase 2a; PLB, phospholamban; TNF-α, tumor necrosis factor-alpha; MCT, monocrotaline; V, vehicle.
3.7 Effects of G1 on pulmonary vascular remodeling, fibrosis, and eNOS expression in PH rats
Figure 7 shows the histological images obtained in this study. MCT injection significantly increased the wall thickness of pulmonary arterioles from 74.2% ± 2.0% (control rats) to 83.5% ± 1.9% (MCT + vehicle rats, Figure 7C). Treatment with G1 (400 μg/kg) reduced the wall thickness of these vessels to 76.8% ± 1.5% (P < 0.05 vs. MCT + vehicle; Figure 7C). After treatment with G1 or vehicle, collagen deposition was measured in the left lungs (Figure 7B) of control and MCT-injected rats, by calculating the relative collagen area fraction (%). This fraction was significantly increased in the MCT + vehicle group compared to the control, and was reduced in the MCT + G1 group compared to MCT + vehicle group (Figure 7D). Western blot analysis showed that MCT significantly decreased eNOS levels in the lungs (Figure 7F), while G1 treatment increased the expression of eNOS in MCT-injured lungs.
Figure 7.

Representative images of lung sections and western blot analysis of control and MCT-injected rats treated with vehicle or G1 (400 μg/kg/day). a and b show vessels at 40x magnification. Each bar represents 20μm. a immunohistochemical staining for alpha-actin, b picrosirius red staining, c vessel wall thickness expressed as a percent of the total area of the vessel ranging between 50–150 μm in external diameter, d collagen volume fraction of pulmonary arterioles in relation to the tissue area, e representative western blot of eNOS expression, and f quantification of eNOS expression levels; GAPDH was used as loading control. Each column and bar represent the mean ± S.E.M. (n = 3–5 rats per group). *P < 0.05 compared with control rats; †P < 0.05 compared with MCT + vehicle rats. eNOS, endothelial nitric oxide synthase; MCT, monocrotaline; V, vehicle.
3.8 Effects of G1 on RV fibrosis in PH rats
Figure 8 reports the results of the histological analysis and representative images of interstitial collagen deposition. As expected, there was a significant increase in interstitial collagen in RV tissues from MCT-injected rats (4.8% ± 1.1%) compared to control rats (1.6 ± 0.2%, P < 0.05, Figure 8B). G1 treatment reduced the interstitial collagen deposition compared to MCT + vehicle rats (1.8% ± 0.3%, P < 0.05, Figure 8B).
Figure 8.

Collagen volume analysis of the right ventricles from MCT-injected rats treated with vehicle or G1 (400 μg/kg/day). a picrosirius red staining under light microscopy (magnification 40×), showing collagen fibers in red, and b collagen volume fraction of right ventricles in relation to the tissue area. Each column and bar represent the mean ± S.E.M. (n = 5 rats per group). *P < 0.05 compared with control rats; †P < 0.05 compared with MCT + vehicle rats. MCT, monocrotaline, V, vehicle.
4. Discussion
The main finding of this study is that GPER, in addition to its salutary effects on systemic circulation, LV function, and LV structure, may exhibit pulmonary vascular and cardioprotective features that prevent the development of RV dysfunction in MCT-induced PH rats.
Although lungs from MCT-treated PH rats do not sufficient generate angioobliteration that recapitulate human pulmonary vascular lesions (Vitali et al., 2014), we have chosen this model because it, similarly to that of hypoxia-induced PH, induces intense RV hypertrophy and dysfunction (Alencar et al., 2014; Alencar et al., 2013; van Suylen et al., 1998; Zapata-Sudo et al., 2012), the major determinants of life expectance in the clinical practice. Furthermore, as we aimed to investigate the specific potential of GPER independent of its activation by endogenous estrogens, the injection of MCT would low plasma E2 levels by its gonadal actions (Tofovic and Jackson, 2013), reducing the physiologic stimulus of GPER and allowing to explore closely the effects of G1.
Our echocardiography data revealed that from 14 to 28 days after MCT injection, PH rats had reduction of PAT and PAVTI due to hypertrophy and stiffness of the PA. Fourteen days of treatment with G1 significantly normalized the PA flow in PH rats, thus preserving the RV-PA coupling and the maintenance of RV hemodynamics and pressure-function relationships. The increased size and pressure overload of the RV in our PH rats were confirmed by echocardiography and invasive experiments. MCT injection induced changes in the structure and function of the pulmonary circulation, which increased the RVSP and RV wall stress, leading to adaptive remodeling and chronic hypertrophy by day 29 of the protocol. Our echocardiographic data also revealed that RVs from MCT-induced PH rats had significantly greater areas than the LVs, which are normally larger. Thus, PH rats developed LV dysfunction, as depicted by the reduced LV stroke volume, ejection fraction, and cardiac output. These LV systolic profile alterations probably influenced our observation of a lower MAP 29 days after MCT injection compared to saline-treated control rats. Daily treatment with G1 for 14 days after disease establishment abolished the increase in RVSP and reduced RV hypertrophy. Cardioprotective effects of G1 were also shown by the improved LV systolic function and normalization of MAP in MCT-injected rats.
When administered at a dose of 400 μg/kg/day for 14 days, G1 did not induce systemic hypotension in male rats. Our results are in agreement with recent reports showing that G1 did not alter systemic blood pressure when administered at a dose of 400 μg/kg/day for 2 weeks in normotensive male and female rats (Lindsey et al., 2013). Exercise intolerance is a cardinal symptom of PH patients, who experience serious limitations in their activities of daily life due to the exertional fatigue and dyspnea associated with RV failure (Manders et al., 2015; Neder et al., 2015). We recently reported that MCT-treated rats developed progressive exercise intolerance (Alencar et al., 2014). Recently, it was found that GPER activation by G1 reduced skeletal muscle dysfunction and exercise intolerance in rats with LV diastolic dysfunction (Wang et al., 2016). Here, we showed that the reduced exercise performance, defined by the time to exhaustion on the treadmill, 28 days after MCT application, was significantly correlated with alterations in PAT, RVSP, and LV cardiac output. G1 attenuated these PH-related alterations on the functional capacity of MCT-injected rats.
Restored RV function in MCT-treated rats was associated with the recovery of SERCA2a and PLB levels in cardiomyocytes (Alencar et al., 2014). Reduction of the expression of SERCA2a and increase of PLB, were responsible for the increased PLB/SERCA2a ratio in RV from MCT-injected animals. These changes suggest that Ca2+ uptake by the sarcoplasmic reticulum is reduced, which contributes to the Ca2+ overload. Consequently, the release of less Ca2+ upon activation reduces force development, corroborating with the lower ejection fraction in MCT-treated group. The administration of G1 recovered the relative ratio of PLB/SERCA2a, in RV from MCT-induced PH rats. This effect likely contributed to the improved heart systolic performance (Dow et al., 1971). Activation of ERs alters cardiomyocyte contraction and Ca2+ handling (Asp et al., 2013), and GPER mediates numerous aspects of cellular signaling, such as Ca2+ mobilization in cells (Ariazi et al., 2010; Brailoiu et al., 2013; Noel et al., 2009). Treatment of rats with G1 attenuated isoproterenol-induced heart failure, by influencing the cardiac β-adrenergic receptor signaling (Kang et al., 2012). GPER long-term activation by G1 normalized SERCA2a and PLB levels in hearts from rats with LV diastolic dysfunction (Alencar et al., 2016).
Many studies have demonstrated that TNF-α expression is elevated in cardiomyocytes and in the plasma of patients with end-stage heart failure (Levine et al., 1990; Mann, 2002). In our PH rats, the RVs showed increased expression of TNF-α 29 days after MCT injection, which might be linked to the depressed contractile activity and remodeling process observed in those hearts (Feldman et al., 2000; Fontoura et al., 2014; Tracey et al., 1989). TNF-α expression was significantly correlated with the expression of Ca2+ handling proteins SERCA2a and PLB in RV samples from our experimental groups. In vivo activation of GPER normalized TNF-α expression in RVs from MCT-treated rats, beneficially reducing the deleterious effects of PH-induced chronic myocardial inflammation and dysfunction.
The discovery of GPER has led to new insights into the cardioprotective and rapid effects of E2, beyond the genomic and nongenomic mechanisms of classic E2 receptors (ERs) (Borbely et al., 2005; Carmeci et al., 1997; Consoli et al., 2013; Filardo et al., 2000; Filardo et al., 2002; Filice et al., 2009; Kang et al., 2012; Weil et al., 2010). GPER activation with G1 preserved the diastolic function and structure, and limited the increase in LV filling pressure, LV mass, wall thickness, cardiomyocyte size, and cardiac fibrosis, in an animal model of left heart dysfunction 17 (Wang et al., 2012). In vitro studies have examined the mechanisms underlying the cardioprotective potential of GPER, focusing on its effects on cardiomyocyte hypertrophy and cardiac fibroblast proliferation. For example, G1 treatment attenuated angiotensin II-induced hypertrophy of H9c2 cardiomyocytes, with the GPER antagonist G15 inhibiting these effects of G1 (Wang et al., 2015). G1 inhibited the proliferation of exogenous GPER-expressing cardiac fibroblasts derived from male adult Sprague-Dawley rats (Wang et al., 2015). This finding rationally explains why RVs from our MCT-injected rats treated chronically with G1 showed reduced levels of collagen deposition compared to MCT + vehicle rats. Taken together, these data reveal the importance of GPER in the maintenance of cardiac structure and function, which likely involves effects on both cardiomyocytes and cardiac fibroblasts in the whole heart.
A single injection of MCT initially leads to chronic inflammation of the lung tissue, which contributes to injury of the vascular endothelium and to dysfunction. This condition results in the decreased production of nitric oxide by endothelial cells of pulmonary arterioles (Alencar et al., 2014; Zhang et al., 2005) and the downregulation of eNOS (Alencar et al., 2014; Pei et al., 2011; Sahara et al., 2012; Walford and Loscalzo, 2003). Injured pulmonary tissue from MCT-injected rats showed reduced eNOS expression in response to inflammation, which was reversed with the GPER activation in G1-treated rats. This finding may explain the improved pulmonary artery flow after G1 treatment. Increased bioavailability of nitric oxide in lungs from MCT-injected rats has promoted antiproliferative effects and prevented the development of fibromuscular hypertrophy and hyperplasia in the pulmonary arteriole walls of PH rats (Alencar et al., 2014).
To the best of our knowledge, this study is the first to demonstrate the role of GPER in the regulation of pulmonary vessel remodeling in rats with MCT-induced PH. Activation of GPER is potentially effective in blocking vascular SMC proliferation in micromolar concentrations (Haas et al., 2009), and inducing antiproliferative effects in microvascular endothelial cells (Holm et al., 2011). It was recently found that anti-remodeling effect of G1 is related to a reduction of glycosaminoglycans content in the aorta medial layer and a decrease in oxidative stress (Liu et al., 2016).
Wright et al., showed the presence of GPER in human PASMCs and that its acute activation with G1 have no proliferative effect (Wright et al., 2015). However, continuous agonism of GPER in our study was protective against PA remodeling in response to the inflammation induced by MCT, as G1 administration for 14 days reduced collagen deposition and the wall thickness in the terminal pulmonary arterioles. We suggest with this finding that GPER is also expressed in pulmonary vascular fibroblasts. Nevertheless, it should be confirmed in additional studies. Furthermore, GPER activation likely induces vasodilation and reduces vascular tone via indirect and poorly understood effects on vasoconstrictors (Meyer et al., 2011a), a beneficial profile for the treatment of exacerbated vasoconstriction during PH development. Despite the benefits of GPER activation parallel that of E2 in various tissues (Han and White, 2014; Meyer et al., 2011b), G1 has no significant genomic and nuclear functions at traditional E2 receptors ERα and ERβ (Bologa et al., 2006; Dennis et al., 2011).
5. Conclusions
Our data indicate that GPER activation by G1 reverses the unfavorable effects of MCT-induced PH on the cardiopulmonary system from male rats, probably by promoting pulmonary endothelial nitric oxide synthesis, Ca2+ handling regulation and reduction of inflammation in cardiomyocytes, and a decrease of collagen deposition by acting in pulmonary and cardiac fibroblasts. This makes G1/GPER an interesting approach for the therapeutic interventions of PH and RV failure in the future.
Supplementary Material
Acknowledgments
This work was supported by Conselho Nacional de Desenvolvimento Cientifico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Programa de Apoio a Núcleos de Experiência (PRONEX), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Instituto Nacional de Ciência e Tecnologia (INCT-INOFAR), and the National Institute on Aging (R01 AG-033727 to L.G) at the National Institutes of Health.
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- “ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS).” Nazzareno Galie, Marc Humbert, Jean-Luc Vachiery, Simon Gibbs, Irene Lang, Adam Torbicki, Gerald Simonneau, Andrew Peacock, Anton Vonk Noordegraaf, Maurice Beghetti, Ardeschir Ghofrani, Miguel Angel Gomez Sanchez, Georg Hansmann, Walter Klepetko, Patrizio Lancellotti, Marco Matucci, Theresa McDonagh, Luc A. Pierard, Pedro T. Trindade, Maurizio Zompatori and Marius Hoeper. Eur Respir J 2015; 46: 903–975. Eur Respir J. 2015;46:1855–1856. doi: 10.1183/13993003.51032-2015. [DOI] [PubMed] [Google Scholar]
- Alencar AK, da Silva JS, Lin M, Silva AM, Sun X, Ferrario CM, Cheng C, Sudo RT, Zapata-Sudo G, Wang H, Groban L. Effect of Age, Estrogen Status, and Late-Life GPER Activation on Cardiac Structure and Function in the Fischer344xBrown Norway Female Rat. J Gerontol A Biol Sci Med Sci. 2016 doi: 10.1093/gerona/glw045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alencar AK, Pereira SL, da Silva FE, Mendes LV, do Cunha VM, Lima LM, Montagnoli TL, Caruso-Neves C, Ferraz EB, Tesch R, Nascimento JH, Sant’anna CM, Fraga CA, Barreiro EJ, Sudo RT, Zapata-Sudo G. N-acylhydrazone derivative ameliorates monocrotaline-induced pulmonary hypertension through the modulation of adenosine AA2R activity. Int J Cardiol. 2014;173:154–162. doi: 10.1016/j.ijcard.2014.02.022. [DOI] [PubMed] [Google Scholar]
- Alencar AK, Pereira SL, Montagnoli TL, Maia RC, Kummerle AE, Landgraf SS, Caruso-Neves C, Ferraz EB, Tesch R, Nascimento JH, de Sant’Anna CM, Fraga CA, Barreiro EJ, Sudo RT, Zapata-Sudo G. Beneficial effects of a novel agonist of the adenosine A2A receptor on monocrotaline-induced pulmonary hypertension in rats. Br J Pharmacol. 2013;169:953–962. doi: 10.1111/bph.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariazi EA, Brailoiu E, Yerrum S, Shupp HA, Slifker MJ, Cunliffe HE, Black MA, Donato AL, Arterburn JB, Oprea TI, Prossnitz ER, Dun NJ, Jordan VC. The G protein-coupled receptor GPR30 inhibits proliferation of estrogen receptor-positive breast cancer cells. Cancer Res. 2010;70:1184–1194. doi: 10.1158/0008-5472.CAN-09-3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asp ML, Martindale JJ, Metzger JM. Direct, differential effects of tamoxifen, 4-hydroxytamoxifen, and raloxifene on cardiac myocyte contractility and calcium handling. PLoS One. 2013;8:e78768. doi: 10.1371/journal.pone.0078768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiker FA, De Windt LJ, van Eickels M, Grohe C, Meyer R, Doevendans PA. Estrogenic hormone action in the heart: regulatory network and function. Cardiovasc Res. 2002;53:709–719. doi: 10.1016/s0008-6363(01)00526-0. [DOI] [PubMed] [Google Scholar]
- Badiani B, Messori A. Targeted Treatments for Pulmonary Arterial Hypertension: Interpreting Outcomes by Network Meta-analysis. Heart Lung Circ. 2016;25:46–52. doi: 10.1016/j.hlc.2015.05.020. [DOI] [PubMed] [Google Scholar]
- Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- Borbely A, van der Velden J, Papp Z, Bronzwaer JG, Edes I, Stienen GJ, Paulus WJ. Cardiomyocyte stiffness in diastolic heart failure. Circulation. 2005;111:774–781. doi: 10.1161/01.CIR.0000155257.33485.6D. [DOI] [PubMed] [Google Scholar]
- Brailoiu GC, Arterburn JB, Oprea TI, Chitravanshi VC, Brailoiu E. Bradycardic effects mediated by activation of G protein-coupled estrogen receptor in rat nucleus ambiguus. Exp Physiol. 2013;98:679–691. doi: 10.1113/expphysiol.2012.069377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. 1997;45:607–617. doi: 10.1006/geno.1997.4972. [DOI] [PubMed] [Google Scholar]
- Consoli C, Gatta L, Iellamo F, Molinari F, Rosano GM, Marlier LN. Severity of left ventricular dysfunction in heart failure patients affects the degree of serum-induced cardiomyocyte apoptosis. Importance of inflammatory response and metabolism. Int J Cardiol. 2013;167:2859–2866. doi: 10.1016/j.ijcard.2012.07.025. [DOI] [PubMed] [Google Scholar]
- Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, Oprea TI, Yamaguchi Y, Hayashi S, Sklar LA, Hathaway HJ, Arterburn JB, Prossnitz ER. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J Steroid Biochem Mol Biol. 2011;127:358–366. doi: 10.1016/j.jsbmb.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschamps AM, Murphy E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am J Physiol Heart Circ Physiol. 2009;297:H1806–1813. doi: 10.1152/ajpheart.00283.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow ML, Kirchhoefer RD, Brower JF. Rapid identification and estimation of gitoxin in digitoxin and digoxin tablets by TLC. J Pharm Sci. 1971;60:298–299. doi: 10.1002/jps.2600600232. [DOI] [PubMed] [Google Scholar]
- Dubey RK, Oparil S, Imthurn B, Jackson EK. Sex hormones and hypertension. Cardiovasc Res. 2002;53:688–708. doi: 10.1016/s0008-6363(01)00527-2. [DOI] [PubMed] [Google Scholar]
- Feldman AM, Combes A, Wagner D, Kadakomi T, Kubota T, Li YY, McTiernan C. The role of tumor necrosis factor in the pathophysiology of heart failure. J Am Coll Cardiol. 2000;35:537–544. doi: 10.1016/s0735-1097(99)00600-2. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14:1649–1660. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Frackelton AR, Jr, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16:70–84. doi: 10.1210/mend.16.1.0758. [DOI] [PubMed] [Google Scholar]
- Filice E, Recchia AG, Pellegrino D, Angelone T, Maggiolini M, Cerra MC. A new membrane G protein-coupled receptor (GPR30) is involved in the cardiac effects of 17beta-estradiol in the male rat. J Physiol Pharmacol. 2009;60:3–10. [PubMed] [Google Scholar]
- Fontoura D, Oliveira-Pinto J, Tavares-Silva M, Leite S, Vasques-Novoa F, Mendes-Ferreira P, Lourenco AP, Leite-Moreira AF. Myocardial and anti-inflammatory effects of chronic bosentan therapy in monocrotaline-induced pulmonary hypertension. Rev Port Cardiol. 2014;33:213–222. doi: 10.1016/j.repc.2013.09.016. [DOI] [PubMed] [Google Scholar]
- Frump AL, Goss KN, Vayl A, Albrecht M, Fisher A, Tursunova R, Fierst J, Whitson J, Cucci AR, Brown MB, Lahm T. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am J Physiol Lung Cell Mol Physiol. 2015;308:L873–890. doi: 10.1152/ajplung.00006.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas E, Bhattacharya I, Brailoiu E, Damjanovic M, Brailoiu GC, Gao X, Mueller-Guerre L, Marjon NA, Gut A, Minotti R, Meyer MR, Amann K, Ammann E, Perez-Dominguez A, Genoni M, Clegg DJ, Dun NJ, Resta TC, Prossnitz ER, Barton M. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res. 2009;104:288–291. doi: 10.1161/CIRCRESAHA.108.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han G, White RE. G-protein-coupled estrogen receptor as a new therapeutic target for treating coronary artery disease. World J Cardiol. 2014;6:367–375. doi: 10.4330/wjc.v6.i6.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm A, Baldetorp B, Olde B, Leeb-Lundberg LM, Nilsson BO. The GPER1 agonist G-1 attenuates endothelial cell proliferation by inhibiting DNA synthesis and accumulating cells in the S and G2 phases of the cell cycle. J Vasc Res. 2011;48:327–335. doi: 10.1159/000322578. [DOI] [PubMed] [Google Scholar]
- Humbert M, Ghofrani HA. The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax. 2016;71:73–83. doi: 10.1136/thoraxjnl-2015-207170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessup JA, Lindsey SH, Wang H, Chappell MC, Groban L. Attenuation of salt-induced cardiac remodeling and diastolic dysfunction by the GPER agonist G-1 in female mRen2.Lewis rats. PLoS One. 2010;5:e15433. doi: 10.1371/journal.pone.0015433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JE, Mendes L, Rudd MA, Russo G, Loscalzo J, Zhang YY. Serial noninvasive assessment of progressive pulmonary hypertension in a rat model. Am J Physiol Heart Circ Physiol. 2002;283(1):H364–71. doi: 10.1152/ajpheart.00979.2001. [DOI] [PubMed] [Google Scholar]
- Kang S, Liu Y, Sun D, Zhou C, Liu A, Xu C, Hao Y, Li D, Yan C, Sun H. Chronic activation of the G protein-coupled receptor 30 with agonist G-1 attenuates heart failure. PLoS One. 2012;7:e48185. doi: 10.1371/journal.pone.0048185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskenvuo JW, Mirsky R, Zhang Y, Angeli FS, Jahn S, Alastalo TP, Schiller NB, Boyle AJ, Chatterjee K, De Marco T, Yeghiazarians Y. A comparison of echocardiography to invasive measurement in the evaluation of pulmonary arterial hypertension in a rat model. Int J Cardiovasc Imaging. 2010;26:509–518. doi: 10.1007/s10554-010-9596-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise J, Solomon S, Spencer KT, St John Sutton M, Stewart W American Society of Echocardiography’s, N., Standards, C., Task Force on Chamber, Q., American College of Cardiology Echocardiography, C., American Heart, A., European Association of Echocardiography, E.S.o.C. Recommendations for chamber quantification. Eur J Echocardiogr. 2006;7:79–108. doi: 10.1016/j.euje.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–241. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- Lindsey SH, Cohen JA, Brosnihan KB, Gallagher PE, Chappell MC. Chronic treatment with the G protein-coupled receptor 30 agonist G-1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology. 2009;150:3753–3758. doi: 10.1210/en.2008-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey SH, da Silva AS, Silva MS, Chappell MC. Reduced vasorelaxation to estradiol and G-1 in aged female and adult male rats is associated with GPR30 downregulation. Am J Physiol Endocrinol Metab. 2013;305:E113–118. doi: 10.1152/ajpendo.00649.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Kashyap S, Murphy B, Hutson DD, Budish RA, Trimmer EH, Zimmerman MA, Trask AJ, Miller KS, Chappell MC, Lindsey SH. GPER activation ameliorates aortic remodeling induced by salt-sensitive hypertension. Am J Physiol Heart Circ Physiol. 2016;310:H953–961. doi: 10.1152/ajpheart.00631.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenco AP, Fontoura D, Henriques-Coelho T, Leite-Moreira AF. Current pathophysiological concepts and management of pulmonary hypertension. Int J Cardiol. 2012;155:350–361. doi: 10.1016/j.ijcard.2011.05.066. [DOI] [PubMed] [Google Scholar]
- Mair KM, Wright AF, Duggan N, Rowlands DJ, Hussey MJ, Roberts S, Fullerton J, Nilsen M, Loughlin L, Thomas M, MacLean MR. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. Am J Respir Crit Care Med. 2014;190:456–467. doi: 10.1164/rccm.201403-0483OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders E, Rain S, Bogaard HJ, Handoko ML, Stienen GJ, Vonk-Noordegraaf A, Ottenheijm CA, de Man FS. The striated muscles in pulmonary arterial hypertension: adaptations beyond the right ventricle. Eur Respir J. 2015;46:832–842. doi: 10.1183/13993003.02052-2014. [DOI] [PubMed] [Google Scholar]
- Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res. 2002;91:988–998. doi: 10.1161/01.res.0000043825.01705.1b. [DOI] [PubMed] [Google Scholar]
- McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114:1417–1431. doi: 10.1161/CIRCULATIONAHA.104.503540. [DOI] [PubMed] [Google Scholar]
- Meyer MR, Prossnitz ER, Barton M. The G protein-coupled estrogen receptor GPER/GPR30 as a regulator of cardiovascular function. Vascul Pharmacol. 2011a;55:17–25. doi: 10.1016/j.vph.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MR, Prossnitz ER, Barton M. GPER/GPR30 and Regulation of Vascular Tone and Blood Pressure. Immunol Endocr Metab Agents Med Chem. 2011b;11:255–261. doi: 10.2174/1871522211108040255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neder JA, Ramos RP, Ota-Arakaki JS, Hirai DM, D’Arsigny CL, O’Donnell D. Exercise intolerance in pulmonary arterial hypertension. The role of cardiopulmonary exercise testing. Ann Am Thorac Soc. 2015;12:604–612. doi: 10.1513/AnnalsATS.201412-558CC. [DOI] [PubMed] [Google Scholar]
- Nishida M, Hasegawa Y, Tanida I, Nakagawa E, Inaji H, Ohkita M, Matsumura Y. Preventive effects of raloxifene, a selective estrogen receptor modulator, on monocrotaline-induced pulmonary hypertension in intact and ovariectomized female rats. Eur J Pharmacol. 2009;614:70–76. doi: 10.1016/j.ejphar.2009.04.016. [DOI] [PubMed] [Google Scholar]
- Noel SD, Keen KL, Baumann DI, Filardo EJ, Terasawa E. Involvement of G protein-coupled receptor 30 (GPR30) in rapid action of estrogen in primate LHRH neurons. Mol Endocrinol. 2009;23:349–359. doi: 10.1210/me.2008-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. 2007;30:104–109. doi: 10.1183/09031936.00092306. [DOI] [PubMed] [Google Scholar]
- Pei Y, Ma P, Wang X, Zhang W, Zhang X, Zheng P, Yan L, Xu Q, Dai G. Rosuvastatin attenuates monocrotaline-induced pulmonary hypertension via regulation of Akt/eNOS signaling and asymmetric dimethylarginine metabolism. Eur J Pharmacol. 2011;666:165–172. doi: 10.1016/j.ejphar.2011.05.035. [DOI] [PubMed] [Google Scholar]
- Sahara M, Sata M, Morita T, Hirata Y, Nagai R. Nicorandil attenuates monocrotaline-induced vascular endothelial damage and pulmonary arterial hypertension. PLoS One. 2012;7:e33367. doi: 10.1371/journal.pone.0033367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol. 2011;8:443–455. doi: 10.1038/nrcardio.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofovic SP, Jackson EK. Complexities of oestradiol pharmacology in pulmonary arterial hypertension. Eur Respir J. 2013;41:1465–1466. doi: 10.1183/09031936.00164912. [DOI] [PubMed] [Google Scholar]
- Tracey KJ, Vlassara H, Cerami A. Cachectin/tumour necrosis factor. Lancet. 1989;1:1122–1126. doi: 10.1016/s0140-6736(89)92394-5. [DOI] [PubMed] [Google Scholar]
- van Suylen RJ, Smits JF, Daemen MJ. Pulmonary artery remodeling differs in hypoxia- and monocrotaline-induced pulmonary hypertension. Am J Respir Crit Care Med. 1998;157:1423–1428. doi: 10.1164/ajrccm.157.5.9709050. [DOI] [PubMed] [Google Scholar]
- Vitali SH, Hansmann G, Rose C, Fernandez-Gonzalez A, Scheid A, Mitsialis SA, Kourembanas S. The Sugen 5416/hypoxia mouse model of pulmonary hypertension revisited: long-term follow-up. Pulm Circ. 2014;4:619–629. doi: 10.1086/678508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, Dupuis J, Long CS, Rubin LJ, Smart FW, Suzuki YJ, Gladwin M, Denholm EM, Gail DB National Heart, L., Blood Institute Working Group on, C., Molecular Mechanisms of Right Heart, F. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation. 2006;114:1883–1891. doi: 10.1161/CIRCULATIONAHA.106.632208. [DOI] [PubMed] [Google Scholar]
- Walford G, Loscalzo J. Nitric oxide in vascular biology. J Thromb Haemost. 2003;1:2112–2118. doi: 10.1046/j.1538-7836.2003.00345.x. [DOI] [PubMed] [Google Scholar]
- Wang H, Alencar A, Lin M, Sun X, Sudo RT, Zapata-Sudo G, Lowe DA, Groban L. Activation of GPR30 improves exercise capacity and skeletal muscle strength in senescent female Fischer344 x Brown Norway rats. Biochem Biophys Res Commun. 2016;475:81–86. doi: 10.1016/j.bbrc.2016.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Jessup JA, Lin MS, Chagas C, Lindsey SH, Groban L. Activation of GPR30 attenuates diastolic dysfunction and left ventricle remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc Res. 2012;94:96–104. doi: 10.1093/cvr/cvs090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhao Z, Lin M, Groban L. Activation of GPR30 inhibits cardiac fibroblast proliferation. Mol Cell Biochem. 2015;405:135–148. doi: 10.1007/s11010-015-2405-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil BR, Manukyan MC, Herrmann JL, Wang Y, Abarbanell AM, Poynter JA, Meldrum DR. Signaling via GPR30 protects the myocardium from ischemia/reperfusion injury. Surgery. 2010;148:436–443. doi: 10.1016/j.surg.2010.03.011. [DOI] [PubMed] [Google Scholar]
- Wright AF, Ewart MA, Mair K, Nilsen M, Dempsie Y, Loughlin L, Maclean MR. Oestrogen receptor alpha in pulmonary hypertension. Cardiovasc Res. 2015;106:206–216. doi: 10.1093/cvr/cvv106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Niu W, Luo Y, Zhang B, Liu M, Dong H, Liu Y, Li Z. Endogenous estrogen attenuates hypoxia-induced pulmonary hypertension by inhibiting pulmonary arterial vasoconstriction and pulmonary arterial smooth muscle cells proliferation. Int J Med Sci. 2013;10:771–781. doi: 10.7150/ijms.5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XP, Reckelhoff JF. Estrogen, hormonal replacement therapy and cardiovascular disease. Curr Opin Nephrol Hypertens. 2011;20:133–138. doi: 10.1097/MNH.0b013e3283431921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapata-Sudo G, Pontes LB, da Silva JS, Lima LM, Nunes IK, Barreiro EJ, Sudo RT. Benzenesulfonamide attenuates monocrotaline-induced pulmonary arterial hypertension in a rat model. Eur J Pharmacol. 2012;690:176–182. doi: 10.1016/j.ejphar.2012.05.043. [DOI] [PubMed] [Google Scholar]
- Zhang TT, Cui B, Dai DZ, Su W. CPU 86017, p-chlorobenzyltetrahydroberberine chloride, attenuates monocrotaline-induced pulmonary hypertension by suppressing endothelin pathway. Acta Pharmacol Sin. 2005;26:1309–1316. doi: 10.1111/j.1745-7254.2005.00214.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.