

Abstract
Reactive oxygen species (ROS) and oxidative stress are closely associated with the development of atherosclerosis, and the most important regulator of ROS production in endothelial cells is NADPH oxidase. Activation of NADPH oxidase requires the assembly of multiple subunits into lipid rafts, which include specific lipid components, including free cholesterol and specific proteins. Disorders of lipid metabolism such as hyperlipidemia affect the cellular lipid components included in rafts, resulting in modification of cellular reactions that produce ROS. In the similar manner, several pathways associating ROS production are affected by the presence of lipid disorder through raft compartments. In this manuscript, we review the pathophysiological implications of hyperlipidemia and lipid rafts in the production of ROS.
Keywords: Lipid raft, Hyperlipidemia, Free cholesterol, Reactive oxygen species, NADPH oxidase
Core tip: Lipid raft is a membrane microdomain in which specific combinations of lipid components such as free cholesterol and proteins function to mediate and amplify a variety of cellular signals. The platform has a significant impact on the cellular reactions such as the production of reactive oxygen species, however, there are limited articles on the clinical relevance of this platform. Lipid disorder, such as hyperlipidemia, is one that significantly affects the platform, with the modification of associating cell functions in various ways. We focused on the effect derived from this platform in hyperlipidemia in this manuscript.
REACTIVE OXYGEN AND VASCULAR INJURY
Reactive oxygen species (ROS) and oxidative stress are considered key mediators of atherosclerosis[1]. ROS are involved in the progression of endothelial-cell dysfunction, which is accompanied by inactivation of endothelial nitric oxide synthase (eNOS) and decrease of nitric oxide (NO) levels[2]. Oxidative stress results from overproduction of ROS, failure of host antioxidant defense, or both. The effects of ROS-associated signal pathways have a meaningful impact on cellular function in endothelial cells. The most important modulator of ROS in endothelial cells is NADPH oxidase[3], and ROS metabolism is constantly modified by the surrounding environment. Pathological conditions associated with hyperlipidemia may be derived from these pathways of ROS, and the suppression of ROS may block the progression of those pathology[4].
RAFT PLATFORMS AS A REGULATOR OF ROS
Lipid rafts or membrane rafts are membrane microdomains in which specific combinations of lipid components and proteins function to mediate and amplify a variety of cellular signals[5]. Rafts are dynamic assemblies of cholesterol and lipids with saturated acyl chains, such as sphingolipids and glycosphingolipids in the exoplasmic leaflet of the membrane bilayer; and cholesterol in the inner leaflet. Intracellular reactions that produce ROS in endothelial cells can occur in lipid rafts, as a plasma membrane-associated NADPH oxidase complex exists within that compartment[6]. Clustering of lipid rafts in the cell membrane of endothelial cells causes the aggregation and activation of NADPH oxidase, thereby forming a redox signaling platform[7].
Raft structure and composition differ in various pathological states. Extracellular free cholesterol can be directly incorporated into the plasma membrane, leading to increase in cellular cholesterol levels[8]. Fang et al[9] showed that hypercholesterolemia increased the level of cellular free cholesterol approximately two-to four-fold in vascular endothelial cells[8]. The presence of very low-density lipoprotein (LDL) can cause a 50%-100% increase in total-cell unesterified cholesterol[10]. Indeed, endothelial cells are more likely to accumulate free rather than esterified cholesterol due to low ratio of hydrolysis to esterification. As a result, an increase in free cholesterol in endothelial cells causes a change in plasma membrane cholesterol content and may contribute to alterations in membrane function[11]. Similarly, hypercholesterolemia is also reported to alter the composition of lipid rafts and affect cell function in smooth muscle cells[12].
These pathological modifications of raft components affect ROS production. For example, a reduction of free cholesterol in rafts attenuates ROS production, leading to the suppression of ROS-associated downstream pathways[13]. By contrast, increase of plasma membrane free cholesterol leads to the modification of associated reactions that enhance ROS production[9]. Other conditions are known to affect the lipid components of rafts. For instance, aging has been associated with changes in sphingolipid and cholesterol, leading to the production of long-chain ceramides in plasma membrane[14] and the resulting enhancement of membrane-associated oxidative stress contributes to the progression of Alzheimer disease.
Not only lipid content of rafts but also specific proteins influence the behavior of associated reactions. Caveolin is an essential protein component of caveolae, which are unique raft compartments in the plasma membrane of endothelial cells[15]. Caveolin interacts with both lipids and lipid anchors on the raft proteins, and it functions as a scaffolding protein to organize and concentrate specific lipids and lipid-modified signaling molecules within the rafts[12,16]. In the presence of hypercholesterolemia, caveolin binding to eNOS is enhanced, leading to eNOS inactivation[17]. The resulting decrease in NO production has a significant impact on ROS metabolism. Hypercholesterolemia thus affects the production of ROS by a caveolin-associated pathway. Lobysheva et al[18] demonstrated that Caveolin-1 modulated the ROS behavior by regulating the balance of eNOS-derived NO. An increase in caveolin and eNOS interactions that occur with hyperlipidemia, may act to decrease NO production and promote endothelial dysfunction and atherosclerotic lesion formation[17].
The spatial compartmentation of eNOS in the raft compartment also has a significant impact of the behavior of ROS, in especially the cross-talk between NO and ROS. Under normal conditions, eNOS is associated with cholesterol-enriched caveolae in endothelial cells, where its activity can be closely regulated. However, in hyperlipidemia, lipoprotein particles modulate the activity and subcellular distribution of eNOS[19]. Incubation of endothelial cells with LDL, particularly oxidized LDL (ox-LDL), causes an increase in the binding of eNOS to CD36, which attenuates its activity and causes displacement of the protein from endothelial caveolae. In addition, the spatial interaction between eNOS and NADPH oxidase determines net NO and ROS production because the NO produced adjacent to NADPH oxidase is scavenged by the ROS[20]. Therefore, the pathological condition affects localization of ROS-associated molecules, resulting a change in the output from these pathways.
Rafts can also be platforms that enhance the production of reactive nitrogen. Yang et al[21] reported that TNF-α enhanced ROS production within these membrane compartments concomitant with recruitment of the p47phox regulatory subunit of NADPH oxidase subunit domains. In addition, TNF-α induced activation and phosphorylation of eNOS present in plasma membrane raft compartments. The dual activation of superoxide-generating and NO-generating systems within the same membrane domains provided a spatially favorable environment for formation of peroxynitrite.
Conversely, raft compartments are also susceptible to the oxidative reactions, resulting the oxidation of lipid components and modifying the associating reactions. For instance, 7-ketocholesterol, one oxidized form of cholesterol, was reported to deplete cholesterol from the raft domains and disrupt it[22,23]. However, the exact results of membrane injury by oxidized lipids are uncertain and are beyond the scope of this manuscript.
RAFT CONDITIONS AND ASSOCIATED REACTIONS
The association of rafts and the actin cytoskeletal network has been reported to affect the endocytic pathway. For instance, when the vacuolating cytotoxin (VacA), a major virulence factor of Helicobacter pylori, was continuously associated with raft compartments it was routed to early endosome antigen 1-sorting endosomes and then sorted to late endosomes[24]. We previously reported that intracellular vesicle structures in endothelial cells act as a raft-like domains that move along the actin cytoskeleton network (Figure 1)[13].
Figure 1.
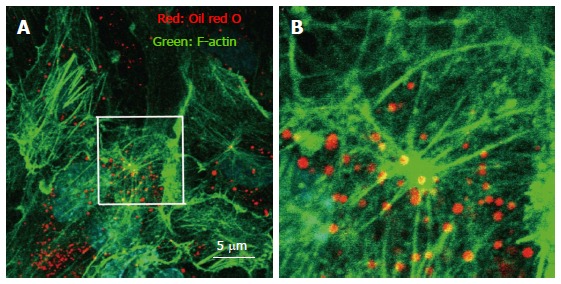
Immunohistochemistry of actin, and visualization of vesicle structures after free cholesterol loading and angiotensin II in cultured human aortic endothelial cells. The cells were loaded by cholesterol-saturated methyl-b-cyclodextrin (Sigma, St. Louis, MO) (Chol/MBCD) and angiotensin II (Wako, Tokyo, Japan) (200 nmol/L). Following treatment, cell were fixed, and stained using Alexa 546-conjugated phalloidin (Invitrogen, Carlsbad, CA) for visualization of F-actin and oil red O for visualization of vesicle structure. Oil red O-positive vesicles formed, and moved along the F-actin filament in the setting of actin remodeling induced by angiotensin II. B is a magnified view of the white square in A.
The most common raft protein, caveolin, can also be found in these endocytic pathways, such as late endosomes and lysosomes. Once it is ubiquitinated, it is transferred into intraluminal vesicles in endosomes for degradation using the endosomal sorting complex required for transport machinery[25]. During this translocation, caveolin is also recruited by accessory membrane compartments that affect its interactions with other intracellular compartments. Changes in lipid raft-based membrane compartmentation can involve movement of key molecules that modify intracellular dynamics. ROS production is one of the activities affected by the translocation of raft compartments. Indeed, NADPH oxidase-dependent ROS production in endosomes is seen as a proinflammatory immune response. Li et al[26] have demonstrated that interleukin-1β (IL-1β) stimulation promotes endocytosis of the IL-1β receptor (IL-1R1), leading to NADPH oxidase-dependent ROS production in early endosomes and subsequent redox-dependent activation of transcription factor NF-κB.
Previous reports demonstrated that visfatin activated lysosomal acid sphingomyelinase (ASM), the formation of raft redox signaling platforms, and consequent local oxidative stress[27]. Lysosome-associated molecular trafficking and the resulting ceramide accumulation in the cell membrane may mediate the assembly of NADPH oxidase subunits and their activation in response to adipokine visfatin in coronary artery endothelial cells, thereby producing endothelial dysfunction in the coronary vasculature.
In addition to intercellular vesicle structures, extracellular vesicle structures have been reported to associate with raft components[28]. Characterization of human B-cell-derived exosomes showed an abundance of membrane raft-associated lipids, including cholesterol and sphingomyelin[29]. Indeed, we found that modification of raft lipid components affected changes of molecules in vesicle structures (unpublished data). In addition, endothelial microparticles induced by angiotensin II through the NADPH oxidase pathway, have been shown to associate with lipid raft[30]. These findings suggest that cholesterol metabolism affects the behavior of extracellular vesicles that can have an effect on pathological conditions. However, the physiological and pathological role of extracellular vesicles had not yet been elucidated. Further study of the mechanisms underlying the relationships of raft compartments and the extracellular vesicles produced by endothelial cells is warranted.
EFFECT OF STATINS ON RAFT COMPLEXES
Statins, inhibitors of HMG-CoA reductase, block cholesterol biosynthesis by inhibiting the mevalonate pathway, thereby producing a dramatic reduction in circulating LDL-cholesterol. Statins also exhibit non-cholesterol-lowering activities, including inhibition of inflammatory responses by immune cells such as macrophages and lymphocytes[31]. Statins also affect intracellular cholesterol pharmacokinetics, leading to other pleiotropic effects.
By interacting with the raft compartment, statins have been reported to inhibit the formation of raft redox signaling platforms and to decrease production of oxidized LDL in endothelial cells stimulated by a proatherogenic factor[32]. The inhibitory effect of statins on raft-redox signaling is associated with their vascular protective effects. Ponce et al[33] demonstrated that small reductions of intracellular cholesterol levels by simvastatin were associated with reduction in neuronal excitotoxity. The mechanism was found to be related to the translocation of NMDA receptors from raft compartment[33]. Other groups have found that statins inhibit OxLDL-induced ASM translocation and ceramide production in human aortic endothelial cells[34]. Previous studies have shown that lysosomal trafficking and translocation of ASM into membrane rafts results in ceramide production, membrane raft clustering, and formation of ceramide-enriched macrodomains[27]. Statins inhibit this ceramide formation, leading to the protection of endothelial function.
Raft cholesterol content affects cell function and changes in raft cholesterol content in response to statins have been shown to impact cell function. Zhuang et al[35] demonstrated that simvastatin lowered raft cholesterol content, leading to inhibition of Akt/PKB pathway signaling and induction of apoptosis in caveolin-negative and phosphatase and tensin homolog-negative LNCaP prostate cancer cells. On the other hand, cholesterol elevation also promoted tumor growth, increased phosphorylation of Akt, and decreased apoptosis in the xenografts.
We also observed that free cholesterol loading-induced vesicle structures were significantly suppressed by statin pretreatment (Figure 2). Intracellular vesicle structure was considered an intracellular raft platform, and statin affected the behavior of these platforms. As a result, the activity of platforms where key ROS-producing molecules are assembled may be decreased, with reduction of intracellular oxidative stress[13]. However, there had been little reports about the clinical effects of raft modifying agents other than statin. Further studies investigating about it is warranted.
Figure 2.
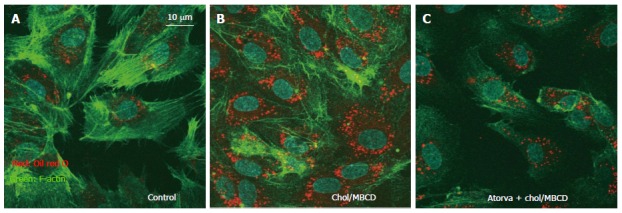
Immunohistochemistry of actin and visualization of vesicle structures after free cholesterol loading and atorvastatin pretreatment in cultured human aortic endothelial cells. The cells were loaded by cholesterol-saturated methyl-b-cyclodextrin (Chol/MBCD) with and without atorvastatin (10 μmol/L) pretreatment. Atorvastatin (Pfizer, New York, NY) pretreatment (C) significantly suppressed formation of vesicles induced by free cholesterol loading, as shown by oil red O as compared with Chol/MBCD loading alone (B); A: Control.
CONCLUSION
This review described how ROS production is affected by the modification of lipid raft compartments in hyperlipidemia. The concept of lipid rafts may stimulate the development of novel therapeutic strategies for hyperlipidemia-associated pathologies. However, there had been little reports that demonstrated the clinical implication and importance of lipid raft compartments in lipid disorder. Further studies investigating about the associations between raft compartment and pathologic changes are needed.
Footnotes
Supported by The Ministry of Education, Culture, Sports, Science and Technology of Japan through grant-in-aid 26461103 (to Amiya E).
Conflict-of-interest statement: The author declares no conflicts of interest.
Manuscript source: Invited manuscript
Specialty type: Cardiac and cardiovascular systems
Country of origin: Japan
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B, B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: July 26, 2016
First decision: September 6, 2016
Article in press: October 9, 2016
P- Reviewer: Cheng TH, Fujiwara N, Kukongviriyapan V, Tonks A S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
References
- 1.Stocker R, Keaney JF. Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 2.Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 3.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 4.Araujo FB, Barbosa DS, Hsin CY, Maranhão RC, Abdalla DS. Evaluation of oxidative stress in patients with hyperlipidemia. Atherosclerosis. 1995;117:61–71. doi: 10.1016/0021-9150(94)05558-z. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Li X, Becker KA, Gulbins E. Ceramide-enriched membrane domains--structure and function. Biochim Biophys Acta. 2009;1788:178–183. doi: 10.1016/j.bbamem.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 6.Frey RS, Ushio-Fukai M, Malik AB. NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxid Redox Signal. 2009;11:791–810. doi: 10.1089/ars.2008.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang AY, Yi F, Zhang G, Gulbins E, Li PL. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension. 2006;47:74–80. doi: 10.1161/10.1161/01.HYP.0000196727.53300.62. [DOI] [PubMed] [Google Scholar]
- 8.Qin C, Nagao T, Grosheva I, Maxfield FR, Pierini LM. Elevated plasma membrane cholesterol content alters macrophage signaling and function. Arterioscler Thromb Vasc Biol. 2006;26:372–378. doi: 10.1161/01.ATV.0000197848.67999.e1. [DOI] [PubMed] [Google Scholar]
- 9.Fang Y, Mohler ER, Hsieh E, Osman H, Hashemi SM, Davies PF, Rothblat GH, Wilensky RL, Levitan I. Hypercholesterolemia suppresses inwardly rectifying K+ channels in aortic endothelium in vitro and in vivo. Circ Res. 2006;98:1064–1071. doi: 10.1161/01.RES.0000218776.87842.43. [DOI] [PubMed] [Google Scholar]
- 10.Ellsworth JL, Erickson SK, Cooper AD. Very low and low density lipoprotein synthesis and secretion by the human hepatoma cell line Hep-G2: effects of free fatty acid. J Lipid Res. 1986;27:858–874. [PubMed] [Google Scholar]
- 11.Kim JA, Maxwell K, Hajjar DP, Berliner JA. Beta-VLDL increases endothelial cell plasma membrane cholesterol. J Lipid Res. 1991;32:1125–1131. [PubMed] [Google Scholar]
- 12.Morikage N, Kishi H, Sato M, Guo F, Shirao S, Yano T, Soma M, Hamano K, Esato K, Kobayashi S. Cholesterol primes vascular smooth muscle to induce Ca2 sensitization mediated by a sphingosylphosphorylcholine-Rho-kinase pathway: possible role for membrane raft. Circ Res. 2006;99:299–306. doi: 10.1161/01.RES.0000235877.33682.e9. [DOI] [PubMed] [Google Scholar]
- 13.Amiya E, Watanabe M, Takeda N, Saito T, Shiga T, Hosoya Y, Nakao T, Imai Y, Manabe I, Nagai R, et al. Angiotensin II impairs endothelial nitric-oxide synthase bioavailability under free cholesterol-enriched conditions via intracellular free cholesterol-rich membrane microdomains. J Biol Chem. 2013;288:14497–14509. doi: 10.1074/jbc.M112.448522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patel HH, Insel PA. Lipid rafts and caveolae and their role in compartmentation of redox signaling. Antioxid Redox Signal. 2009;11:1357–1372. doi: 10.1089/ars.2008.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Labrecque L, Royal I, Surprenant DS, Patterson C, Gingras D, Béliveau R. Regulation of vascular endothelial growth factor receptor-2 activity by caveolin-1 and plasma membrane cholesterol. Mol Biol Cell. 2003;14:334–347. doi: 10.1091/mbc.E02-07-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feron O, Dessy C, Moniotte S, Desager JP, Balligand JL. Hypercholesterolemia decreases nitric oxide production by promoting the interaction of caveolin and endothelial nitric oxide synthase. J Clin Invest. 1999;103:897–905. doi: 10.1172/JCI4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lobysheva I, Rath G, Sekkali B, Bouzin C, Feron O, Gallez B, Dessy C, Balligand JL. Moderate caveolin-1 downregulation prevents NADPH oxidase-dependent endothelial nitric oxide synthase uncoupling by angiotensin II in endothelial cells. Arterioscler Thromb Vasc Biol. 2011;31:2098–2105. doi: 10.1161/ATVBAHA.111.230623. [DOI] [PubMed] [Google Scholar]
- 19.Blair A, Shaul PW, Yuhanna IS, Conrad PA, Smart EJ. Oxidized low density lipoprotein displaces endothelial nitric-oxide synthase (eNOS) from plasmalemmal caveolae and impairs eNOS activation. J Biol Chem. 1999;274:32512–32519. doi: 10.1074/jbc.274.45.32512. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Q, Malik P, Pandey D, Gupta S, Jagnandan D, Belin de Chantemele E, Banfi B, Marrero MB, Rudic RD, Stepp DW, et al. Paradoxical activation of endothelial nitric oxide synthase by NADPH oxidase. Arterioscler Thromb Vasc Biol. 2008;28:1627–1633. doi: 10.1161/ATVBAHA.108.168278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang B, Oo TN, Rizzo V. Lipid rafts mediate H2O2 prosurvival effects in cultured endothelial cells. FASEB J. 2006;20:1501–1503. doi: 10.1096/fj.05-5359fje. [DOI] [PubMed] [Google Scholar]
- 22.Gaus K, Kritharides L, Schmitz G, Boettcher A, Drobnik W, Langmann T, Quinn CM, Death A, Dean RT, Jessup W. Apolipoprotein A-1 interaction with plasma membrane lipid rafts controls cholesterol export from macrophages. FASEB J. 2004;18:574–576. doi: 10.1096/fj.03-0486fje. [DOI] [PubMed] [Google Scholar]
- 23.Rentero C, Zech T, Quinn CM, Engelhardt K, Williamson D, Grewal T, Jessup W, Harder T, Gaus K. Functional implications of plasma membrane condensation for T cell activation. PLoS One. 2008;3:e2262. doi: 10.1371/journal.pone.0002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gauthier NC, Monzo P, Kaddai V, Doye A, Ricci V, Boquet P. Helicobacter pylori VacA cytotoxin: a probe for a clathrin-independent and Cdc42-dependent pinocytic pathway routed to late endosomes. Mol Biol Cell. 2005;16:4852–4866. doi: 10.1091/mbc.E05-05-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayer A, Stoeber M, Ritz D, Engel S, Meyer HH, Helenius A. Caveolin-1 is ubiquitinated and targeted to intralumenal vesicles in endolysosomes for degradation. J Cell Biol. 2010;191:615–629. doi: 10.1083/jcb.201003086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Q, Harraz MM, Zhou W, Zhang LN, Ding W, Zhang Y, Eggleston T, Yeaman C, Banfi B, Engelhardt JF. Nox2 and Rac1 regulate H2O2-dependent recruitment of TRAF6 to endosomal interleukin-1 receptor complexes. Mol Cell Biol. 2006;26:140–154. doi: 10.1128/MCB.26.1.140-154.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia M, Zhang C, Boini KM, Thacker AM, Li PL. Membrane raft-lysosome redox signalling platforms in coronary endothelial dysfunction induced by adipokine visfatin. Cardiovasc Res. 2011;89:401–409. doi: 10.1093/cvr/cvq286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. 2014:3. doi: 10.3402/jev.v3.24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wubbolts R, Leckie RS, Veenhuizen PT, Schwarzmann G, Möbius W, Hoernschemeyer J, Slot JW, Geuze HJ, Stoorvogel W. Proteomic and biochemical analyses of human B cell-derived exosomes. Potential implications for their function and multivesicular body formation. J Biol Chem. 2003;278:10963–10972. doi: 10.1074/jbc.M207550200. [DOI] [PubMed] [Google Scholar]
- 30.Burger D, Montezano AC, Nishigaki N, He Y, Carter A, Touyz RM. Endothelial microparticle formation by angiotensin II is mediated via Ang II receptor type I/NADPH oxidase/ Rho kinase pathways targeted to lipid rafts. Arterioscler Thromb Vasc Biol. 2011;31:1898–1907. doi: 10.1161/ATVBAHA.110.222703. [DOI] [PubMed] [Google Scholar]
- 31.Chyu KY, Lio WM, Dimayuga PC, Zhou J, Zhao X, Yano J, Trinidad P, Honjo T, Cercek B, Shah PK. Cholesterol lowering modulates T cell function in vivo and in vitro. PLoS One. 2014;9:e92095. doi: 10.1371/journal.pone.0092095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li D, Chen H, Romeo F, Sawamura T, Saldeen T, Mehta JL. Statins modulate oxidized low-density lipoprotein-mediated adhesion molecule expression in human coronary artery endothelial cells: role of LOX-1. J Pharmacol Exp Ther. 2002;302:601–605. doi: 10.1124/jpet.102.034959. [DOI] [PubMed] [Google Scholar]
- 33.Ponce J, de la Ossa NP, Hurtado O, Millan M, Arenillas JF, Dávalos A, Gasull T. Simvastatin reduces the association of NMDA receptors to lipid rafts: a cholesterol-mediated effect in neuroprotection. Stroke. 2008;39:1269–1275. doi: 10.1161/STROKEAHA.107.498923. [DOI] [PubMed] [Google Scholar]
- 34.Wei YM, Li X, Xiong J, Abais JM, Xia M, Boini KM, Zhang Y, Li PL. Attenuation by statins of membrane raft-redox signaling in coronary arterial endothelium. J Pharmacol Exp Ther. 2013;345:170–179. doi: 10.1124/jpet.112.201442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J Clin Invest. 2005;115:959–968. doi: 10.1172/JCI200519935. [DOI] [PMC free article] [PubMed] [Google Scholar]