

Abstract
Pulmonary hypertension (PH), a serious disorder with a high morbidity and mortality rate, is known to occur in a number of unrelated systemic diseases. Several hematological disorders such as sickle cell disease, thalassemia and myeloproliferative diseases develop PH which worsens the prognosis. Associated oxidant injury and vascular inflammation cause endothelial damage and dysfunction. Pulmonary vascular endothelial damage/dysfunction is an early event in PH resulting in the loss of vascular reactivity, activation of proliferative and antiapoptotic pathways leading to vascular remodeling, elevated pulmonary artery pressure, right ventricular hypertrophy and premature death. Hemolysis observed in hematological disorders leads to free hemoglobin which rapidly scavenges nitric oxide (NO), limiting its bioavailability, and leading to endothelial dysfunction. In addition, hemolysis releases arginase into the circulation which converts L-arginine to ornithine, thus bypassing NO production. Furthermore, treatments for hematological disorders such as immunosuppressive therapy, splenectomy, bone marrow transplantation, and radiation have been shown to contribute to the development of PH. Recent studies have shown deregulated iron homeostasis in patients with cardiopulmonary diseases including pulmonary arterial hypertension (PAH). Several studies have reported low iron levels in patients with idiopathic PAH, and iron deficiency is an important risk factor. This article reviews PH associated with hematological disorders and its mechanism; and iron homeostasis and its relevance to PH.
Keywords: Anemia, Hemolysis, Iron homeostasis, Myelofibrosis, Pulmonary hypertension
Core tip: Oxidant injury, inflammation, impaired nitric oxide bioavailability and coagulopathy that occur in hematological diseases lead to endothelial dysfunction and thrombo-embolism with subsequent development of pulmonary hypertension (PH). In addition, treatment used for these disorders such as immunosuppressive drugs, splenectomy, bone marrow transplantation and radiation therapy are also known to cause endothelial damage and thrombo-embolism leading to PH. Furthermore, there is a causal relationship between vascular and hematopoietic systems. Patients with chronic myeloproliferative diseases are at a risk of developing PH; and the occurrence of myelofibrosis contributing to impaired hematopoiesis is not uncommon in PH.
INTRODUCTION
Pulmonary hypertension (PH) is a devastating sequela of a number of diverse systemic diseases including cardiopulmonary, autoimmune, inflammatory and myeloproliferative diseases, drug toxicity, acquired immunodeficiency syndrome, portal hypertension, and hemolytic anemia. Based on the clinical diagnosis, PH is classified into 5 major groups, which was updated in 2013[1]. Group 1 is labeled pulmonary arterial hypertension (PAH). Included in this group are idiopathic and heritable PAH, PAH associated with human immunodeficiency viral infection, schistosomiasis, congenital heart defect, connective tissue diseases, portal hypertension and drug-induced PAH. In the current updated classification, PH associated with hematological disorders, myeloproliferative diseases and splenectomy has been moved to Group 5. Pulmonary veno-occlusive disease (PVOD)/pulmonary capillary hemangioma and persistent PH of the newborn are in Group 1 as subcategories (1′ and 1″ respectively). Group 2 comprises PH associated with congenital and acquired left heart diseases, Group 3 includes PH due to lung diseases and/or hypoxia, Group 4 includes chronic thromboembolic pulmonary hypertension (CTEPH). PH associated with hematological disorders, myeloproliferative diseases, splenectomy and a number of miscellaneous systemic and metabolic disorders are included in group 5. PH is defined as a mean pulmonary artery (PA) pressure of ≥ 25 mmHg at rest as measured by cardiac catheterization. Right heart catheterization is considered the gold standard for the diagnosis of PH. Echocardiography is a useful noninvasive tool to estimate right ventricular systolic pressure (in the absence of right heart obstruction) for screening and monitoring the patients with PH[2].
Pulmonary vascular endothelial injury/disruption is considered to be an important initiating factor in the development of PH. The severity, the extent and the site of endothelial damage may determine the type of PH and the irreversibility of the disease. Endothelial cells (EC), a non-thrombogenic monocellular layer function as an interface between the circulating blood and the underlying tissue. EC produce vasorelaxants such as nitric oxide (NO), prostacyclin, and endothelium-derived hyperpolarizing factor. In addition, EC inhibit cell proliferation, and participate in inflammation, thrombosis, barrier function, cell cycle and apoptosis; EC control vascular tone and structure, maintain homeostasis, thus, participate in vascular pathobiology. NO, generated from L-arginine by catalytic activity of endothelial NO synthase (eNOS) in vascular EC is a short-lived free radical; it stimulates soluble guanylate cyclase that catalyzes guanosine triphosphate to cyclic guanosine monophopshate (cGMP). Increase in cGMP results in a decrease in Ca2+ levels that mediates NO functions including vascular relaxation[3]. eNOS is localized in special cellular domains in EC including Golgi bodies and plasmalemmal caveolae, and is tightly regulated by a variety of transcriptional, post-transcriptional and post-translational mechanisms. The proteins that modulate the eNOS activity include caveolin-1, heat shock protein 90, cationic amino acid transporter 1 (arginine transporter), Ca2+-calmodulin, and others. Caveolin-1 is a scaffolding protein of caveolae found on the plasma membrane of a variety of cells including EC, smooth muscle cells (SMC) and fibroblasts. Caveolin-1 interacts with transducing molecules in caveolae and maintains these molecules in an inhibitory state. It has a dynamic relationship with eNOS. In EC, caveolin-1 inhibits NO signaling by binding to eNOS. In response to various stimuli, eNOS is dissociated from caveolin-1, and generates NO. However, caveolin-1 is essential for agonist-induced eNOS activation[3,4]. In addition, the eNOS activity is controlled by endogenous circulating inhibitors; the most important being the L-arginine analog, asymmetric dimethylarginine (ADMA). ADMA inhibits eNOS-mediated production of NO from L-arginine. A large portion of circulating ADMA is metabolized by dimethylarginine dimethylaminohydrolase (DDAH) to L-citrulline and dimethylamine. DDAH is inhibited by oxidative stress, thereby leading to ADMA accumulation and resulting EC dysfunction[5]. Recent studies have shown that erythrocytes take up and store ADMA. Following lysis of erythrocytes, proteolysis of methylated proteins generate free ADMA which then can inhibit NO production leading to EC dysfunction, and contribute to vascular disease[6]. In a group of 34 healthy individuals (age 2 d-24 years), plasma levels of ADMA has been shown to decrease with age[7].
Hemolysis is a common occurrence in a number of hematological disorders. Released free hemoglobin (Hb) as a result of hemolysis reacts with NO and forms inactive nitrate and methemoglobin, thus leading to endothelial dysfunction. In addition, arginase 1 released during hemolysis alters arginine metabolism, further reducing NO bioavailability[8,9]. Arginase 1 converts L-arginine to ornithine, a precursor of proline. Proline is an amino acid involved in collagen formation, lung fibrosis and SMC proliferation. Low arginine/ornithine ratio has been reported to be associated with high mortality. Under conditions of low arginine and tetrahydrobiopterin, eNOS is uncoupled generating reactive oxygen species[10]. These changes lead to pulmonary vascular remodeling and increased pressure. Furthermore, therapeutic measures used in patients with hemolytic disorders have been shown to be associated with PH[11]. Figure 1 depicts the alterations observed in hematological disorders that can lead to PH.
Figure 1.
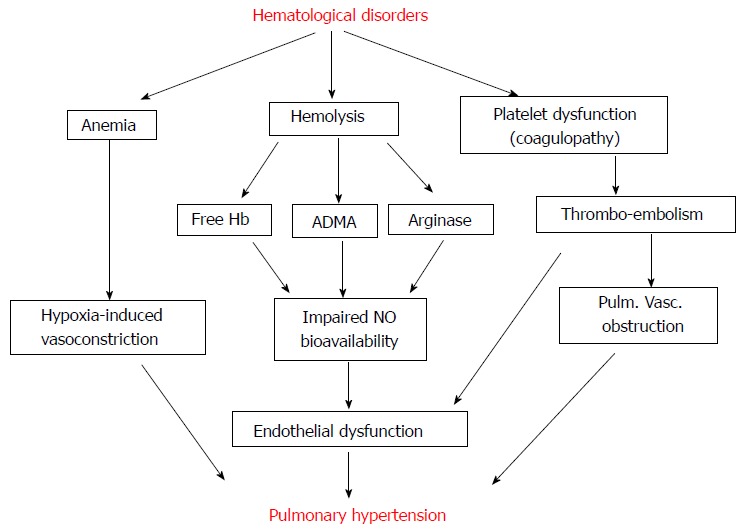
Various pathways of hematological disturbances leading to pulmonary hypertension. ADMA: Asymmetric dimethylarginine; Hb: Hemoglobin; NO: Nitric oxide; Pulm. Vasc.: Pulmonary vascular.
Iron is an essential trace element required for a number of biological processes including cellular response to hypoxia, cell proliferation, immune responses and mitochondrial function. It also has the ability to generate free radicals, which cause deleterious effects. Mitochondria use iron for heme synthesis and in iron-sulfur cluster biogenesis. Hepcidin expressed in the liver is thought to be a key regulator of iron homeostasis. Dietary iron is absorbed through the duodenal enterocytes and exported to circulation via ferroportin, an iron transporter. Increased levels of hepcidin degrade ferroportin, thus inhibit iron uptake; whereas low levels allow increased iron absorption. Hepcidin is upregulated by BMP6, and inflammatory cytokines including IL-6, IL-1β through JAK2/STAT3 pathway. It is downregulated by iron deficiency, erythropoiesis and hypoxia in order to increase iron levels. Major portion of iron is in erythroid marrow, and erythropoiesis is the major regulator of hepcidin. Erythropoiesis releases erythroferrone that in turn inhibits hepcidin transcription to increase iron absorption. Excess intracellular iron is stored by ferritin that prevents iron-mediated free radical formation[12-15]. Iron circulates bound to a glycoprotein, transferrin, which keeps it soluble; iron is delivered into cells through transferrin receptor (TfR1)[16]. Physiological iron saturation range for transferrin is 20%-45%. Less saturation is indicative of iron deficiency and saturation above 80% is associated with non-transferrin-bound iron which has toxic effect on the tissue[17]. Intracellular iron regulates TfR1 via iron responsive elements that are recognized by iron regulatory proteins (IRPs) which bind to iron responsive elements of TfR1, and prevent degradation when the intracellular iron levels are low. Increased cellular iron levels inactivate IRP1 resulting in degradation of TfR. Furthermore, IRP1 and IRP2 are required for mitochondrial iron supply and function[18,19]. Deregulation of iron homeostasis plays an important role in the pathophysiology of hematological disorders and several cardiovascular diseases including PAH. Deregulated iron metabolism can result in iron overload as seen in some of the hematological disorders leading to toxic effects, or to deficiency as seen in anemia. Several recent studies have reported low iron levels in patients with idiopathic PAH, that is considered to be an important risk factor[20].
HEMATOLOGICAL DISORDERS AND PH
Persistent pulmonary hypertension of the newborn associated with anemia
Persistent pulmonary hypertension of the newborn (PPHN) is the result of failure of cardiopulmonary transition at birth. It is associated with cardiovascular anomalies, meconium aspiration syndrome, lung hypoplasia, sepsis, respiratory distress syndrome, or it could be idiopathic. In addition, maternal factors such as diabetes, obesity, elective cesarean section; and maternal drug use such as aspirin, nonsteroidal inflammatory agents and serotonin reuptake inhibitors are known to be associated with PPHN. The incidence of PPHN is about 1.9 per 1000 live births, and the mortality is reported to be 10%. The major findings of PPHN are elevated pulmonary artery pressure, right to left shunt at the foramen ovale or at the ductus level, and hypoxemia[21,22]. Recent studies have shown that PPHN can also be associated with severe neonatal anemia. However, anemia as a potential cause of PPHN is not well recognized. In a series of 12 infants, 7 were reported to have congenital dysrythropoietic anemia; and three with ε-γ-δ β-thalassemia, one with HbH disease and another one with Diamond-Blackfan anemia[23]. Another report described 3 siblings with dysrythropoietic anemia and PPHN. Two infants survived after blood transfusion, oxygen; and one infant in addition, had received inhaled NO[24]. Others have reported PPHN associated with anemia; one infant with fetal anemia associated with maternal trophoblastic tumor, two infants with fetal anemia due to massive feto-maternal hemorrhage and in the fourth case the reason for anemia was not known. All these infants had received blood transfusion for anemia[25,26]. In addition, neonates with twin-to-twin transfusion syndrome are at a risk of developing PPHN[27]. The reason for PPHN associated with anemia is not clear. Hypoxia secondary to low Hb level could be a contributing factor to PPHN. Interestingly, booster packed red blood cells (RBCs) transfusion has been shown to improve tissue oxygenation in premature infants[25,28]. The increase in plasma Hb levels following transfusion could be an additional factor contributing to high pulmonary artery pressure. Cell-free Hb scavenges NO, thus, leading to vasoconstriction and increased pulmonary artery pressure. Experimental studies have shown transient increase in pulmonary artery pressure following blood transfusion[29]. Furthermore, transfusion with aged stored blood results in increased cell free plasma Hb levels, higher levels of arginase, endothelial dysfunction and increased pulmonary artery pressure[30,31]. Recently, significant reduction in flow-mediated dilatation was reported in adult patients who received old blood (> 21 d) compared with the ones who received fresh blood (< 14 d old)[32]. Inhaled NO prevents the elevation of pulmonary artery pressure induced by aged blood transfusion[31,32]. The possibility of PPHN needs to be considered in the presence of severe anemia in newborns. In addition to blood transfusion, inhaled NO may be necessary to ameliorate PH.
Hemolytic disorders and PH
Hb disorders include sickle cells disease and thalassemia; and RBC membrane diseases include spherocytosis, stomatocytosis and paroxysmal nocturnal hemoglobinurea. PH is one of the leading causes of morbidity and mortality in patients with hemolytic disorders. Major causes of PH in hemolytic disorders are hemolysis, hypercoagulabilty and iron overload resulting from transfusions and splenectomy[9,33-35]. Recently, in a murine model of hemolysis, significant reduction in NO bioavailability due to free Hb was shown to be accompanied by platelet activation and the activation of coagulation pathway resulting in thrombosis, PH, right ventricular failure and death. Interestingly, treatment with sildenafil reduced the mortality rate[36]. Furthermore, Hb has been shown to interact with superoxide and hydrogen peroxide, thus increasing reactive oxygen species formation, lipid peroxidation, and increase inflammatory response. Interestingly, in an experimental model, treatment with haptoglobin, a Hb scavenger was shown to decrease oxidative and inflammatory response and attenuate PH[37]. Free Hb plays a significant role in the pathogenesis of PH in hemolytic disorders; therefore, treatment with Hb scavengers appears to be an attractive therapeutic option.
Sickle cell disease: Hb in patients with sickle cell disease (SCD) is structurally different; valine is substituted for glutamic acid in the 6th position of β-globulin subunit of Hb[38]. This mutation produces abnormal and insoluble HbS. The major genotypes of SCD are homozygous SS, heterozygous SC and S/β thalassemia. In the United States, 0.15% of African-Americans are homozygous for SCD, and 8% have sickle trait. SCD is characterized by anemia, severe pain, potentially life-threatening complications such as bacterial sepsis, splenic sequestration, acute chest syndrome, stroke, chronic organ damage resulting from chronic hemolysis and intermittent ischemia. Vasculopathy in SCD results in irreversible organ damage, a frequent cause of death beyond childhood. Recent studies have shown that chemically-induced RBC stiffness leads to increased pulmonary artery pressure and pulmonary vascular resistance[39]. Importantly, sickled RBCs are stiffer than controls[40], which may partly contribute to PH in SCD. Furthermore, RBCs from SCD patients have an abnormal tendency to adhere to vascular endothelium. This abnormal adhesion plays an important role in facilitating the trapping of sickle cells in post-capillary venules and causing vascular obstruction which is the underlying factor for the characteristic features of SCD such as painful vascular occlusive crises and acute chest syndrome. In addition, the sickle cell adherence to EC results in the activation of EC and a chronic state of inflammation. Endothelial activation is a critical component of the microvascular responses accompanying SCD resulting in inflammatory response, increased expression of cell adhesion molecules and reactive oxygen species, and altered vasomotor tone leading to vasculopathy including PH. Interestingly, hypoxia/reperfusion injury causes inflammatory response in sickle cell transgenic mice[41-43].
Morbidity and mortality in SCD are high, and PH is a serious complication in SCD. Sudden death in patients with SCD and PH is not uncommon[44,45]. In a small series of autopsy cases (12 patients), 75% of patients had right ventricular hypertrophy and 50% revealed large thrombus in pulmonary artery, and 40% exhibited pulmonary vascular remodeling. The mortality in patients with catheterization-confirmed PH is 50% within 2 years compared to 7% at 10 years in SCD patients without PH[46-49]. In adult population with SCD, echocardiography revealed high incidence of PH (27%) as assessed by a tricuspid regurgitation jet velocity (TRJV) of > 2.5 m/s, however, the incidence was confirmed to be 6%-10% by cardiac catheterization, and > 50% of these patients had post-capillary PH[50-52]. A recent study showed increased TRJV in children to be associated with an increased PA pressure, increased cardiac output due to anemia and normal pulmonary vascular resistance[53]. The incidence of PH in patients with SCD, however, is relatively high (6%-10%), compared with the normal population (2.4-7.6 people/million per year). It is noteworthy that SCD patients with lower pulmonary artery pressure are at a higher risk compared with idiopathic PAH with equivalent pressure. Recent experimental studies in rodents reveal that it is the Hb-induced inflammation and to a lesser extent the Hb-induced oxidant injury leads to vascular injury[54]. Thus, RBC sickling, rheological abnormalities, hypoxemia, heme-induced oxidant injury and resulting inflammatory response leading to endothelial dysfunction play a major role in vasculopathy leading to vaso-occlusive disease including PH.
Thalassemia: Thalassemia diseases are an inherited Hb disorders associated with chronic anemia, impaired erythropoiesis and dysregulated iron metabolism; resulting from defective synthesis of α and β subunits of HbA. Absence or impaired production of α globulin results in β thalassemia and vice versa. PH is quite rare in α thalassemia. β thalassemia is characterized by impaired erythropoiesis and dysregulated iron metabolism. Two types of β thalassemia have been described; thalassemia major (TM) and thalassemia intermedia (TI). Patients at birth are asymptomatic because of the presence of HbF. Diagnosis of TM is usually made during infancy because of anemia. They require frequent transfusion and chelation therapy which have improved their survival. Furthermore, well transfused patients with TM are at a lower risk of developing PH. In contrast, the TI patients remain transfusion-independent for a longer period; the incidence of PH is higher in this group[34,55-57]. Pathophysiology of PH in thalassemia is similar to other hemoglobinopathies. Chronic hemolysis, iron overload, splenectomy, hypercoagulability, vascular inflammation and left ventricular dysfunction contribute to the pathogenesis of PH. Dysregulated arginine metabolism[58] and elevated levels of ADMA[59] have been reported in patients with β-thalassemia associated with PH. Higher incidence of PH was noted in patients with E/β-thalassemia who had more severe hemolysis and had had splenectomy; in addition, inflammatory markers were increased[60]. Increased non-transferrin bound iron and increased transferrin saturation indicative of iron overload increase the risk of cardiopulmonary damage[61]. Interestingly, in a mouse model of β thalassemia, transferrin treatment normalized labile plasma iron levels and RBC survival, and increased hepcidin expression[62]. In addition, increased hepcidin levels were accompanied by increased BMP2 expression in the liver and concomitant decrease in extracellular-signal related kinase (ERK) activation[63].
Compared to β thalassemia, SCD patients do not have iron overload. This difference is thought to be due to the presence of chronic inflammation in SCD which could block iron release from reticulo-endothelial system. In addition, unlike SCD, hepcidin levels are low in β thalassemia, which can further enhance iron absorption[64]. In β thalassemia, transfusion not only improves anemia but also suppresses erythropoiesis and increases hepcidin levels[65]. Globin chain imbalance leads to ineffective erythropoiesis, and erythroferrone suppresses hepcidin production during increased erythropoiesis, resulting in low hepcidin levels and increased iron absorption. In a mouse model of β-thalassemia, ablation of erythroferrone restored hepcidin expression and reduced iron accumulation without affecting anemia[66]. Furthermore, thalassemia carriers have been reported to have abnormal iron metabolism[67].
RBC membrane disorders: RBC membrane-associated abnormalities are found in inherited disorders such as spherocytosis and stomatocytosis. A defect in one or several proteins such as ankyrin, spectrin (α and β), band 3 has been reported. Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired RBC membrane defect. RBCs play a role in regulating membrane properties to undergo reversible deformation while maintaining integrity. In addition, RBCs have a pivotal role in regulating cell volume homeostasis. Inability to regulate cell volume is a feature of hemoglobinopathies[68-70].
Hereditary spherocytosis (HS) is considered not to be associated with thrombo-embolic risk. In a recent study, 26 children who underwent splenectomy, no evidence of PH or coagulation defect was observed during a follow-up period of median 4.5 years[71]. In another study that included 36 patients with HS (28 with splenectomy and 8 without), no evidence of PH was found[72]. However, arterial and venous thrombo-embolic events in patients with HS have been observed after splenectomy[73]; and several cases of CTEPH have been reported in patients with HS several years after splenectomy[74-76]. In a review of 22 patients with CTEPH following splenectomy, 3 patients with HS had had splenectomy 17-35 years before the diagnosis of CTEPH was made[77].
In hereditary stomatocytosis, the RBC membrane shows a leak of univalent cations (Na+ and K+). Two clinical variants have been recognized; hydrocytosis (overhydrated) and xerocytosis (dehydrated). Stewart et al[78] described 11 patients with stomatocytosis after splenectomy. Most of them had thrombo-embolic episodes, and 3 of them developed PH. Other case reports have described PH in patients with stomatocytosis several years (approx 6-30 years) after splenectomy. One patient underwent successful pulmonary endarterectomy for CTEPH. He had undergone splenectomy as a child because of the family history of spherocytosis[79]. Another patient with dehydrated hereditary stomatocytosis underwent splenectomy because of splenic infarct following air travel. Approximately 12 years later she developed CTEPH. Because of the worsening condition she underwent successful heart-lung transplantation[80]. The third case of stomatocytosis had splenectomy done for traumatic rupture of the spleen. About 6 years later he developed PH[81]. Splenectomy is not recommended for stomatocytosis, however, stomatocytosis is often mistaken for spherocytosis, and splenectomy is performed. At times it is difficult to distinguish RBC morphology; therefore, intracellular electrolyte measurements or flux studies may be required to make the correct diagnosis[78].
PNH is a progressive hemolytic disorder. It is an acquired clonal genetic deficiency of glycosylphosphatidylinositol-linked protein on the RBC surface that leads to complement-mediated hemolysis[35,82]. One case of PNH was diagnosed to have PH 5 years after splenectomy and associated chronic thrombo-embolism[83]. In one study, 41% patients with PNH and associated hemolysis (total 29 patients) had echocardiographic evidence of PH. Treatment with eculizumab reduced hemolysis[82,84]. In another study, 23 patients with PNH and hemolysis were examined before and after eculizumab therapy. Importantly, markers of endothelial dysfunction (sVCAM1, vWF) and coagulation activation were significantly reduced after eculizumab therapy[85].
Chronic myeloproliferative diseases and PH
Evidence is accumulating to suggest a link between PH and chronic myeloproliferative diseases (CMPD). CMPD originate in multipotent hematopoietic progenitor cells that are characterized by increases in one or more types of blood cells. CMPD include polycythemia vera, essential thrombocythemia, idiopathic myelofibrosis and chronic myeloid leukemia (CML)[86]. Dingli et al[87] examined 26 patients with CMPD and echocardiography based diagnosis of PH (estimated systolic pulmonary artery pressure 35-100 mmHg); 24 patients had symptoms related to PH and 4 had had splenectomy. The mortality rate among these patients was high. Another report[88] described 6 patients with myeloproliferative disease who developed PH (echocardiographic diagnosis, and in 4 confirmed with cardiac catheterization), and all had had splenectomy; 5 patients died within 1-6 mo of PH diagnosis. Lung histology in 3 patients revealed pulmonary myeloid metaplasia and fibrosis. A 72-year-old patient developed PH, right ventricular failure and thrombocytosis after splenectomy. The peripheral blood smear revealed megakaryoblasts. Interestingly, treatment with hydroxyurea not only decreased the platelet counts but also improved right heart failure. It was considered possible that megakaryocytes created obstruction in the pulmonary capillaries leading to PH[89]. In a group of 30 patients with a past history of thromboembolism, high incidence of valve disease (aortic and mitral valve with vegetation) was noted; 13% of patients had PH secondary to venous obstruction[90]. In another study, 46 patients with essential thrombocytosis were compared with 40 patients with reactive thrombocytosis secondary to anemia. In the essential thrombocytosis group, elevated platelet levels and 43% thrombo-embolic events were recorded; and 47.8% (22/46) had echocardiographic evidence of PH. In contrast, the reactive thrombocytosis secondary to anemia group did not have increased platelet levels, thrombo-embolic events or PH[91]. Garypidou et al[92] reported incidence of PH by echocardiography to be 41.7% in 24 patients with CMPD. In another report, among 103 patients with various CMPD, echocardiographic diagnosis of PH was made about 15 mo after the initial diagnosis of CMPD. The incidence of PH was found in less than 5%[93]. A 50 years old individual was diagnosed to have PH (confirmed by cardiac catheterization) 15 years after the diagnosis of latent myeloproliferative disorder and portal hypertension. Portal hypertension is a known complication of CMPD[94]. PVOD also has been reported in CMPD. A patient with myeloproliferative and myelodysplastic syndrome was treated with hydroxyurea for 4 years. Because of refractory thrombocythemia and hydroxyurea-induced neutropenia, anagrelide was started. Six weeks later, the patient was admitted with severe dyspnea at rest and was diagnosed to have PVOD[95]. Guilpain et al[96], reviewed 10 cases of CMPD (8 polycythemia vera and 2 essential thrombocythemia) and PH; 6 patients developed CTEPH and 4 patients had PAH. Importantly, CTEPH occurred early in the course of the disease and PAH occurred several years after the diagnosis of CMPD. All patients with PAH revealed myeloid metaplasia but none in the CTEPH group.
The patients with CMPD are at a risk of developing PH; and the occurrence of myelofibrosis in patients with PAH is not uncommon and is thought to contribute to impaired hematopoiesis. Popat et al[97] reported moderate to severe myelofibrosis in 14/17 patients with PAH. However, platelets and granulocytes in PAH patients were polyclonal unlike monoclonal cells that were found in patients with polycythemia vera and essential thrombocythemia. Erythropoietin facilitates erythroid lineage and proliferation. Erythropoietin has also been shown to induce tyrosine phosphorylation of JAK2 and to associate with it for biological activities including mitogenesis[98]. In a number of patients with CMPD, an acquired somatic JAK2V617F mutation has been observed, which confers a selective growth advantage. Interestingly, a small molecule inhibitor of JAK2 has been shown to attenuate myeloproliferative disease in a mouse model[99,100]. However, the patients with PAH (13 Familial PAH, 24 Idiopathic PAH, and 15 Associated PAH) and the controls did not reveal JAK2 mutation[101], nor was the JAK2 mutation noted in 19 patients with myelofibrosis secondary to PH[102]. Circulating CD34+CD133+ cells were higher in familial PAH compared with idiopathic PAH and the control subjects; interestingly, in non-affected family members, the CD34+CD133+ cell counts were comparable to that observed in Familial PAH group[101]. Furthermore, patients with PAH and myelofibrosis have blood vessels morphologically similar to what is observed in myeloproliferative myelofibrosis such as, microvascular density, distended lumina and irregular branching. In addition, VEGF levels are much higher in patients with primary myelofibrosis compared with the controls; and even higher in patients with primary myelofibrosis associated with PH. However, in PH associated with myeloproliferative diseases, the levels of circulating endothelial progenitor cells and the bone marrow pericytes were lower[103,104]. Almost a century ago it was thought that EC and hematopoietic cells have a common progenitor, hemangioblasts. Furthermore, EC and hematopoietic cells affect each other[105], which may explain the increased incidence of PH in CMPD and myelofibrosis accompanying PH. Transplantation of bone marrow-derived CD133+ cells from PAH patients into mice has been shown to result in endothelial injury, angioproliferative remodeling of pulmonary vasculature and right ventricular failure; CD133+ cells from control subjects, however, had no effect[106]. Recent studies have shown that bone marrow cells from BMPR2 mutant mice when transplanted into control mice induce PH, whereas bone marrow cells from the control mice protect mutant mice from developing PH[107]. These results further support a causal relationship between vascular and hematopoietic systems.
Autoimmunity, PH and hematological disorders
Autoimmunity is a well-known underlying feature of hematological disorders as well as of PH. Autoimmune diseases such as systemic sclerosis, systemic lupus erythematosus (SLE), Sjogren’s disease, and mixed connective tissue diseases are known to be associated with PH[108-110]. Loss of CD4+CD25+ cells, the T regulatory (Treg) cell population has been reported in several forms of PAH[110]. Furthermore, normal Treg function has been shown to limit the vascular injury and provide protection from developing PH[111]. In 132 patients with SLE, the incidence of PH was 12.9%. PH patients had longer duration of anemia; oxygen delivery was inversely related to PA pressure, indicating that tissue hypoxia may play a greater role in the lupus-associated PH[112]. Another patient with SLE and associated lupus anticoagulant and clotting disorder was described to have PH[113].
Autoimmunity is also important in thyroid diseases and thyroid disease-associated PH. Scicchitano et al[114] in a recent review article have discussed the prevalence of PH in hypothyroid state as well in hyperthyroid state. Interestingly, approximately half of the patients with PAH have been shown to have autoimmune thyroid disease[115]. Coagulation abnormalities associated with thyroiditis[116] may lead to chronic embolism and eventually CTEPH. Furthermore, thyroid hormone participates in EC proliferation and facilitates angiogenesis. Recent studies with an angio-proliferative model (Sugen + hypoxia) of PH have shown that thyrodectomy inhibits angioproliferation and reduces the expression of p-ERK1/2, integrin receptor αvβ3, fibroblast growth factor (FGF) 2 and FGF receptor[117]. These results suggest that the status of thyroid function in PH is important and it may affect the progression of the disease adversely.
Evan’s syndrome includes immune thrombocytopenia and associated autoimmune hemolytic anemia. Connor et al[118] reported 2 children with Evan’s syndrome and associated PH; both with the evidence of perivascular lymphoid infiltration indicative of vasculitis. Both improved with steroid and rituximab treatment. The incidence of PH in Evans’s syndrome, however, is not known. PH has also been reported in an adult patient with autoimmune hemolytic anemia who improved significantly on regular steroid therapy[119].
Therapy-associated PH
A number of alkylating agents including cyclophosphamide, bleomycin, mitomycin used for hematological diseases have been shown to lead to PVOD and PH[11,120]. Other therapeutic measures used for hematological disorders such as tyrosine kinase inhibitor dasatinib, interferon, splenectomy, bone marrow transplantation (BMT) and radiation also contribute to PH as discussed below.
Dasatinib: CML is caused by active BCR/ABL tyrosine kinase. Tyrosine kinase inhibitor, imatinib inhibits BCR/ABL and platelet-derived growth factor (PDGF), and has been used as a first line treatment for CML with good results. However about 29% of patients do not recover completely with imatinib, therefore, newer tyrosine kinase inhibitor, dasatinib is used as a second line treatment. Dasatinib inhibits Src kinase in addition to BCR/ABL and PDGF. Several case reports have appeared showing the development of precapillary PH after about 8-48 mo of dasatinib therapy[121-127]. In the French experience, the incidence of dasatinib-associated PH is 0.45%. The patients, however, did not recover fully after having been taken off dasatinib treatment. Interestingly, in the monocrotaline (MCT) and hypoxia-induced PH models, the pretreatment with dasatinib, unlike imatinib induced increased pulmonary artery pressure and increased inflammatory cells in the perivascular area. Furthermore, in vitro studies with human pulmonary EC, dasatinib induced apoptosis in a dose dependent manner through mitochondrial reactive oxygen species generation[128,129]. Interestingly a number of patients with dasatinib-induced PH is accompanied by pleural effusion (as high as 68%), which is not observed in classical PH. In most cases, discontinuing the medication appeared to have reversed PH; however, in a few cases prolonged PH therapy might be required[130]. Recent studies have shown that the inhibition of Src tyrosine kinase or dasatinib increases pulmonary artery pressure, and depolarizes PA SMC by altering potassium channels[131]. Thus, dasatinib-associated Src inhibition and the alterations in potassium channels may be responsible for the increased vasoconstriction and PH. It is noteworthy that decreased expression of Src tyrosine kinase has been reported in the lungs of patients with PAH[132]. It is suggested that Src function may depend on the state of vascular SMC[133].
Interferon: Interferon (IFN) α and β are used for various hematological disorders, cancer and infection especially hepatitis C. Evidence is accumulating to suggest that IFN pathway may have a role in the pathobiology of PH. INF therapy has been shown to be complicated by vasculopathy. IFN therapy has been shown to lead to reversible PH and in some cases irreversible PH[134-136]. Infusion of IFN-α into sheep has been shown to elevate pulmonary artery pressure associated with increased expression of thromboxane B2, a stable byproduct of thromboxane A2, a vasoconstrictor; that is attenuated by a selective thromboxane A2 synthetase inhibitor, OKY-046[137]. Interestingly, a subgroup of patients treated with INF exhibit increased levels of endothelin-1 (ET-1), which is known to play an important role in PH. Recent studies have shown that IFN induces ET1 gene and IFN-inducible protein IP10, a mediator of inflammation in vascular SMC; and the combination of IFN and TNF-α produce the highest amount of ET1. These cytokines have direct effect on ET1 transcription and also on increased translocation of NF-κB and STAT1[138]. Importantly, recent studies have shown increased levels of IP10 and ET1 in patients with PAH which correlated positively with serum brain natriuretic peptide and the status of the disease. These Authors have further shown increased type 1 IFN receptor (IFNR1) protein levels in the lungs of patients with PAH compared with the controls. Furthermore, IFNR1 knockout mice exhibit attenuated response to hypoxia[139]. These studies strongly indicate a role for IFN in the pathobiology of PAH.
Splenectomy: A number of patients who undergo splenectomy following trauma or for various hematological disorders develop PH, associated with histological changes in pulmonary arteries such as intimal fibrosis, plexiform lesions and thrombo-embolic lesions. The prevalence of PH in patients in the presence of asplenia is reported to be 11.5%[140]. In another study, 22 out of 257 patients with CTEPH (8.6%) had a prior history of splenectomy, compared with the positive history of splenectomy in 2.5% of idiopathic PAH patients and 0.4% in general population[77]. PH has been shown to occur several years after splenectomy for hereditary spherocytosis[74,75], stomatocytosis[78], thalassemia[141] and Hb Mainz hemolytic anemia[142]. Splenectomy is associated with deep vein thrombosis and un-resolving recurrent thrombosis eventually leading to CTEPH. Loss of spleen results in a loss of filtering function leading to abnormal circulating erythrocytes and the activation of coagulation. The activation of platelets enhances thrombin generation as well as cytokine activation. Human thrombi obtained after pulmonary endarterectomy revealed increased platelet-derived micro-particles and increased anionic phospholipids (phosphotidylserine, phosphotidylethanol and phosphotidylglycerine), reduced angiogenesis related gene expression, and reduced vascular canalization. These micro-particles are pro-coagulant. In addition, in a murine model of CTEPH, inhibition of angiogenesis was associated with delay in thrombus resolution[143,144]. In a rabbit model with splenic artery ligation, transfusion of sonicated blood resulted in platelet rich thrombi in pulmonary circulation; in contrast, transfusion of normal blood did not have any effect[145].
BMT: BMT is used for a number of blood disorders and cancer. Hepatic veno-occlusive disease is a well-established complication of BMT and cytotoxic drugs. In 1984, Troussard et al[146] were the first ones to report a child who developed PVOD a few years after having received BMT for a relapse of acute lymphoblastic leukemia. Since then, PVOD following BMT have been reported in several adults and children[147-152]. Hepatic veno-occlusive disease is a recognized complication of cytotoxic therapy used concomitantly with BMT. BMT in combination with cytotoxic drugs and radiation increases the chances of EC damage and PH. Another possibility that has been considered is that malignancy itself may cause PH[151]. Transplantation-associated thrombotic microangiopathy (TM-TMA), a known complication of BMT is caused by EC injury resulting in thrombin and fibrin deposition in microcirculation with ensuing organ damage. Jodele et al[153] have described 5 children who developed severe PH 71-205 d after having undergone hemopoietic stem cell transplantation. These children did have TM-TMA 56-101 d before the diagnosis of PH was made. PH can occur from a few months to several years after transplantation. In addition, PH without any evidence of PVOD was reported to occur in an adult almost a year after BMT[154]. A 5.25-year-old child underwent BMT after conditioning with cyclophosphamide and antithymocyte globulin; and he was treated with cyclosporine A and a short course of methotrexate to prevent graft-vs-host disease. Within a month of BMT, he developed respiratory distress, anemia and thrombocytopenia. Approximately 1.5 mo later, he was diagnosed to have microangiopathic changes. His condition, however, stabilized after cyclosporine A was discontinued and treatment with mycophenolate mofetil was started. About a year or so later he started to have vague respiratory symptoms which was subsequently diagnosed as severe PH[155]. These cases illustrate that PH can occur early or late after BMT. Cytotoxic drugs and radiation used to prepare the patient for BMT and to prevent graft-vs-host disease can contribute to EC damage leading to pulmonary vasculopathy. These patients need to be carefully monitored and PH should be considered a possibility when they present with pulmonary symptoms.
Radiation injury: Lung radiation leads to pneumonitis, fibrosis and vascular injury. Thoracic or whole body radiation is used for several types of lung cancer; and at times radiation in combination with immunosuppressive drugs is used before BMT. PVOD and pulmonary insufficiency have been reported to occur several months to years following therapy for cancer that included chemotherapy and radiation therapy. Histopathological changes in the lungs comprised interstitial fibrosis, thromboemboli, veno-occlusive lesions, and medial hypertrophy of pulmonary arteries, consistent with PVOD[156,157]. In addition, a 14-year-old was reported to have developed PH after receiving radiation therapy during infancy following the surgical removal of neuroblastoma arising from the left of the thoracic spine. At cardiac catheterization significant PH was noted. In addition, the branches of left pulmonary artery were described as hypoplastic, and the pulmonary veins from the left lung were underdeveloped[158].
EC play a pivotal role in radiation-induced vascular injury. Irradiated EC from rectal adenocarcinoma have been shown to induce fibrogenic phenotype in vascular SMC, and increase proliferation and migration[159]. Furthermore, several experimental studies have shown radiation injury resulting in elevated pulmonary artery pressure, and structural remodeling of the small pulmonary arteries. In a sheep model, several weeks after the whole lung exposure to radiation resulted in abnormal vascular reactivity, PH and pulmonary vascular remodeling[160]. In a mouse model, low dose radiation resulted in EC injury, followed by rapid recovery. However, a higher dose resulted not only in EC injury, but also a delay in recovery followed by prolonged EC proliferation, fibroblast proliferation and collagen secretion indicative of significant vascular damage[161]. In a rat model, radiation injury induced pulmonary vascular EC damage followed by medial wall and adventitial thickening, neointima formation and obliteration of vessels similar to what is observed in PAH[162].
These studies underscore the fact that vascular EC are susceptible to radiation injury. The patients who receive radiation therapy with or without alkylating drugs are at a risk of developing PH. PH has been shown to occur several years after the cessation of therapy; therefore these patients need a long careful follow-up.
IRON HOMEOSTASIS AND PAH
Deregulation of iron homeostasis and resulting alterations in iron availability plays an important role in the pathogenesis of cardiovascular diseases including PH. Both iron deficiency and iron overload have deleterious effect on cardiovascular system. Iron deficiency has been shown to have an adverse effect on survival in patients with chronic heart failure[163]. Anemia in PH is associated with worse function and poor survival[164]. Iron deficiency is being recognized as an important factor in the prognosis of PAH. Low transferrin saturation, an indicator of iron deficiency has been reported in PAH patients, particularly the ones with BMPR2 mutation, but not in the CTEPH group. In this group of PAH patients, 72% of iron deficient patients had anemia, whereas only 4% in non-iron deficient patients[20]. In another study, iron deficiency was found in 43% of 70 patients with idiopathic PAH accompanied by low exercise capacity. However, anemia did not affect the exercise intolerance. Interestingly, 8 out of 18 patients did not respond to oral iron therapy[165]. Red cell distribution width (RDW), a biomarker of anemia has a better survival predictive value independent of NT-proBNP levels and 6 min walk distance. Increased RDW was accompanied by other indicators of iron deficiency such as decreased ferritin levels and low transferrin saturation. Patients with increased soluble TfR (sTfR) had higher mortality independent of WHO class or exercise capacity. sTfR levels are a sensitive marker of tissue iron availability, unaffected by inflammation. Interestingly, hepcidin levels were increased in PAH despite iron deficiency. Hepcidin which restricts iron absorption is stimulated by cytokines and BMP6; however, hepcidin levels did not correlate with IL-6 levels. Since a number of patients have BMPR2 mutation and loss of function, it is likely that increased BMP6 levels secondary to BMPR2 loss may increase hepcidin levels. Furthermore, erythropoietin levels are increased in idiopathic PAH despite the fact that these patients were not anemic. The hematocrit and Hb levels were not different compared with the controls. Erythropoietin is known to reduce hepcidin levels in order to increase iron uptake. Increased levels of hepcidin in the presence of increased erythropoietin indicates deregulated erythropoiesis in idiopathic PAH[166,167]. In 29 patients with idiopathic PAH, 46.2% of iron deficient patients belonged to NYHA functional class 3 or higher compared with 12.5% in non-iron deficient. There were no differences in the hematocrit or Hb levels between the two groups. The iron deficiency was related to the severity[168]. In addition, zinc protoporphyrin (ZnPP) levels, indicative of iron deficiency was significantly higher in patients with idiopathic PAH associated with increased RDW; however, ZnPP levels were not altered in “Associated” PAH. Iron containing protein is also required for mitochondrial electron transport and catalyzes reactions that form NO[169]. Intravenous iron therapy in patients with idiopathic PAH was well tolerated and it improved endurance capacity; however, it did not alter cardiac function[170]. Thus, iron deficiency seems to be a more important prognosticator compared with anemia.
Iron deficiency is common in patients with systemic sclerosis (SSc) associated with PH than in the non-PH group. PH was present in 27.8% of patients with SSc. Iron deficiency was associated with poor exercise tolerance and survival. Hepcidin levels were high in the SSc population, but did not correlate with IL-6 levels. Hb levels, however, were not altered. Soluble transferrin receptor (sTfR) levels in both groups were significantly increased associated with iron deficiency[171]. Interestingly, iron-depletion by desferrioxamine infusion in normal individuals resulted in higher systolic pulmonary artery pressure during 8 h hypoxia compared with the iron-repleted individuals. Thus, the alterations in iron availability affect the pulmonary vascular response to hypoxia. HIF is implicated in hypoxia; it is likely that increased iron potentiates HIF hydroxylation and its degradation[172]. Sufficient iron availability is required for adjustment to high-altitude hypoxia. There is a close connection between oxygen and iron homeostasis[173].
Recently it was reported that iron-deficient diet in rats resulted in elevated PA pressure, right ventricular hypertrophy, vascular remodeling, and increased expression of HIF1α, HIF2α, STAT3 activation and aerobic glycolysis, which could be reversed by iron therapy[174]. Furthermore, deletion of iron regulatory protein 1 (IRP1) in mice leads to PH and polycythemia that is exacerbated by low iron diet, resulting in increased HIF2α levels and ET1 in EC. Iron deficiency can stabilize HIF2α by diminishing activity of iron-dependent prolyl hydroxylases involved in HIF2α degradation[175]. In contrast, dietary iron restriction attenuated monocrotaline-induced PH, although, the serum iron concentration in MCT group was not different from the control group. However, the expression of TfR1 in pulmonary arteries was increased. Interestingly, TfR1 hetero-knockout mice showed attenuated hypoxia-induced PH, right ventricular hypertrophy and vascular remodeling[176,177]. Iron chelation has been shown to attenuate hypoxia-induced PH, pulmonary vascular remodeling and right ventricular hypertrophy in rats. In addition, carbonylation of proteins was increased in hypoxia-induced rats as well in the plasma of the patients with PAH indicative of oxidative stress[178]. Furthermore, PH in patients with idiopathic pulmonary fibrosis was shown to correlate with iron deposition in alveolar spaces[179]. These foregoing results show opposite effects of iron levels on pulmonary vasculature. Iron homeostasis is intricately balanced and maintained; any injury and/or stress can alter this balance resulting in iron overload or iron deficiency. Mitochondria play a pivotal role in energy and iron metabolism[180]. The opposing effects of iron levels observed in different forms of PH may depend on the level of non-transferrin-bound iron and on the status/health of mitochondria.
In summary, hemopoietin system, pulmonary vasculature and iron metabolism are intricately related. Hematological disorders affect pulmonary vasculature and PH can cause myelofibrosis. Deregulated iron homeostasis and resulting status and function of mitochondria in PH may have an important effect on prognosis.
Footnotes
Conflict-of-interest statement: None of the authors has conflict of interest.
Manuscript source: Invited manuscript
Specialty type: Cardiac and cardiovascular systems
Country of origin: United States
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B, B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: July 1, 2016
First decision: August 5, 2016
Article in press: October 9, 2016
P- Reviewer: Ciccone MM, Nakhoul FM, Ueda H, Wang F S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
References
- 1.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–D41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 2.Bossone E, D’Andrea A, D’Alto M, Citro R, Argiento P, Ferrara F, Cittadini A, Rubenfire M, Naeije R. Echocardiography in pulmonary arterial hypertension: from diagnosis to prognosis. J Am Soc Echocardiogr. 2013;26:1–14. doi: 10.1016/j.echo.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 3.Mathew R. Sulica R and Preston I editors. Publishers: Intech; 2011. Pulmonary Hypertension: Endothelial cell Function. In Pulmonary hypertension: From Bench Research to Clinical Challenge (pp 1-24) [Google Scholar]
- 4.Mathew R. Pathogenesis of pulmonary hypertension: a case for caveolin-1 and cell membrane integrity. Am J Physiol Heart Circ Physiol. 2014;306:H15–H25. doi: 10.1152/ajpheart.00266.2013. [DOI] [PubMed] [Google Scholar]
- 5.Lin KY, Ito A, Asagami T, Tsao PS, Adimoolam S, Kimoto M, Tsuji H, Reaven GM, Cooke JP. Impaired nitric oxide synthase pathway in diabetes mellitus: role of asymmetric dimethylarginine and dimethylarginine dimethylaminohydrolase. Circulation. 2002;106:987–992. doi: 10.1161/01.cir.0000027109.14149.67. [DOI] [PubMed] [Google Scholar]
- 6.Davids M, van Hell AJ, Visser M, Nijveldt RJ, van Leeuwen PA, Teerlink T. Role of the human erythrocyte in generation and storage of asymmetric dimethylarginine. Am J Physiol Heart Circ Physiol. 2012;302:H1762–H1770. doi: 10.1152/ajpheart.01205.2011. [DOI] [PubMed] [Google Scholar]
- 7.Lücke T, Kanzelmeyer N, Kemper MJ, Tsikas D, Das AM. Developmental changes in the L-arginine/nitric oxide pathway from infancy to adulthood: plasma asymmetric dimethylarginine levels decrease with age. Clin Chem Lab Med. 2007;45:1525–1530. doi: 10.1515/CCLM.2007.300. [DOI] [PubMed] [Google Scholar]
- 8.Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA. 2005;293:1653–1662. doi: 10.1001/jama.293.13.1653. [DOI] [PubMed] [Google Scholar]
- 9.Morris CR, Gladwin MT, Kato GJ. Nitric oxide and arginine dysregulation: a novel pathway to pulmonary hypertension in hemolytic disorders. Curr Mol Med. 2008;8:620–632. doi: 10.2174/156652408786241447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morris CR. Mechanisms of vasculopathy in sickle cell disease and thalassemia. Hematology Am Soc Hematol Educ Program. 2008:177–185. doi: 10.1182/asheducation-2008.1.177. [DOI] [PubMed] [Google Scholar]
- 11.Ranchoux B, Günther S, Quarck R, Chaumais MC, Dorfmüller P, Antigny F, Dumas SJ, Raymond N, Lau E, Savale L, et al. Chemotherapy-induced pulmonary hypertension: role of alkylating agents. Am J Pathol. 2015;185:356–371. doi: 10.1016/j.ajpath.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 12.Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of Mammalian iron metabolism. Cell. 2010;142:24–38. doi: 10.1016/j.cell.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 13.Camaschella C. Iron and hepcidin: a story of recycling and balance. Hematology Am Soc Hematol Educ Program. 2013;2013:1–8. doi: 10.1182/asheducation-2013.1.1. [DOI] [PubMed] [Google Scholar]
- 14.Camaschella C, Pagani A, Nai A, Silvestri L. The mutual control of iron and erythropoiesis. Int J Lab Hematol. 2016;38 Suppl 1:20–26. doi: 10.1111/ijlh.12505. [DOI] [PubMed] [Google Scholar]
- 15.Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108:3204–3209. doi: 10.1182/blood-2006-06-027631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andrews NC. Molecular control of iron metabolism. Best Pract Res Clin Haematol. 2005;18:159–169. doi: 10.1016/j.beha.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 17.Ganz T, Nemeth E. Iron homeostasis in host defence and inflammation. Nat Rev Immunol. 2015;15:500–510. doi: 10.1038/nri3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kohgo Y, Torimoto Y, Kato J. Transferrin receptor in tissue and serum: updated clinical significance of soluble receptor. Int J Hematol. 2002;76:213–218. doi: 10.1007/BF02982790. [DOI] [PubMed] [Google Scholar]
- 19.Galy B, Ferring-Appel D, Sauer SW, Kaden S, Lyoumi S, Puy H, Kölker S, Gröne HJ, Hentze MW. Iron regulatory proteins secure mitochondrial iron sufficiency and function. Cell Metab. 2010;12:194–201. doi: 10.1016/j.cmet.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 20.Soon E, Treacy CM, Toshner MR, MacKenzie-Ross R, Manglam V, Busbridge M, Sinclair-McGarvie M, Arnold J, Sheares KK, Morrell NW, et al. Unexplained iron deficiency in idiopathic and heritable pulmonary arterial hypertension. Thorax. 2011;66:326–332. doi: 10.1136/thx.2010.147272. [DOI] [PubMed] [Google Scholar]
- 21.Sharma V, Berkelhamer S, Lakshminrusimha S. Persistent pulmonary hypertension of the newborn. Matern Health Neonatol Perinatol. 2015;1:14. doi: 10.1186/s40748-015-0015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cabral JE, Belik J. Persistent pulmonary hypertension of the newborn: recent advances in pathophysiology and treatment. J Pediatr (Rio J) 2013;89:226–242. doi: 10.1016/j.jped.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 23.Landau D, Kapelushnik J, Harush MB, Marks K, Shalev H. Persistent pulmonary hypertension of the newborn associated with severe congenital anemia of various etiologies. J Pediatr Hematol Oncol. 2015;37:60–62. doi: 10.1097/MPH.0000000000000064. [DOI] [PubMed] [Google Scholar]
- 24.Shalev H, Moser A, Kapelushnik J, Karplus M, Zucker N, Yaniv I, Tamary H. Congenital dyserythropoietic anemia type I presenting as persistent pulmonary hypertension of the newborn. J Pediatr. 2000;136:553–555. doi: 10.1016/s0022-3476(00)90025-8. [DOI] [PubMed] [Google Scholar]
- 25.Shah P, Thompson K, Rao S. Fetal Anemia With Persistent Pulmonary Hypertension: A Report of 3 Cases. J Pediatr Hematol Oncol. 2015;37:e204–e205. doi: 10.1097/MPH.0000000000000267. [DOI] [PubMed] [Google Scholar]
- 26.Parveen V, Patole SK, Whitehall JS. Massive fetomaternal hemorrhage with persistent pulmonary hypertension in a neonate. Indian Pediatr. 2002;39:385–388. [PubMed] [Google Scholar]
- 27.Delsing B, Lopriore E, Blom N, Te Pas AB, Vandenbussche FP, Walther FJ. Risk of persistent pulmonary hypertension of the neonate in twin-to-twin transfusion syndrome. Neonatology. 2007;92:134–138. doi: 10.1159/000101433. [DOI] [PubMed] [Google Scholar]
- 28.Mintzer JP, Parvez B, Chelala M, Alpan G, LaGamma EF. Monitoring regional tissue oxygen extraction in neonates & lt; 1250 g helps identify transfusion thresholds independent of hematocrit. J Neonatal Perinatal Med. 2014;7:89–100. doi: 10.3233/NPM-1477213. [DOI] [PubMed] [Google Scholar]
- 29.Baron DM, Yu B, Lei C, Bagchi A, Beloiartsev A, Stowell CP, Steinbicker AU, Malhotra R, Bloch KD, Zapol WM. Pulmonary hypertension in lambs transfused with stored blood is prevented by breathing nitric oxide. Anesthesiology. 2012;116:637–647. doi: 10.1097/ALN.0b013e318246ef77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Risbano MG, Kanias T, Triulzi D, Donadee C, Barge S, Badlam J, Jain S, Belanger AM, Kim-Shapiro DB, Gladwin MT. Effects of Aged Stored Autologous Red Blood Cells on Human Endothelial Function. Am J Respir Crit Care Med. 2015;192:1223–1233. doi: 10.1164/rccm.201501-0145OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berra L, Pinciroli R, Stowell CP, Wang L, Yu B, Fernandez BO, Feelisch M, Mietto C, Hod EA, Chipman D, et al. Autologous transfusion of stored red blood cells increases pulmonary artery pressure. Am J Respir Crit Care Med. 2014;190:800–807. doi: 10.1164/rccm.201405-0850OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neuman R, Hayek S, Rahman A, Poole JC, Menon V, Sher S, Newman JL, Karatela S, Polhemus D, Lefer DJ, et al. Effects of storage-aged red blood cell transfusions on endothelial function in hospitalized patients. Transfusion. 2015;55:782–790. doi: 10.1111/trf.12919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 34.Farmakis D, Aessopos A. Pulmonary hypertension associated with hemoglobinopathies: prevalent but overlooked. Circulation. 2011;123:1227–1232. doi: 10.1161/CIRCULATIONAHA.110.988089. [DOI] [PubMed] [Google Scholar]
- 35.Machado RF, Farber HW. Pulmonary hypertension associated with chronic hemolytic anemia and other blood disorders. Clin Chest Med. 2013;34:739–752. doi: 10.1016/j.ccm.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu W, Jin R, Zhang J, You T, Peng Z, Ge X, Bronson RT, Halperin JA, Loscalzo J, Qin X. The critical roles of platelet activation and reduced NO bioavailability in fatal pulmonary arterial hypertension in a murine hemolysis model. Blood. 2010;116:1613–1622. doi: 10.1182/blood-2010-01-267112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irwin DC, Baek JH, Hassell K, Nuss R, Eigenberger P, Lisk C, Loomis Z, Maltzahn J, Stenmark KR, Nozik-Grayck E, et al. Hemoglobin-induced lung vascular oxidation, inflammation, and remodeling contribute to the progression of hypoxic pulmonary hypertension and is attenuated in rats with repeated-dose haptoglobin administration. Free Radic Biol Med. 2015;82:50–62. doi: 10.1016/j.freeradbiomed.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ingram VM. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature. 1956;178:792–794. doi: 10.1038/178792a0. [DOI] [PubMed] [Google Scholar]
- 39.Schreier DA, Forouzan O, Hacker TA, Sheehan J, Chesler N. Increased Red Blood Cell Stiffness Increases Pulmonary Vascular Resistance and Pulmonary Arterial Pressure. J Biomech Eng. 2016;138:021012. doi: 10.1115/1.4032187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brandão MM, Fontes A, Barjas-Castro ML, Barbosa LC, Costa FF, Cesar CL, Saad ST. Optical tweezers for measuring red blood cell elasticity: application to the study of drug response in sickle cell disease. Eur J Haematol. 2003;70:207–211. doi: 10.1034/j.1600-0609.2003.00027.x. [DOI] [PubMed] [Google Scholar]
- 41.Hoppe C, Kuypers F, Larkin S, Hagar W, Vichinsky E, Styles L. A pilot study of the short-term use of simvastatin in sickle cell disease: effects on markers of vascular dysfunction. Br J Haematol. 2011;153:655–663. doi: 10.1111/j.1365-2141.2010.08480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest. 2000;106:411–420. doi: 10.1172/JCI9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hebbel RP, Vercellotti GM. The endothelial biology of sickle cell disease. J Lab Clin Med. 1997;129:288–293. doi: 10.1016/s0022-2143(97)90176-1. [DOI] [PubMed] [Google Scholar]
- 44.Powars DR, Chan LS, Hiti A, Ramicone E, Johnson C. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine (Baltimore) 2005;84:363–376. doi: 10.1097/01.md.0000189089.45003.52. [DOI] [PubMed] [Google Scholar]
- 45.Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, Brown B, Coles WA, Nichols JS, Ernst I, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350:886–895. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- 46.Ataga KI, Moore CG, Jones S, Olajide O, Strayhorn D, Hinderliter A, Orringer EP. Pulmonary hypertension in patients with sickle cell disease: a longitudinal study. Br J Haematol. 2006;134:109–115. doi: 10.1111/j.1365-2141.2006.06110.x. [DOI] [PubMed] [Google Scholar]
- 47.Manci EA, Culberson DE, Yang YM, Gardner TM, Powell R, Haynes J, Shah AK, Mankad VN. Causes of death in sickle cell disease: an autopsy study. Br J Haematol. 2003;123:359–365. doi: 10.1046/j.1365-2141.2003.04594.x. [DOI] [PubMed] [Google Scholar]
- 48.Graham JK, Mosunjac M, Hanzlick RL, Mosunjac M. Sickle cell lung disease and sudden death: a retrospective/prospective study of 21 autopsy cases and literature review. Am J Forensic Med Pathol. 2007;28:168–172. doi: 10.1097/01.paf.0000257397.92466.50. [DOI] [PubMed] [Google Scholar]
- 49.Haque AK, Gokhale S, Rampy BA, Adegboyega P, Duarte A, Saldana MJ. Pulmonary hypertension in sickle cell hemoglobinopathy: a clinicopathologic study of 20 cases. Hum Pathol. 2002;33:1037–1043. doi: 10.1053/hupa.2002.128059. [DOI] [PubMed] [Google Scholar]
- 50.Parent F, Bachir D, Inamo J, Lionnet F, Driss F, Loko G, Habibi A, Bennani S, Savale L, Adnot S, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med. 2011;365:44–53. doi: 10.1056/NEJMoa1005565. [DOI] [PubMed] [Google Scholar]
- 51.Fonseca GH, Souza R, Salemi VM, Jardim CV, Gualandro SF. Pulmonary hypertension diagnosed by right heart catheterisation in sickle cell disease. Eur Respir J. 2012;39:112–118. doi: 10.1183/09031936.00134410. [DOI] [PubMed] [Google Scholar]
- 52.Mehari A, Gladwin MT, Tian X, Machado RF, Kato GJ. Mortality in adults with sickle cell disease and pulmonary hypertension. JAMA. 2012;307:1254–1256. doi: 10.1001/jama.2012.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chaudry RA, Cikes M, Karu T, Hutchinson C, Ball S, Sutherland G, Rosenthal M, Bush A, Crowley S. Paediatric sickle cell disease: pulmonary hypertension but normal vascular resistance. Arch Dis Child. 2011;96:131–136. doi: 10.1136/adc.2010.184028. [DOI] [PubMed] [Google Scholar]
- 54.Buehler PW, Baek JH, Lisk C, Connor I, Sullivan T, Kominsky D, Majka S, Stenmark KR, Nozik-Grayck E, Bonaventura J, et al. Free hemoglobin induction of pulmonary vascular disease: evidence for an inflammatory mechanism. Am J Physiol Lung Cell Mol Physiol. 2012;303:L312–L326. doi: 10.1152/ajplung.00074.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fraidenburg DR, Machado RF. Pulmonary hypertension associated with thalassemia syndromes. Ann N Y Acad Sci. 2016;1368:127–139. doi: 10.1111/nyas.13037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meloni A, Detterich J, Pepe A, Harmatz P, Coates TD, Wood JC. Pulmonary hypertension in well-transfused thalassemia major patients. Blood Cells Mol Dis. 2015;54:189–194. doi: 10.1016/j.bcmd.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ginzburg Y, Rivella S. β-thalassemia: a model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood. 2011;118:4321–4330. doi: 10.1182/blood-2011-03-283614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morris CR, Kim HY, Klings ES, Wood J, Porter JB, Trachtenberg F, Sweeters N, Olivieri NF, Kwiatkowski JL, Virzi L, et al. Dysregulated arginine metabolism and cardiopulmonary dysfunction in patients with thalassaemia. Br J Haematol. 2015;169:887–898. doi: 10.1111/bjh.13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mohamed el-S, Ibrahim B, Amr D, Noha el-K, Mokhtar M. Asymmetric dimethylarginine levels in children with β-thalassemia and their correlations to tricuspid regurgitant jet velocity. Pediatr Blood Cancer. 2014;61:1540–1543. doi: 10.1002/pbc.25076. [DOI] [PubMed] [Google Scholar]
- 60.Atichartakarn V, Chuncharunee S, Archararit N, Udomsubpayakul U, Lee R, Tunhasiriwet A, Aryurachai K. Prevalence and risk factors for pulmonary hypertension in patients with hemoglobin E/β-thalassemia disease. Eur J Haematol. 2014;92:346–353. doi: 10.1111/ejh.12242. [DOI] [PubMed] [Google Scholar]
- 61.Piga A, Longo F, Duca L, Roggero S, Vinciguerra T, Calabrese R, Hershko C, Cappellini MD. High nontransferrin bound iron levels and heart disease in thalassemia major. Am J Hematol. 2009;84:29–33. doi: 10.1002/ajh.21317. [DOI] [PubMed] [Google Scholar]
- 62.Li H, Rybicki AC, Suzuka SM, von Bonsdorff L, Breuer W, Hall CB, Cabantchik ZI, Bouhassira EE, Fabry ME, Ginzburg YZ. Transferrin therapy ameliorates disease in beta-thalassemic mice. Nat Med. 2010;16:177–182. doi: 10.1038/nm.2073. [DOI] [PubMed] [Google Scholar]
- 63.Chen H, Choesang T, Li H, Sun S, Pham P, Bao W, Feola M, Westerman M, Li G, Follenzi A, et al. Increased hepcidin in transferrin-treated thalassemic mice correlates with increased liver BMP2 expression and decreased hepatocyte ERK activation. Haematologica. 2016;101:297–308. doi: 10.3324/haematol.2015.127902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koren A, Fink D, Admoni O, Tennenbaum-Rakover Y, Levin C. Non-transferrin-bound labile plasma iron and iron overload in sickle-cell disease: a comparative study between sickle-cell disease and beta-thalassemic patients. Eur J Haematol. 2010;84:72–78. doi: 10.1111/j.1600-0609.2009.01342.x. [DOI] [PubMed] [Google Scholar]
- 65.Pasricha SR, Frazer DM, Bowden DK, Anderson GJ. Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with β-thalassemia major: a longitudinal study. Blood. 2013;122:124–133. doi: 10.1182/blood-2012-12-471441. [DOI] [PubMed] [Google Scholar]
- 66.Kautz L, Jung G, Du X, Gabayan V, Chapman J, Nasoff M, Nemeth E, Ganz T. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of β-thalassemia. Blood. 2015;126:2031–2037. doi: 10.1182/blood-2015-07-658419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guimarães JS, Cominal JG, Silva-Pinto AC, Olbina G, Ginzburg YZ, Nandi V, Westerman M, Rivella S, de Souza AM. Altered erythropoiesis and iron metabolism in carriers of thalassemia. Eur J Haematol. 2015;94:511–518. doi: 10.1111/ejh.12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Da Costa L, Galimand J, Fenneteau O, Mohandas N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 2013;27:167–178. doi: 10.1016/j.blre.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 69.An X, Mohandas N. Disorders of red cell membrane. Br J Haematol. 2008;141:367–375. doi: 10.1111/j.1365-2141.2008.07091.x. [DOI] [PubMed] [Google Scholar]
- 70.Gallagher PG. Red cell membrane disorders. Hematology Am Soc Hematol Educ Program. 2005:13–18. doi: 10.1182/asheducation-2005.1.13. [DOI] [PubMed] [Google Scholar]
- 71.Das A, Bansal D, Ahluwalia J, Das R, Rohit MK, Attri SV, Trehan A, Marwaha RK. Risk factors for thromboembolism and pulmonary artery hypertension following splenectomy in children with hereditary spherocytosis. Pediatr Blood Cancer. 2014;61:29–33. doi: 10.1002/pbc.24766. [DOI] [PubMed] [Google Scholar]
- 72.Crary SE, Ramaciotti C, Buchanan GR. Prevalence of pulmonary hypertension in hereditary spherocytosis. Am J Hematol. 2011;86:E73–E76. doi: 10.1002/ajh.22182. [DOI] [PubMed] [Google Scholar]
- 73.Schilling RF, Gangnon RE, Traver MI. Delayed adverse vascular events after splenectomy in hereditary spherocytosis. J Thromb Haemost. 2008;6:1289–1295. doi: 10.1111/j.1538-7836.2008.03024.x. [DOI] [PubMed] [Google Scholar]
- 74.Smedema JP, Louw VJ. Pulmonary arterial hypertension after splenectomy for hereditary spherocytosis. Cardiovasc J Afr. 2007;18:84–89. [PubMed] [Google Scholar]
- 75.Jardine DL, Laing AD. Delayed pulmonary hypertension following splenectomy for congenital spherocytosis. Intern Med J. 2004;34:214–216. doi: 10.1111/j.1444-0903.2004.00580.x. [DOI] [PubMed] [Google Scholar]
- 76.Hayag-Barin JE, Smith RE, Tucker FC. Hereditary spherocytosis, thrombocytosis, and chronic pulmonary emboli: a case report and review of the literature. Am J Hematol. 1998;57:82–84. doi: 10.1002/(sici)1096-8652(199801)57:1<82::aid-ajh15>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 77.Jaïs X, Ioos V, Jardim C, Sitbon O, Parent F, Hamid A, Fadel E, Dartevelle P, Simonneau G, Humbert M. Splenectomy and chronic thromboembolic pulmonary hypertension. Thorax. 2005;60:1031–1034. doi: 10.1136/thx.2004.038083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stewart GW, Amess JA, Eber SW, Kingswood C, Lane PA, Smith BD, Mentzer WC. Thrombo-embolic disease after splenectomy for hereditary stomatocytosis. Br J Haematol. 1996;93:303–310. doi: 10.1046/j.1365-2141.1996.4881033.x. [DOI] [PubMed] [Google Scholar]
- 79.Murali B, Drain A, Seller D, Dunning J, Vuylsteke A. Pulmonary thromboendarterectomy in a case of hereditary stomatocytosis. Br J Anaesth. 2003;91:739–741. doi: 10.1093/bja/aeg237. [DOI] [PubMed] [Google Scholar]
- 80.Jaïs X, Till SJ, Cynober T, Ioos V, Garcia G, Tchernia G, Dartevelle P, Simonneau G, Delaunay J, Humbert M. An extreme consequence of splenectomy in dehydrated hereditary stomatocytosis: gradual thrombo-embolic pulmonary hypertension and lung-heart transplantation. Hemoglobin. 2003;27:139–147. doi: 10.1081/hem-120023377. [DOI] [PubMed] [Google Scholar]
- 81.Yoshimoto A, Fujimura M, Nakao S. Pulmonary hypertension after splenectomy in hereditary stomatocytosis. Am J Med Sci. 2005;330:195–197. doi: 10.1097/00000441-200510000-00008. [DOI] [PubMed] [Google Scholar]
- 82.Hill A, Sapsford RJ, Scally A, Kelly R, Richards SJ, Khurisgara G, Sivananthan MU, Hillmen P. Under-recognized complications in patients with paroxysmal nocturnal haemoglobinuria: raised pulmonary pressure and reduced right ventricular function. Br J Haematol. 2012;158:409–414. doi: 10.1111/j.1365-2141.2012.09166.x. [DOI] [PubMed] [Google Scholar]
- 83.Heller PG, Grinberg AR, Lencioni M, Molina MM, Roncoroni AJ. Pulmonary hypertension in paroxysmal nocturnal hemoglobinuria. Chest. 1992;102:642–643. doi: 10.1378/chest.102.2.642. [DOI] [PubMed] [Google Scholar]
- 84.Hill A, Rother RP, Wang X, Morris SM, Quinn-Senger K, Kelly R, Richards SJ, Bessler M, Bell L, Hillmen P, et al. Effect of eculizumab on haemolysis-associated nitric oxide depletion, dyspnoea, and measures of pulmonary hypertension in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2010;149:414–425. doi: 10.1111/j.1365-2141.2010.08096.x. [DOI] [PubMed] [Google Scholar]
- 85.Helley D, de Latour RP, Porcher R, Rodrigues CA, Galy-Fauroux I, Matheron J, Duval A, Schved JF, Fischer AM, Socié G. Evaluation of hemostasis and endothelial function in patients with paroxysmal nocturnal hemoglobinuria receiving eculizumab. Haematologica. 2010;95:574–581. doi: 10.3324/haematol.2009.016121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Adir Y, Humbert M. Pulmonary hypertension in patients with chronic myeloproliferative disorders. Eur Respir J. 2010;35:1396–1406. doi: 10.1183/09031936.00175909. [DOI] [PubMed] [Google Scholar]
- 87.Dingli D, Utz JP, Krowka MJ, Oberg AL, Tefferi A. Unexplained pulmonary hypertension in chronic myeloproliferative disorders. Chest. 2001;120:801–808. doi: 10.1378/chest.120.3.801. [DOI] [PubMed] [Google Scholar]
- 88.García-Manero G, Schuster SJ, Patrick H, Martinez J. Pulmonary hypertension in patients with myelofibrosis secondary to myeloproliferative diseases. Am J Hematol. 1999;60:130–135. doi: 10.1002/(sici)1096-8652(199902)60:2<130::aid-ajh8>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 89.Marvin KS, Spellberg RD. Pulmonary hypertension secondary to thrombocytosis in a patient with myeloid metaplasia. Chest. 1993;103:642–644. doi: 10.1378/chest.103.2.642. [DOI] [PubMed] [Google Scholar]
- 90.Reisner SA, Rinkevich D, Markiewicz W, Tatarsky I, Brenner B. Cardiac involvement in patients with myeloproliferative disorders. Am J Med. 1992;93:498–504. doi: 10.1016/0002-9343(92)90576-w. [DOI] [PubMed] [Google Scholar]
- 91.Altintas A, Karahan Z, Pasa S, Cil T, Boyraz T, Iltumur K, Ayyildiz O. Pulmonary hypertension in patients with essential thrombocythemia and reactive thrombocytosis. Leuk Lymphoma. 2007;48:1981–1987. doi: 10.1080/10428190701493928. [DOI] [PubMed] [Google Scholar]
- 92.Garypidou V, Vakalopoulou S, Dimitriadis D, Tziomalos K, Sfikas G, Perifanis V. Incidence of pulmonary hypertension in patients with chronic myeloproliferative disorders. Haematologica. 2004;89:245–246. [PubMed] [Google Scholar]
- 93.Chebrek S, Aïssi K, Francès Y, Mercier C, Farnault L, Sébahoun G, Costello R. Pulmonary hypertension in patients with chronic myeloproliferative neoplasms. Leuk Lymphoma. 2014;55:223–225. doi: 10.3109/10428194.2013.797083. [DOI] [PubMed] [Google Scholar]
- 94.Ito H, Adachi Y, Arimura Y, Endo T, Hinoda Y, Imai K. A 25-year clinical history of portopulmonary hypertension associated with latent myeloproliferative disorder. J Gastroenterol. 2003;38:488–492. doi: 10.1007/s00535-002-1086-3. [DOI] [PubMed] [Google Scholar]
- 95.Willems E, Canivet JL, Ghaye B, de Leval L, Radermecker M, Preiser JC, Beguin Y. Pulmonary veno-occlusive disease in myeloproliferative disorder. Eur Respir J. 2009;33:213–216. doi: 10.1183/09031936.00157707. [DOI] [PubMed] [Google Scholar]
- 96.Guilpain P, Montani D, Damaj G, Achouh L, Lefrère F, Le Pavec J, Marfaing-Koka A, Dartevelle P, Simonneau G, Humbert M, et al. Pulmonary hypertension associated with myeloproliferative disorders: a retrospective study of ten cases. Respiration. 2008;76:295–302. doi: 10.1159/000112822. [DOI] [PubMed] [Google Scholar]
- 97.Popat U, Frost A, Liu E, May R, Bag R, Reddy V, Prchal JT. New onset of myelofibrosis in association with pulmonary arterial hypertension. Ann Intern Med. 2005;143:466–467. doi: 10.7326/0003-4819-143-6-200509200-00017. [DOI] [PubMed] [Google Scholar]
- 98.Witthuhn BA, Quelle FW, Silvennoinen O, Yi T, Tang B, Miura O, Ihle JN. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell. 1993;74:227–236. doi: 10.1016/0092-8674(93)90414-l. [DOI] [PubMed] [Google Scholar]
- 99.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 100.Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE, Gozo M, McDowell EP, Levine RL, Doukas J, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13:311–320. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 101.Farha S, Asosingh K, Xu W, Sharp J, George D, Comhair S, Park M, Tang WH, Loyd JE, Theil K, et al. Hypoxia-inducible factors in human pulmonary arterial hypertension: a link to the intrinsic myeloid abnormalities. Blood. 2011;117:3485–3493. doi: 10.1182/blood-2010-09-306357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Popat U, Frost A, Liu E, Guan Y, Durette A, Reddy V, Prchal JT. High levels of circulating CD34 cells, dacrocytes, clonal hematopoiesis, and JAK2 mutation differentiate myelofibrosis with myeloid metaplasia from secondary myelofibrosis associated with pulmonary hypertension. Blood. 2006;107:3486–3488. doi: 10.1182/blood-2005-08-3319. [DOI] [PubMed] [Google Scholar]
- 103.Zetterberg E, Popat U, Hasselbalch H, Prchal J, Palmblad J. Angiogenesis in pulmonary hypertension with myelofibrosis. Haematologica. 2008;93:945–946. doi: 10.3324/haematol.12426. [DOI] [PubMed] [Google Scholar]
- 104.Cortelezzi A, Gritti G, Del Papa N, Pasquini MC, Calori R, Gianelli U, Cortiana M, Parati G, Onida F, Sozzi F, et al. Pulmonary arterial hypertension in primary myelofibrosis is common and associated with an altered angiogenic status. Leukemia. 2008;22:646–649. doi: 10.1038/sj.leu.2404943. [DOI] [PubMed] [Google Scholar]
- 105.Ribatti D, Vacca A, Roncali L, Dammacco F. Hematopoiesis and angiogenesis: a link between two apparently independent processes. J Hematother Stem Cell Res. 2000;9:13–19. doi: 10.1089/152581600319577. [DOI] [PubMed] [Google Scholar]
- 106.Asosingh K, Farha S, Lichtin A, Graham B, George D, Aldred M, Hazen SL, Loyd J, Tuder R, Erzurum SC. Pulmonary vascular disease in mice xenografted with human BM progenitors from patients with pulmonary arterial hypertension. Blood. 2012;120:1218–1227. doi: 10.1182/blood-2012-03-419275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yan L, Chen X, Talati M, Nunley BW, Gladson S, Blackwell T, Cogan J, Austin E, Wheeler F, Loyd J, et al. Bone Marrow-derived Cells Contribute to the Pathogenesis of Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2016;193:898–909. doi: 10.1164/rccm.201502-0407OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen CH, Chen HA, Wang HP, Liao HT, Chou CT, Huang DF. Pulmonary arterial hypertension in autoimmune diseases: an analysis of 19 cases from a medical center in northern Taiwan. J Microbiol Immunol Infect. 2006;39:162–168. [PubMed] [Google Scholar]
- 109.Launay D, Hachulla E, Hatron PY, Jais X, Simonneau G, Humbert M. Pulmonary arterial hypertension: a rare complication of primary Sjögren syndrome: report of 9 new cases and review of the literature. Medicine (Baltimore) 2007;86:299–315. doi: 10.1097/MD.0b013e3181579781. [DOI] [PubMed] [Google Scholar]
- 110.Nicolls MR, Taraseviciene-Stewart L, Rai PR, Badesch DB, Voelkel NF. Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J. 2005;26:1110–1118. doi: 10.1183/09031936.05.00045705. [DOI] [PubMed] [Google Scholar]
- 111.Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, Patterson AJ, Agrawal R, Rabinovitch M, Ambler K, et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res. 2011;109:867–879. doi: 10.1161/CIRCRESAHA.110.236927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim KJ, Baek IW, Yoon CH, Kim WU, Cho CS. Association of Anemic Hypoxia and Increased Pulmonary Artery Systolic Pressure in Patients With Systemic Lupus Erythematosus. Arthritis Care Res (Hoboken) 2015;67:1702–1711. doi: 10.1002/acr.22630. [DOI] [PubMed] [Google Scholar]
- 113.Mackworth-Young CG, Gharavi AE, Boey ML, Hughes GR. Portal and pulmonary hypertension in a case of systematic lupus erythematosus: possible relationship with a clotting abnormality. Eur J Rheumatol Inflamm. 1984;7:71–74. [PubMed] [Google Scholar]
- 114.Scicchitano P, Dentamaro I, Tunzi F, Ricci G, Carbonara S, Devito F, Zito A, Ciampolillo A, Ciccone MM. Pulmonary hypertension in thyroid diseases. Endocrine. 2016;54:578–587. doi: 10.1007/s12020-016-0923-8. [DOI] [PubMed] [Google Scholar]
- 115.Chu JW, Kao PN, Faul JL, Doyle RL. High prevalence of autoimmune thyroid disease in pulmonary arterial hypertension. Chest. 2002;122:1668–1673. doi: 10.1378/chest.122.5.1668. [DOI] [PubMed] [Google Scholar]
- 116.Erem C, Ucuncu O, Yilmaz M, Kocak M, Nuhoglu İ, Ersoz HO. Increased thrombin-activatable fibrinolysis inhibitor and decreased tissue factor pathway inhibitor in patients with hyperthyroidism. Endocrine. 2009;36:473–478. doi: 10.1007/s12020-009-9271-2. [DOI] [PubMed] [Google Scholar]
- 117.Al Husseini A, Bagnato G, Farkas L, Gomez-Arroyo J, Farkas D, Mizuno S, Kraskauskas D, Abbate A, Van Tassel B, Voelkel NF, et al. Thyroid hormone is highly permissive in angioproliferative pulmonary hypertension in rats. Eur Respir J. 2013;41:104–114. doi: 10.1183/09031936.00196511. [DOI] [PubMed] [Google Scholar]
- 118.Connor P, Veys P, Amrolia P, Haworth S, Ashworth M, Moledina S. Pulmonary hypertension in children with Evans syndrome. Pediatr Hematol Oncol. 2008;25:93–98. doi: 10.1080/08880010801888253. [DOI] [PubMed] [Google Scholar]
- 119.Zhang Y, Qui Y, Zhu J, Gao D. Pulmonary hypertension associated with autoimmune hemolytic anemia: a case report. Int J Cardiol. 2007;115:e1–e2. doi: 10.1016/j.ijcard.2006.05.053. [DOI] [PubMed] [Google Scholar]
- 120.Perros F, Günther S, Ranchoux B, Godinas L, Antigny F, Chaumais MC, Dorfmüller P, Hautefort A, Raymond N, Savale L, et al. Mitomycin-Induced Pulmonary Veno-Occlusive Disease: Evidence From Human Disease and Animal Models. Circulation. 2015;132:834–847. doi: 10.1161/CIRCULATIONAHA.115.014207. [DOI] [PubMed] [Google Scholar]
- 121.Dumitrescu D, Seck C, ten Freyhaus H, Gerhardt F, Erdmann E, Rosenkranz S. Fully reversible pulmonary arterial hypertension associated with dasatinib treatment for chronic myeloid leukaemia. Eur Respir J. 2011;38:218–220. doi: 10.1183/09031936.00154210. [DOI] [PubMed] [Google Scholar]
- 122.Sano M, Saotome M, Urushida T, Katoh H, Satoh H, Ohnishi K, Hayashi H. Pulmonary arterial hypertension caused by treatment with dasatinib for chronic myeloid leukemia -critical alert- Intern Med. 2012;51:2337–2340. doi: 10.2169/internalmedicine.51.7472. [DOI] [PubMed] [Google Scholar]
- 123.Mattei D, Feola M, Orzan F, Mordini N, Rapezzi D, Gallamini A. Reversible dasatinib-induced pulmonary arterial hypertension and right ventricle failure in a previously allografted CML patient. Bone Marrow Transplant. 2009;43:967–968. doi: 10.1038/bmt.2008.415. [DOI] [PubMed] [Google Scholar]
- 124.Orlandi EM, Rocca B, Pazzano AS, Ghio S. Reversible pulmonary arterial hypertension likely related to long-term, low-dose dasatinib treatment for chronic myeloid leukaemia. Leuk Res. 2012;36:e4–e6. doi: 10.1016/j.leukres.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 125.Rasheed W, Flaim B, Seymour JF. Reversible severe pulmonary hypertension secondary to dasatinib in a patient with chronic myeloid leukemia. Leuk Res. 2009;33:861–864. doi: 10.1016/j.leukres.2008.09.026. [DOI] [PubMed] [Google Scholar]
- 126.Yun S, Anwer F, Vincelette ND. Dasatinib-induced pulmonary hypertension in chronic myelogenous leukaemia. BMJ Case Rep. 2014;2014:pii: bcr2014204477. doi: 10.1136/bcr-2014-204477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hong JH, Lee SE, Choi SY, Kim SH, Jang EJ, Bang JH, Park JE, Jeon HR, Oh YJ, Yi JE, et al. Reversible Pulmonary Arterial Hypertension Associated with Dasatinib for Chronic Myeloid Leukemia. Cancer Res Treat. 2015;47:937–942. doi: 10.4143/crt.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Montani D, Bergot E, Günther S, Savale L, Bergeron A, Bourdin A, Bouvaist H, Canuet M, Pison C, Macro M, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation. 2012;125:2128–2137. doi: 10.1161/CIRCULATIONAHA.111.079921. [DOI] [PubMed] [Google Scholar]
- 129.Guignabert C, Phan C, Seferian A, Huertas A, Tu L, Thuillet R, Sattler C, Le Hiress M, Tamura Y, Jutant EM, et al. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J Clin Invest. 2016;126:3207–3218. doi: 10.1172/JCI86249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shah NP, Wallis N, Farber HW, Mauro MJ, Wolf RA, Mattei D, Guha M, Rea D, Peacock A. Clinical features of pulmonary arterial hypertension in patients receiving dasatinib. Am J Hematol. 2015;90:1060–1064. doi: 10.1002/ajh.24174. [DOI] [PubMed] [Google Scholar]
- 131.Nagaraj C, Tang B, Bálint Z, Wygrecka M, Hrzenjak A, Kwapiszewska G, Stacher E, Lindenmann J, Weir EK, Olschewski H, et al. Src tyrosine kinase is crucial for potassium channel function in human pulmonary arteries. Eur Respir J. 2013;41:85–95. doi: 10.1183/09031936.00211811. [DOI] [PubMed] [Google Scholar]
- 132.Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, Cool CD, Bishop AE, Geraci M, Semenza GL, et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol. 2001;195:367–374. doi: 10.1002/path.953. [DOI] [PubMed] [Google Scholar]
- 133.Guignabert C, Montani D. Key roles of Src family tyrosine kinases in the integrity of the pulmonary vascular bed. Eur Respir J. 2013;41:3–4. doi: 10.1183/09031936.00091912. [DOI] [PubMed] [Google Scholar]
- 134.Fruehauf S, Steiger S, Topaly J, Ho AD. Pulmonary artery hypertension during interferon-alpha therapy for chronic myelogenous leukemia. Ann Hematol. 2001;80:308–310. doi: 10.1007/s002770100298. [DOI] [PubMed] [Google Scholar]
- 135.Savale L, Sattler C, Günther S, Montani D, Chaumais MC, Perrin S, Jaïs X, Seferian A, Jovan R, Bulifon S, et al. Pulmonary arterial hypertension in patients treated with interferon. Eur Respir J. 2014;44:1627–1634. doi: 10.1183/09031936.00057914. [DOI] [PubMed] [Google Scholar]
- 136.Dhillon S, Kaker A, Dosanjh A, Japra D, Vanthiel DH. Irreversible pulmonary hypertension associated with the use of interferon alpha for chronic hepatitis C. Dig Dis Sci. 2010;55:1785–1790. doi: 10.1007/s10620-010-1220-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hanaoka M, Kubo K, Hayano T, Koizumi T, Kobayashi T. Interferon-alpha elevates pulmonary blood pressure in sheep--the role of thromboxane cascade. Eur J Pharmacol. 1999;370:145–151. doi: 10.1016/s0014-2999(99)00107-7. [DOI] [PubMed] [Google Scholar]
- 138.Woods M, Wood EG, Bardswell SC, Bishop-Bailey D, Barker S, Wort SJ, Mitchell JA, Warner TD. Role for nuclear factor-kappaB and signal transducer and activator of transcription 1/interferon regulatory factor-1 in cytokine-induced endothelin-1 release in human vascular smooth muscle cells. Mol Pharmacol. 2003;64:923–931. doi: 10.1124/mol.64.4.923. [DOI] [PubMed] [Google Scholar]
- 139.George PM, Oliver E, Dorfmuller P, Dubois OD, Reed DM, Kirkby NS, Mohamed NA, Perros F, Antigny F, Fadel E, et al. Evidence for the involvement of type I interferon in pulmonary arterial hypertension. Circ Res. 2014;114:677–688. doi: 10.1161/CIRCRESAHA.114.302221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hoeper MM, Niedermeyer J, Hoffmeyer F, Flemming P, Fabel H. Pulmonary hypertension after splenectomy? Ann Intern Med. 1999;130:506–509. doi: 10.7326/0003-4819-130-6-199903160-00014. [DOI] [PubMed] [Google Scholar]
- 141.Phrommintikul A, Sukonthasarn A, Kanjanavanit R, Nawarawong W. Splenectomy: a strong risk factor for pulmonary hypertension in patients with thalassaemia. Heart. 2006;92:1467–1472. doi: 10.1136/hrt.2005.079970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lode HN, Krings G, Schulze-Neick I, Dähmlow S, Schroeder U, Bonnet R, DaPalma J, Luck W, Strauss G, Berger F, et al. Pulmonary hypertension in a case of Hb-Mainz hemolytic anemia. J Pediatr Hematol Oncol. 2007;29:173–177. doi: 10.1097/MPH.0b013e318032568c. [DOI] [PubMed] [Google Scholar]
- 143.Kimmig LM, Palevsky HI. Review of the Association between Splenectomy and Chronic Thromboembolic Pulmonary Hypertension. Ann Am Thorac Soc. 2016;13:945–954. doi: 10.1513/AnnalsATS.201512-826FR. [DOI] [PubMed] [Google Scholar]
- 144.Frey MK, Alias S, Winter MP, Redwan B, Stübiger G, Panzenboeck A, Alimohammadi A, Bonderman D, Jakowitsch J, Bergmeister H, et al. Splenectomy is modifying the vascular remodeling of thrombosis. J Am Heart Assoc. 2014;3:e000772. doi: 10.1161/JAHA.113.000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kisanuki A, Kietthubthew S, Asada Y, Marutsuka K, Funahara Y, Sumiyoshi A. Intravenous injection of sonicated blood induces pulmonary microthromboembolism in rabbits with ligation of the splenic artery. Thromb Res. 1997;85:95–103. doi: 10.1016/s0049-3848(96)00226-5. [DOI] [PubMed] [Google Scholar]
- 146.Troussard X, Bernaudin JF, Cordonnier C, Fleury J, Payen D, Briere J, Vernant JP. Pulmonary veno-occlusive disease after bone marrow transplantation. Thorax. 1984;39:956–957. doi: 10.1136/thx.39.12.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schechter T, Leucht S, Bouffet E, Cutz E, Gassas A, Huang A, Bartels U, Humpl T, Doyle J. Pulmonary hypertensive vasculopathy following tandem autologous transplantation in pediatric patients with central nervous system tumors. Biol Blood Marrow Transplant. 2013;19:235–239. doi: 10.1016/j.bbmt.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 148.Trobaugh-Lotrario AD, Greffe B, Deterding R, Deutsch G, Quinones R. Pulmonary veno-occlusive disease after autologous bone marrow transplant in a child with stage IV neuroblastoma: case report and literature review. J Pediatr Hematol Oncol. 2003;25:405–409. doi: 10.1097/00043426-200305000-00011. [DOI] [PubMed] [Google Scholar]
- 149.Özyörük D, Kibar AE, Sürücü M, Azak E, Emir S, Çetin İİ, Tunç B, Özbek NY. Pulmonary arterial hypertension in a child with stage-IV neuroblastoma after autologous hematopoietic stem cell transplantation and review of the literature. Pediatr Transplant. 2015;19:E185–E188. doi: 10.1111/petr.12576. [DOI] [PubMed] [Google Scholar]
- 150.Seguchi M, Hirabayashi N, Fujii Y, Azuno Y, Fujita N, Takeda K, Sato Y, Nishimura M, Yamada K, Oka Y. Pulmonary hypertension associated with pulmonary occlusive vasculopathy after allogeneic bone marrow transplantation. Transplantation. 2000;69:177–179. doi: 10.1097/00007890-200001150-00030. [DOI] [PubMed] [Google Scholar]
- 151.Salzman D, Adkins DR, Craig F, Freytes C, LeMaistre CF. Malignancy-associated pulmonary veno-occlusive disease: report of a case following autologous bone marrow transplantation and review. Bone Marrow Transplant. 1996;18:755–760. [PubMed] [Google Scholar]
- 152.Bunte MC, Patnaik MM, Pritzker MR, Burns LJ. Pulmonary veno-occlusive disease following hematopoietic stem cell transplantation: a rare model of endothelial dysfunction. Bone Marrow Transplant. 2008;41:677–686. doi: 10.1038/sj.bmt.1705990. [DOI] [PubMed] [Google Scholar]
- 153.Jodele S, Hirsch R, Laskin B, Davies S, Witte D, Chima R. Pulmonary arterial hypertension in pediatric patients with hematopoietic stem cell transplant-associated thrombotic microangiopathy. Biol Blood Marrow Transplant. 2013;19:202–207. doi: 10.1016/j.bbmt.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 154.Grigg A, Buchanan M, Whitford H. Late-onset pulmonary arterial hypertension in association with graft-versus-host disease after allogeneic stem-cell transplantation. Am J Hematol. 2005;80:38–42. doi: 10.1002/ajh.20373. [DOI] [PubMed] [Google Scholar]
- 155.Mathew R, Huang J, Katta US, Krishnan U, Sandoval C, Gewitz MH. Immunosuppressant-induced endothelial damage and pulmonary arterial hypertension. J Pediatr Hematol Oncol. 2011;33:55–58. doi: 10.1097/MPH.0b013e3181ec0ede. [DOI] [PubMed] [Google Scholar]
- 156.Lombard CM, Churg A, Winokur S. Pulmonary veno-occlusive disease following therapy for malignant neoplasms. Chest. 1987;92:871–876. doi: 10.1378/chest.92.5.871. [DOI] [PubMed] [Google Scholar]
- 157.Kramer MR, Estenne M, Berkman N, Antoine M, de Francquen P, Lipski A, Jacobovitz D, Lafair J. Radiation-induced pulmonary veno-occlusive disease. Chest. 1993;104:1282–1284. doi: 10.1378/chest.104.4.1282. [DOI] [PubMed] [Google Scholar]
- 158.Butler P, Chahal P, Hudson NM, Hubner PJ. Pulmonary hypertension after lung irradiation in infancy. Br Med J (Clin Res Ed) 1981;283:1365. doi: 10.1136/bmj.283.6303.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Milliat F, François A, Isoir M, Deutsch E, Tamarat R, Tarlet G, Atfi A, Validire P, Bourhis J, Sabourin JC, et al. Influence of endothelial cells on vascular smooth muscle cells phenotype after irradiation: implication in radiation-induced vascular damages. Am J Pathol. 2006;169:1484–1495. doi: 10.2353/ajpath.2006.060116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Perkett EA, Brigham KL, Meyrick B. Increased vasoreactivity and chronic pulmonary hypertension following thoracic irradiation in sheep. J Appl Physiol (1985) 1986;61:1875–1881. doi: 10.1152/jappl.1986.61.5.1875. [DOI] [PubMed] [Google Scholar]
- 161.Adamson IY, Bowden DH. Endothelial injury and repair in radiation-induced pulmonary fibrosis. Am J Pathol. 1983;112:224–230. [PMC free article] [PubMed] [Google Scholar]
- 162.Ghobadi G, Bartelds B, van der Veen SJ, Dickinson MG, Brandenburg S, Berger RM, Langendijk JA, Coppes RP, van Luijk P. Lung irradiation induces pulmonary vascular remodelling resembling pulmonary arterial hypertension. Thorax. 2012;67:334–341. doi: 10.1136/thoraxjnl-2011-200346. [DOI] [PubMed] [Google Scholar]
- 163.Jankowska EA, Rozentryt P, Witkowska A, Nowak J, Hartmann O, Ponikowska B, Borodulin-Nadzieja L, Banasiak W, Polonski L, Filippatos G, et al. Iron deficiency: an ominous sign in patients with systolic chronic heart failure. Eur Heart J. 2010;31:1872–1880. doi: 10.1093/eurheartj/ehq158. [DOI] [PubMed] [Google Scholar]
- 164.Krasuski RA, Hart SA, Smith B, Wang A, Harrison JK, Bashore TM. Association of anemia and long-term survival in patients with pulmonary hypertension. Int J Cardiol. 2011;150:291–295. doi: 10.1016/j.ijcard.2010.04.038. [DOI] [PubMed] [Google Scholar]
- 165.Ruiter G, Lankhorst S, Boonstra A, Postmus PE, Zweegman S, Westerhof N, van der Laarse WJ, Vonk-Noordegraaf A. Iron deficiency is common in idiopathic pulmonary arterial hypertension. Eur Respir J. 2011;37:1386–1391. doi: 10.1183/09031936.00100510. [DOI] [PubMed] [Google Scholar]
- 166.Rhodes CJ, Wharton J, Howard LS, Gibbs JS, Wilkins MR. Red cell distribution width outperforms other potential circulating biomarkers in predicting survival in idiopathic pulmonary arterial hypertension. Heart. 2011;97:1054–1060. doi: 10.1136/hrt.2011.224857. [DOI] [PubMed] [Google Scholar]
- 167.Rhodes CJ, Howard LS, Busbridge M, Ashby D, Kondili E, Gibbs JS, Wharton J, Wilkins MR. Iron deficiency and raised hepcidin in idiopathic pulmonary arterial hypertension: clinical prevalence, outcomes, and mechanistic insights. J Am Coll Cardiol. 2011;58:300–309. doi: 10.1016/j.jacc.2011.02.057. [DOI] [PubMed] [Google Scholar]
- 168.van Empel VP, Lee J, Williams TJ, Kaye DM. Iron deficiency in patients with idiopathic pulmonary arterial hypertension. Heart Lung Circ. 2014;23:287–292. doi: 10.1016/j.hlc.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 169.Decker I, Ghosh S, Comhair SA, Farha S, Tang WH, Park M, Wang S, Lichtin AE, Erzurum SC. High levels of zinc-protoporphyrin identify iron metabolic abnormalities in pulmonary arterial hypertension. Clin Transl Sci. 2011;4:253–258. doi: 10.1111/j.1752-8062.2011.00301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ruiter G, Manders E, Happé CM, Schalij I, Groepenhoff H, Howard LS, Wilkins MR, Bogaard HJ, Westerhof N, van der Laarse WJ, et al. Intravenous iron therapy in patients with idiopathic pulmonary arterial hypertension and iron deficiency. Pulm Circ. 2015;5:466–472. doi: 10.1086/682217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ruiter G, Lanser IJ, de Man FS, van der Laarse WJ, Wharton J, Wilkins MR, Howard LS, Vonk-Noordegraaf A, Voskuyl AE. Iron deficiency in systemic sclerosis patients with and without pulmonary hypertension. Rheumatology (Oxford) 2014;53:285–292. doi: 10.1093/rheumatology/ket331. [DOI] [PubMed] [Google Scholar]
- 172.Smith TG, Balanos GM, Croft QP, Talbot NP, Dorrington KL, Ratcliffe PJ, Robbins PA. The increase in pulmonary arterial pressure caused by hypoxia depends on iron status. J Physiol. 2008;586:5999–6005. doi: 10.1113/jphysiol.2008.160960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Gassmann M, Muckenthaler MU. Adaptation of iron requirement to hypoxic conditions at high altitude. J Appl Physiol (1985) 2015;119:1432–1440. doi: 10.1152/japplphysiol.00248.2015. [DOI] [PubMed] [Google Scholar]
- 174.Cotroneo E, Ashek A, Wang L, Wharton J, Dubois O, Bozorgi S, Busbridge M, Alavian KN, Wilkins MR, Zhao L. Iron homeostasis and pulmonary hypertension: iron deficiency leads to pulmonary vascular remodeling in the rat. Circ Res. 2015;116:1680–1690. doi: 10.1161/CIRCRESAHA.116.305265. [DOI] [PubMed] [Google Scholar]
- 175.Ghosh MC, Zhang DL, Jeong SY, Kovtunovych G, Ollivierre-Wilson H, Noguchi A, Tu T, Senecal T, Robinson G, Crooks DR, et al. Deletion of iron regulatory protein 1 causes polycythemia and pulmonary hypertension in mice through translational derepression of HIF2α. Cell Metab. 2013;17:271–281. doi: 10.1016/j.cmet.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Naito Y, Hosokawa M, Hao H, Sawada H, Hirotani S, Iwasaku T, Okuhara Y, Eguchi A, Hirota S, Ohyanagi M, et al. Impact of dietary iron restriction on the development of monocrotaline-induced pulmonary vascular remodeling and right ventricular failure in rats. Biochem Biophys Res Commun. 2013;436:145–151. doi: 10.1016/j.bbrc.2013.05.059. [DOI] [PubMed] [Google Scholar]
- 177.Naito Y, Hosokawa M, Sawada H, Oboshi M, Hirotani S, Iwasaku T, Okuhara Y, Morisawa D, Eguchi A, Nishimura K, et al. Transferrin Receptor 1 in Chronic Hypoxia-Induced Pulmonary Vascular Remodeling. Am J Hypertens. 2016;29:713–718. doi: 10.1093/ajh/hpv163. [DOI] [PubMed] [Google Scholar]
- 178.Wong CM, Preston IR, Hill NS, Suzuki YJ. Iron chelation inhibits the development of pulmonary vascular remodeling. Free Radic Biol Med. 2012;53:1738–1747. doi: 10.1016/j.freeradbiomed.2012.08.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kim KH, Maldonado F, Ryu JH, Eiken PW, Hartman TE, Bartholmai BJ, Decker PA, Yi ES. Iron deposition and increased alveolar septal capillary density in nonfibrotic lung tissue are associated with pulmonary hypertension in idiopathic pulmonary fibrosis. Respir Res. 2010;11:37. doi: 10.1186/1465-9921-11-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Richardson DR, Lane DJ, Becker EM, Huang ML, Whitnall M, Suryo Rahmanto Y, Sheftel AD, Ponka P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci USA. 2010;107:10775–10782. doi: 10.1073/pnas.0912925107. [DOI] [PMC free article] [PubMed] [Google Scholar]