

Abstract
Tissues are the organizational units of function in metazoan organisms. Tissues comprise an assortment of cellular building blocks, soluble factors, and extracellular matrix (ECM) that are composed into specific three dimensional (3D) structures. The capacity to reconstitute tissues in vitro with the structural complexity observed in vivo is key to understanding processes such as morphogenesis, homeostasis, and disease. In this unit, we describe DNA-programmed Assembly of Cells (DPAC), a method to fabricate viable, functional arrays of organoid-like tissues within 3D ECM gels. In DPAC, dissociated cells are chemically functionalized with degradable oligonucleotide “velcro,” allowing rapid, specific, and reversible cell adhesion to a two-dimensional (2D) template patterned with complementary DNA. An iterative assembly process builds up organoids, layer-by-layer, from this initial 2D template and into the third dimension. Cleavage of the DNA releases the completed array of tissues that are captured and fully embedded in ECM gels for culture and observation. DPAC controls the size, shape, composition, and spatial heterogeneity of organoids, and permits positioning constituent cells with single-cell resolution even within cultures several centimeters long.
Keywords: tissue array, DNA, synthetic biology, cell-cell interactions, tissue engineering, patterning, organotypic, organoid, 3D culture
INTRODUCTION
This Unit details a suite of protocols, termed DNA-programmed Assembly of Cells (DPAC), for fabricating arrays of mammalian organoids (Fig. 1). DPAC uses Watson-Crick base-pairing of DNA-functionalized cells to drive organoid assembly. DPAC-based organoids can contain arbitrary combinations of cell types, are endowed with three-dimensional structure at cell-diameter resolution, span centimeters, and are compatible with a variety of extracellular matrix (ECM) gels. Organoids assembled using DPAC can be cultured long-term in vitro, and they are amenable to live cell imaging, immunofluorescence, and other analytical techniques. DPAC has utility as a tool for tissue engineering, for studying development and morphogenesis, and as a drug-screening platform.
Figure 1. Overall scheme of DPAC.

In Basic Protocol 1, DNA droplets are patterned across a surface with microscale direct writing. In Basic Protocol 2, this DNA is covalently linked to the surface, and the surface is otherwise passivated to cell adhesion. In Basic Protocol 3, cells are functionalized with DNA. In Basic Protocol 4, these cells are assembled into organoids on the surface via DNA hybridization. In Basic Protocol 5, these organoids are released from the surface and into extracellular matrix hydrogel.
Basic Protocol 1 describes methods for preparing a tissue template on a glass slide using microscale direct writing, a droplet-based patterning method. Basic Protocol 2 describes chemical manipulations to render the slide non-adhesive. Basic Protocol 3 describes chemically functionalizing mammalian cells with the lipid-DNA necessary for interaction with the template, and a supporting protocol describes the synthesis of the lipid-DNA itself. Basic Protocol 4 describes the iterative assembly of DNA-functionalized cells via hybridization with the template slide. Basic Protocol 5 describes the 3D transfer process necessary for embedding into hydrogels suitable for mammalian cell culture.
STRATEGIC PLANNING
Choice of DNA Patterning Methods
All of the protocols described in this unit require a glass slide patterned with DNA. A variety of methods are available for patterning glass slides with DNA, including microscale direct writing, microcontact printing, and photolithography. The best method for use is application-dependent. A discussion of patterning methods is given below.
Microscale direct writing is a process wherein aqueous droplets of DNA are deposited one-at-a-time onto a substrate using a purpose-built cantilever. Complex patterns are designed with no lead-time, subcellular resolution, and with facile multiplexing of multiple DNA sequences. However, with current instrumentation, feature deposition is limited to 2500–3500 features/hour. Additionally, it is necessary to have access to the appropriate equipment, which is not widely available. We recommend microscale direct writing if the equipment is available and if no more than several thousand DNA droplets are desired per-slide. Microscale direct writing using the BioForce Nano eNabler is described in-depth in Basic Protocol 1.
Microcontact printing is a dry transfer process wherein DNA adsorbed onto a PDMS stamp is transferred to a glass slide (Lange et al., 2004) (Kumar et al., 1994). This method allows all features to be deposited simultaneously across the area of the stamp and therefore scales efficiently. However, patterns are prone to a variety of failure modes, especially for sparse patterns or patterns with subcellular-resolution features, and the probability of failure is multiplicative when multiplexing several DNA sequences. There is also lead-time associated with fabricating a silicon master and casting PDMS stamps. We recommend microcontact printing in the absence of microscale direct writing equipment, or if the desired pattern could only be printed with ten thousand or more DNA droplets.
Photolithographic techniques, in principle, have the highest resolution, print faster than direct writing, and multiplex better than microcontact printing. Although photolithographic techniques have been reported for DNA patterning (Onoe et al., 2012) (El Muslemany et al., 2014), the authors have not sufficiently tested and can not yet recommend any easily adoptable protocols at this time.
Choice of DNA Strands
We use 20mer DNA strands with approximately 50% GC content. Keeping the GC content fixed across strands ensures that the melting temperature and hybridization thermodynamics are roughly the same across strands. Due to the size of the cellular glycocalyx, lipid-anchored oligonucleotides (incorporated into the cell as per Basic Protocol 3) require spacer nucleotides to provide access for hybridization of the adhesive 20mer component. This is not necessary when anchoring DNA strands to cell surface amines (using NHS-DNA) (Hsiao et al., 2009) or glycans (using metabolic engineering and biorthogonal chemistry) (Gartner and Bertozzi, 2009). A 60mer polyT sequence is used as a spacer. The polyT sequence lacks secondary structure and its 60mer length is long enough to allow accessibility but short enough to be easily synthesized. If using a library of multiple strands, individual sequences should be designed to minimize secondary structure and intra-library hybridization, even at low temperatures (4 °C). The DINAMelt web server (Markham and Zuker, 2005) is well-suited for modeling intra-library hybridization when designing strands. The library of four strands (and their complements) shown below has been extensively tested by the authors and allows encoding up to four discrete intercellular interactions:
| A: | 5'-ACTGACTGACTGACTGACTG-3' |
| Aprime: | 5'-CAGTCAGTCAGTCAGTCAGT-3' |
| B: | 5'-TCATACGACTCACTCTAGGG-3' |
| Bprime: | 5'-CCCTAGAGTGAGTCGTATGA-3' |
| C: | 5'-GTAACGATCCAGCTGTCACT-3' |
| Cprime: | 5'-AGTGACAGCTGGATCGTTAC-3' |
| D: | 5'-AGAAGAAGAACGAAGAAGAA-3' |
| Dprime: | 5'-TTCTTCTTCGTTCTTCTTCT-3' |
Additionally, when using fatty acid DNA (see Support Protocol 1), an internal hybridizing sequence is required, called the Co-anchor sequence. This sequence is:
| Co-anchor: | 5’-AGTGACAGCTGGATCGTTAC-3’ |
| Co-anchor-prime: | 5’-GTAACGATCCAGCTGTCACT-3’ |
Organoid Geometry Considerations
One DNA pattern can produce a variety of organoid geometries, depending on the synthetic scheme used during programmed assembly (Basic Protocol 4). Successive rounds of assembly elaborate a single cell into progressively larger hemispheres of cells. Correspondingly, lines of cells expand into half-cylinders, and a variety of three-dimensional geometries can be achieved when combining multiple cell types. Appropriately choosing a synthetic scheme allows forming tissue with sharp internal boundaries and distinct tissue compartments (Todhunter et al., 2015).
DPAC manipulations are performed within flow cells, and these flow cells constrain feasible tissue geometries. It is not feasible to produce organoids taller than half the height of the flow cell due to shear flow considerations. For flow cells ~200 µm tall, this translates to organoids with a maximum height of 4–6 cell diameters. It is worth noting that some cells, such as epithelial cells, compact into smaller aggregates upon one or two days' culture. Taller structures will fall apart under flow. It should be feasible to fabricate taller flow cells, but such flow cells have not been extensively tested by the authors. Furthermore, tall features create downstream "flow shadows" within the flow cell, regions where it becomes difficult to wash. To minimize this, synthetic schemes should account for all small features before assembling tall features. Typical flow cells have length and width of 18 mm×4.5 mm. Patterns must fit within these bounds, preferably with margins such that the printable area does not exceed 16 mm×3.5 mm. Other flow cell dimensions, such as 18 mm×9 mm, have been successfully used but not extensively tested by the authors.
BASIC PROTOCOL 1
Patterning Aqueous DNA using Microscale Direct Writing
The first step in any DPAC experiment is producing a DNA-patterned glass slide onto which DNA-functionalized cells can attach. Microscale direct writing offers a useful combination of speed and flexibility. This process begins with a raster-based digital image, which is used to define the DNA pattern. The Nano eNabler accepts 24-bit bitmap files for patterns, which are binarized into on-or-off pixels. Patterns should fit within a 16 mm×3.5 mm area as described in the Organoid Geometry Considerations section, above.
The higher the concentration of patterned DNA, the more efficiently the surface will capture cells in Basic Protocol 4. However, for the 20mer single-stranded DNA typically used, viscosity becomes a limiting factor above 1 mM. As viscosity increases, the droplets deposited by microscale direct writing shrink. Although we have successfully patterned with DNA concentrations as high as 3 mM, 1.5 mM seems to be a more effective trade-off.
When designing patterns using multiple DNA sequences, include a calibration grid offset from the main pattern (Fig. 2). This is a sacrificial pattern component that aids the alignment of multiple sequences. For patterns with three or more sequences, multiple calibration grids are required.
Figure 2. Sample bitmap pattern with calibration grid.

Checkerboard-like calibration grids, at left, permit manual alignment of multiplexed pattern components.
Materials
2 mM 20mer 5'-amine-DNA in dH2O (e.g., available from Eurofins MWG Operon)
Aldehyde-silanized glass slide (e.g., Nexterion AL, Applied Microarrays cat. #1064874)
Diamond scribe (Fisher, cat. #17-467-634)
Surface patterning tool (SPT) (BioForce Nanosciences cat. #SPT-S-C30S)
UV/Ozone apparatus (e.g., BioForce ProCleaner)
Microscale direct writing apparatus (e.g., BioForce Nano eNabler)
Steps
Mix 1.5 µL 5'-amine-DNA with 0.5 µL 4× spotting buffer and pipet to mix.
- Take a fresh aldehyde slide from storage. On the reverse of the slide, use a diamond scribe to etch origins for each of the desired patterns.Although there are many ways to mark slides, marks from a diamond scribe resist the solvent treatments in Basic Protocol 2.
- Using polymer forceps, place a clean SPT in the BioForce ProCleaner. Expose the SPT to ozone for at least 15 minutes.If a BioForce ProCleaner is not available, it is also possible to oxidize SPTs in a plasma oxidizer. Settings vary by instrument and would need to be established empirically, but if, after oxidation, the DNA solution, loaded in Step 4, completely wets the liquid channel on the SPT cantilever, then the oxidiation is sufficient.
Take the SPT out of the ProCleaner and load 0.4 µL DNA solution into the SPT’s reservoir. Place the SPT on an SPT Holder and mount on the Nano eNabler. Mount the aldehyde slide on the stage.
- Launch NanoWare and print DNA droplets as described by BioForce Application Note 203 (Application Note 203 - “Speed Printing”; or, printing in “No Laser Mode” with the Nano eNabler System™). Adjust settings as needed to yield droplets with 8 µm diameter. A good starting point is 0.1 s Contact Time, 0.0 s Wait Time, and 65% Target Humidity.When optimizing droplet size, several variables should be considered. Droplet size correlates strongly with Contact Time from 0.1 s to 1.0 s, with droplet size mattering less above 1.0 s. Droplet size correlates strongly with Target Humidity from 45% to 85%. Below this range crystallization of the buffer typically occurs, and above this range fog forms within the instrument. Increasing Wait Time is typically unnecessary unless droplet size decreases between successive droplets, in which case Wait Time should be increased until the phenomenon stops. Reformulating the spotting buffer strongly affects the droplet size, but the relationship between buffer components and droplet size is complex, requiring optimization beyond the scope of this Unit.
- Once patterning is complete, bake the slide at 120 °C for 15 minutes then store in a vacuum desiccator until use. Patterned slides can be stored for at least one month before proceeding to downstream protocols.The interaction of the DNA's 5'-amine and the slide's aldehyde forms an imine, a covalent bond that is reversible in water. Baking the slide removes water that could destabilize this linkage.
BASIC PROTOCOL 2
Passivation of Patterned Slides
It is necessary to minimize non-specific cell attachment to the patterned slides before proceeding with DPAC experiments. This is a two-part process: first, any unreacted aldehydes must be quenched by reduction to alcohols (a process which simultaneously links the DNA to the surface via an irreversible amine bond rather than a reversible imine bond), and, second, the surface must be rendered non-adhesive by treatment with a perfluorosilane.
Passivating the slide ensures that when cells are exposed to the surface they attach only where complementary DNA is patterned. Mammalian cells are, to varying degrees, adhesive, which can severely interfere with DNA-directed cell patterning. Additionally, in Basic Protocol 5, the 3D transfer of gel-embedded cells is facilitated by minimizing the adhesion between bulk hydrogel and the slide. For these reasons, surface passivation is indispensible.
Passivated slides are stable under dry vacuum for at least one month before proceeding to subsequent protocols.
Materials
Patterned slides (Basic Protocol 1)
Sodium borohydride (Fisher, cat. #AC41947-1000)
Phosphate-buffered saline (e.g. Fisher, cat. #R58190001A)
Absolute ethanol (Fisher, cat. #BP2818-4)
0.1% SDS Solution (see recipe)
All-glass Coplin jar (Fisher, cat. #E94)
Dichloromethane (Fisher, cat. #AC40692-0040)
(Tridecafluoro-1,1,2,2-tetrahydrooctyl)dimethylchlorosilane (Gelest, cat. #SIT8170.0)
Triethylamine (Fisher, cat. #O4884-100)
Glacial acetic acid (Fisher, cat. #BP1185-500)
Perform reductive amination
-
1.
Prepare fresh 50 mL of 0.25% NaBH4 (w/v, 125 mg) in 25% ethanol in PBS (v/v, 12.5 mL ethanol, 37.5 mL PBS).
-
2.
Place up to four patterned slides (from Basic Protocol 1) in a 15-cm Petri dish. Immerse in NaBH4 solution and shake at 120 rpm for 15 minutes on an orbital shaker.
-
3.Aspirate NaBH4 solution and set aside for disposal.WARNING: NaBH4 evolves H2 gas. Perform this step in a fume hood. Do not tightly cap any containers of this solution, and do not store near an open flame. Do not dispose of fresh NaBH4 solutions down the drain. Either inactivate with dilute acid or wait at least 24 hours for the solution to spontaneously decompose.
-
4.
In the same dish, immerse the slides in 0.1% SDS solution. Agitate 10 times then dispose of the wash. Repeat once.
-
5.
In the same dish, immerse the slides in distilled water. Agitate 10 times then dispose of the wash. Repeat twice.
-
6.
Dry the slide under an air or nitrogen nozzle.
Perform silanization
-
7.In a glass Coplin jar, mix 60 mL dichloromethane, 600 µL tridecafluoro-1,1,2,2-tetrahydrooctyl)dimethylchlorosilane, and 600 µL triethylamine. Mix with a metal spatula.WARNING: Chlorosilanes and their vapors react readily with moisture to produce hydrochloric acid. Work in a fume hood and use appropriate personal protective equipment (PPE).Ideally, the perfluorosilane and triethylamine should be dispensed using a glass syringe. However, we regularly use 1 mL disposable plastic syringes, which are more convenient and readily available.Dichloromethane is the solvent of choice because chlorosilanes will react with protic solvents or the moisture in water-miscible solvents. The chlorosilane and triethylamine appear to form a complex that precipitates from other candidate solvents such as toluene. Triethylamine is added to greatly increase the rate of the silanization reaction (Kinkel and Unger, 1984).
-
8.Prepare a solution of 10% acetic acid in water in a 50 mL conical tube. Place the slide in the tube, cap, and invert 20 times. Remove the slide from the tube with tweezers and dry under an air or nitrogen nozzle.Washing with acetic acid immediately before silanization has been empirically determined to increase the rate of the reaction.
-
9.Place the slide in the Coplin jar containing the perfluorosilane mixture and shake at 120 rpm for 15 minutes on an orbital shaker.The silane solution can be used for up to several hours. Old silane solution develops a straw to brown color and must be discarded. Dispose of used reagents in a compatible chemical waste stream.
-
10.Prepare four 50 mL conical tubes. Fill the first and second with dichloromethane, the third with absolute ethanol, and the fourth with dH2O.Dichloromethane leaches plasticizers and dissolves plastics such as polycarbonate and polystyrene rapidly and is not suitable for long-term storage in polypropylene or polyethylene tubes.
-
11.
Remove the slide from the Coplin jar with tweezers.
-
12.Place the slide in the first conical tube, cap, and invert ten times. Remove the slide from the tube and place it in the next tube. Repeat, washing the slide in all four tubes.It is necessary to remove any traces of chlorosilane before washing with water, otherwise blemishing will occur. The ethanol wash is necessary due to the immiscibility of dichloromethane and water. Additionally, note that the same four wash tubes can be used for multiple slides on the same day.
-
13.
Dry the slide under an air or nitrogen nozzle. Store in a vacuum desiccator until ready to begin the next Protocol.
BASIC PROTOCOL 3
DNA Labeling of Mammalian Cells
The principle of DPAC is to attach cells to surfaces or to other cells by DNA hybridization. This requires that cells first be decorated with oligonucleotides. A variety of methods have been described to decorate cells with oligonucleotides including conjugation to sialic acid via the Staudinger ligation (Gartner and Bertozzi, 2009), conjugation to proteins via N-hydroxysuccinimide chemistry (Hsiao et al., 2009), hydrazone conjugation (Twite et al., 2012), or incorporation of dialkyl-DNA into the cell membrane (Selden et al., 2012). In the authors' experience, the easiest, most effective means to display DNA on cells is the use of fatty-acid-conjugated duplex DNA (Weber et al., 2014). This is a combination of an “Adhesion Strand” and a “Co-anchor Strand,” detailed in Support Protocol 1. Other methods of DNA functionalization may be desirable if a particular cell type proves recalcitrant to functionalization by fatty-acid DNA.
A variety of cell types are compatible with DPAC. We have successfully used epithelial cells, endothelial cells, fibroblasts, leukocytes, and neurons, including primary human cells (Todhunter et al., 2015) (Cerchiari et al., 2014) (Chen et al., 2016). DNA functionalization of cells and subsequent cell-surface attachment have been demonstrated in embryonic stem cells (Weber et al., 2014) and myoblasts (Hsiao et al., 2009). In our experience, the only cells that have been recalcitrant to DNA functionalization have been freshly dissociated primary epithelial cells. It may be possible to functionalize even these cells through optimization of tissue digestion protocols. For delicate cells such as neurons, it is essential to move through the procedures as quickly as possible and keep the cells ice-cold at all times.
Materials
Cells (1×106 per reaction)
Sterile calcium-and-magnesium-free PBS (PBS-CMF) (Fisher, cat. #BW17512F12)
5’-Fatty-acid DNA (Adhesion Strand) (see Support Protocol 1)
3’-Fatty-acid DNA (Co-anchor Strand) (see Support Protocol 1)
Flow buffer (see recipe)
Steps
- Prepare a monodisperse suspension of cells. For adherent cells, lift from culture vessels with PBS+EDTA, trypsin, or using other protocols, as appropriate. Inspect cells under microscope and triturate as appropriate to ensure monodispersity.Care must be taken when trypsinizing cells, particularly if using amine- or glycan-reactive oligonucleotides to functionalize cell surfaces. Excessive trypsinization removes cell surface amines and glycans and limits the extent of subsequent functionalization.
Prepare separate 50 µM solutions of both Adhesion Strand and Co-anchor Strand.
- Count the cells. Pipet 1×106 cells into an Eppendorf tube. Pellet (as appropriate for your cell type) and wash cells 3× with 1 mL PBS-CMF to remove media components, particularly BSA, that might interfere with fatty-acid DNA labeling. Resuspend into a final volume of 48 µL PBS-CMF.Care must be taken when resuspending cells. Some cell types are prone to lysis under vigorous resuspension. If necessary, trim the pipet tip with a razor blade to widen the nozzle.
Add 1 µL of the 50 µM Adhesion Strand solution to the cell suspension. Pipet up and down 5–10 times, pipetting half the volume of the solution (25 µL) to ensure the fatty-acid DNA is fully mixed.
Let the tube containing cells and Adhesion Strand sit on ice for 5–10 minutes.
After 5–10 minutes, add 1 µL of the 50 µM Co-anchor solution. Pipet up and down 5–10 times, pipetting half the volume of the solution (25 µL) to ensure the Co-anchor Strand is fully mixed.
Let the cell suspension now containing both Adhesion and Co-anchor Strands sit on ice for 5–10 minutes.
After 5 minutes, add 1 mL PBS-CMF to the Eppendorf tube and pellet the cells. Pellet and resuspend the cells three times in order to dilute out any residual fatty-acid DNA.
After the final pelleting step, resuspend the cells into 1 mL Flow Buffer and store on ice until use.
SUPPORT PROTOCOL 1
Synthesis of Fatty-acid DNA
Support Protocol 1 describes the synthesis of fatty-acid DNA for Basic Protocol 3 (Fig. 3). The following protocol requires some specialized equipment (see Materials), especially a DNA synthesizer. If a DNA synthesizer is unavailable, fully protected oligonucleotides with 5’ or 3’-amine modifications can be custom-ordered from DNA synthesis vendors and delivered unpurified and attached to controlled-pore glass (CPG) beads. When ordering and using beads in this manner, the Synthesize DNA steps 6–8 are skipped.
Figure 3. Synthetic scheme for fatty acid-DNA.
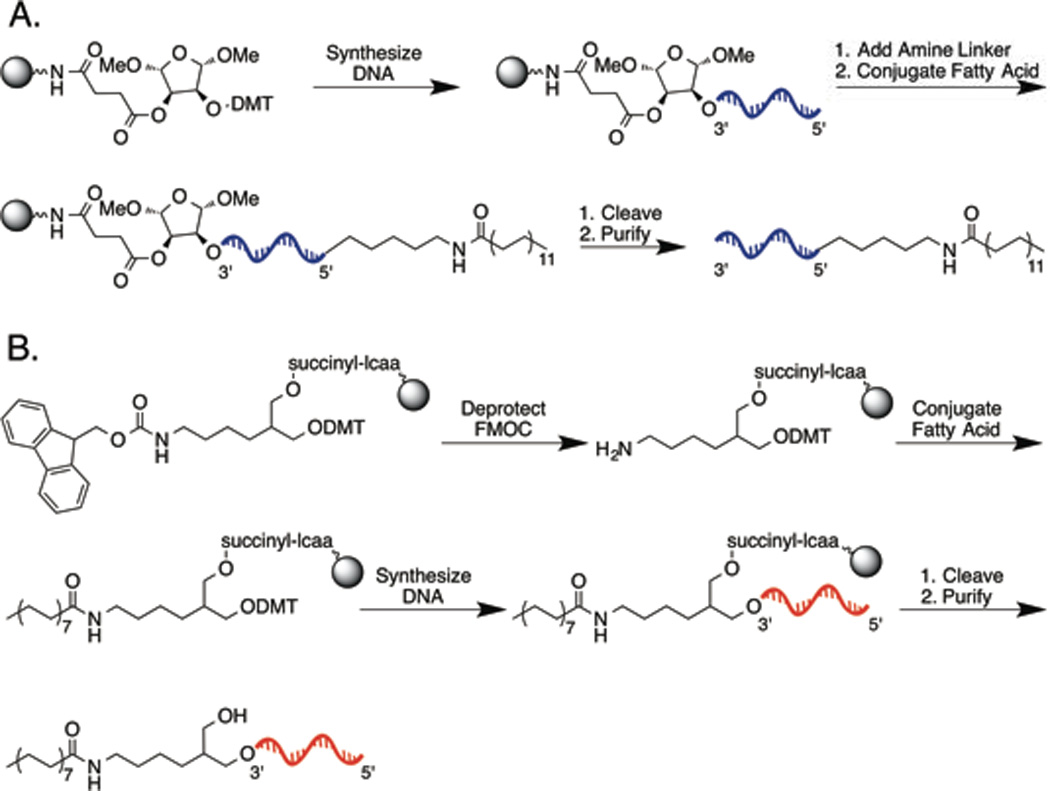
(A) Synthesis of 3’-lipid DNA. FMOC-protected CPG beads are deprotected. Fatty acid is conjugated to the beads via carbodiimide coupling. DNA is synthesized off the beads, and finally cleaved and purified. (B) Synthesis of 5’-lipid DNA. DNA is synthesized off CPG beads. An amino phosphoramidite is reacted to the 5’ terminus. Fatty acid is conjugated via carbodiimide coupling. Finally, the DNA is cleaved from the beads and purified.
Materials
FMOC-protected 3’ amino-modified CPG 1000 Å (Glen Research, cat. #20-2958-10)
20% piperidine in dimethylformamide (Sigma, cat. #80645-500ML)
Polyethylene wash bottles (Sigma, cat. #Z177024-6EA)
Dimethylformamide (DMF) (Sigma, cat. #227056-2L)
Dicholoromethane (DCM) (Sigma, cat. #270997-2L)
Vacuum concentrator (e.g., SpeedVac system)
Lignoceric acid (Sigma, cat. #L6641-1G)
N,N-Diisopropylethylamine (DIPEA) (Sigma, cat. #387649-100ML)
N,N’-Diisopropylcarbodiimide (DIC) (Sigma, cat. #D125407-25G)
Empty synthesis columns (Glen Research, cat. #20-0021-01)
Synthesis column frits (Glen Research, cat. # 20-0021-0F)
DNA synthesizer (e.g., Expedite 8909 or Biolytic 3900)
Ammonium hydroxide, 28% in water (Sigma, cat. #221228-1L-A)
Methylamine, 40% in water (Sigma, cat. #426466-1L)
1:1 triethylamine:acetic acid (TEAA) (Sigma, cat. #09748-100ML)
0.2 µm Ultrafree-MC Centrifugal Filter Units (Millipore, cat. #UFC30GV0S)
HPLC system (e.g., Agilent 1200 Series)
C8 Column (Hypersil Gold, Thermo Scientific, cat. #25205254630)
Lyophilizer (e.g., Labconco FreeZone)
Spectrophotometer (e.g., Thermo NanoDrop 2000)
Universal solid support DNA synthesis resin (Glen Research, cat. #20-5041-10)
5’-Amino-Modifier C6 (Glen Research, cat. #10-1906-90)
Palmitic acid (Sigma, cat. #P0500-10G)
3’-Lipid Co-anchor Synthesis
Deprotect FMOC
-
1.
Weigh out 1.25 µmoles of 3’-amino-modified CPG beads (mass varies with lot of beads) and transfer to an Eppendorf tube.
-
2.
Incubate CPG beads in 1 mL of 20% piperidine for 10 minutes. Pellet beads by brief table-top centrifugation and remove supernatant, disposing in a compatible chemical waste stream. Repeat this step two more times to fully deprotect FMOC.
-
3.
Rinse the beads 3× with 1 mL DMF and 3× with 1 mL DCM, using DCM as the final wash to aid in drying. Dispose the solvent in a compatible chemical waste stream. Dry the beads on a vacuum concentrator.
Conjugate Fatty Acid
-
4.
Mix 50 mg palmitic acid (200 mM) with 900 µL DCM in an Eppendorf tube. Add 66 µL DIPEA (400 mM) and gently agitate, dissolving the lignoceric acid and turning the solution pale yellow. Rapidly add 32 µL DIC (200 mM). Resuspend the dry, deprotected beads in 1 mL of this mixture in an Eppendorf tube. Secure the lid with a cap lock and agitate overnight at room temperature.
-
5.
Pellet the beads by brief centrifugation and remove the supernatant. Rinse the beads 3× with DCM, 3× with DMF and 1 final rinse with DCM. Dry the beads on a vacuum concentrator. Most of the bead sites are now modified with palmitic acid.
Synthesize DNA
-
6.
Prepare an empty DNA synthesis column compatible with your DNA synthesis instrument.
-
7.
Load the beads into an empty DNA synthesis column. We typically transfer the dried beads to piece of weigh paper folded in half and gently pour them into the synthesis column, or resuspend the beads in acetonitrile and transfer them via a 1 mL pipet with the tip cut off to make a snug seal with the synthesis vial. The transfer is imperfect but the goal is to get about 1 millimole of functionalized bead sites into the synthesis vial.
-
8.
Perform DNA synthesis on the beads which proceeds in the 3’-5’ direction. Be sure to remove the final DMT protecting group at the end of the synthesis.
Cleave Oligonucleotides From the Solid Support
-
9.Prepare a solution of AMA by carefully mixing ammonium hydroxide and 40% methylamine in a 1:1 ratio in a glass vessel with a magnetic stir bar. We typically prepare 500 mL at a time in a 1 L Pyrex bottle and store at −20 °C indefinitely.AMA is highly volatile and a severe irritant. Perform this step in a fume hood and take caution opening bottles that have warmed to room temperature.
-
10.
Remove the beads from the synthesis vial and transfer to an Eppendorf tube once again.
-
11.
Cleave the fatty acid DNA from the beads by adding approximately 1 mL of AMA per Eppendorf tube and incubating them for 1 hour at 65 °C with a cap lock. Note: without a caplock, the volatile AMA will build up pressure inside the sealed Eppendorf tube and explode. Not only is this dangerous but it also adversely affects final product yield.
-
12.Place the Eppendorf tube on ice long enough for the tube to come to room temperature. Remove the caplock, open carefully, and dry the beads on a vacuum concentrator.WARNING: Failure to let the tube come to room temperature may cause heated and pressurized AMA to boil violently and burst from the tube when opened.
-
13.
Resuspend and thoroughly mix the dried beads and cleaved DNA in 750 µL of 0.1 M TEAA.
Purify Fatty-acid DNA from Unmodified DNA by HPLC
-
14.
Transfer the beads and TEAA to a 0.2 µm Ultrafree-MC Centrifugal Filter Unit and centrifuge at maximum speed for 2 minutes to ensure any CPG beads and insoluble debris are removed from the solution.
-
15.
Use the filtered solution for HPLC purification. The point of this purification is to remove oligonucleotides that did not get modified with the fatty acid in step 5. For the mobile phase we use 0.1 M triethylamine acetate (TEAA, pH 7) as solvent A and acetonitrile as solvent B. A C8 column is used for the stationary phase. More hydrophobic columns (e.g. C18) will retain the highly hydrophobic lipid-modified DNA when using this solvent system and are not suitable for this step. We have purified at both analytic and semi-preparative scales with similar success. At the analytical scale, we use a gradient elution system (Table 2) at a 1.0 mL/min flow rate. Detect at 230 nm and 260 nm (DNA absorption maximum). The elution time varies with the hydrophobicity of the fatty acid as well as the length of the oligonucleotide, but the typical fatty acid oligonucleotide elutes around 13 minutes in our system. In contrast, DNA without a fatty acid modification elutes around 7 minutes.
-
16.
Collect the fatty acid modified fraction and lyophilize it at least 3 times to remove the volatile TEAA, resuspending in several milliliters of MilliQ water each time. After the first lyophilization, fatty acid DNA appears as a viscous gel due to the relatively high concentration of TEAA. After subsequent lyophilizations, fatty acid DNA begins to appear as a white powder.
-
17.
After lyophilization, resuspend the fatty acid DNA in MilliQ water to a concentration of 250 µM. Measure 260 nm absorption on a Nanodrop to calculate the appropriate resuspension volume. The fatty-acid DNA can be stored up to three years at −20 °C. It is recommended to make 50 µM aliquots for cell labeling and to avoid freeze-thaw cycles.
Table 2.
HPLC table.
| Step | Time (min) | % A (TEAA/H2O) | % B (ACN) |
|---|---|---|---|
| 1 | 0 | 92 | 8 |
| 2 | 4 | 80 | 20 |
| 3 | 17 | 5 | 95 |
| 4 | 27 | 5 | 95 |
| 5 | 27.5 | 92 | 8 |
| 6 | 32 | 92 | 8 |
5’-Lipid Adhesion Strand Synthesis
-
18.
Perform DNA synthesis on a 1 millimole Universal support DNA synthesis column.
-
19.For the final base, add the 5’-Amino-Modifier C6. Be sure to remove the MMT protecting group after synthesis is complete.The MMT protecting group is more difficult to remove than the DMT protecting group. It may be advisable to flow additional deblocking solution over the DNA column until you can confirm that no additional yellow color, indicative of MMT, is eluting from the column.
-
20.
To conjugate fatty acid to the 5’-amine, follow the protocol in Conjugate Fatty Acid, then Cleave Oligonucleotides from the Solid Support, then Purify Fatty-acid DNA from Unmodified DNA by HPLC. Substitute lignoceric acid for palmitic acid during the fatty acid conjugation.
BASIC PROTOCOL 4
DNA-programmed Assembly of Cells on Patterned Glass
In a DPAC experiment, a single patterned slide can template a variety of tissues based on the Scheme specified in Basic Protocol 4. Changing which cell types are labeled with which DNA strands, and the order in which these cells are flowed over the surface, changes the resultant tissue structure.
For the entirety of Basic Protocol 4, cells are held in suspension. To maximize viability, it is essential that this Protocol be performed as rapidly and as close to 0 °C as possible. Mammalian cells have varying susceptibility to being held in suspension, and this must be empirically determined on a cell-line-to-cell-line basis.
Proceeding to Basic Protocol 5 permits embedding the DPAC-patterned cells into hydrogel for 3D culture experiments. Terminating at Basic Protocol 4 permits performing experiments in-solution for as long as the DNA hybridization is maintained (typically 12–24 hours) (Selden et al., 2012). If the cells can anchor to the surface independently of DNA, then it may be possible to culture them indefinitely in this manner.
Materials
Patterned slides (Basic Protocol 1)
PDMS flow cells (see Support Protocol 5)
Scotch tape (3M, "Magic" Tape 810)
microcrystalline wax (Douglas & Sturgess, cat. #SC-1159)
Mounted toggle clamp (see Support Protocol 6)
Priming buffer (see recipe)
Flow buffer (see recipe)
Steps
Gather a number of PDMS flow cells (Fig. 4A) (see Support Protocol 5) equal to the number of patterns on the slide (up to four). Using your fingers, press a piece of Scotch tape against both the top and bottom faces of the flow cell to clean it of any dust.
- Place each flow cell channel-side-up. Take a chunk of microcrystalline wax and, using it like a crayon, draw a line of wax along the underside of each side wall (Fig. 4B).The purpose of the wax is to destabilize the interface between the PDMS and the glass slide. While typical microfluidics applications require a tightly and permanently sealed interface, DPAC is different in that the flow cells must be precisely disassembled in Basic Protocol 5. This unique requirement is facilitated by the wax.As an alternative to wax, it may be feasible to strategically abrade the flow cell master (see Support Protocol 5), using coarse sandpaper or a riffler, such that the sidewalls of the flow cells are no longer smooth.
- Place the flow cells on the slide, taking care to center them over the patterns. The diamond-scribe-etched markings from Basic Protocol 1 facilitate proper placement. Apply gentle pressure to each flow cell to secure it in place.If the slide is on a hard surface (like a benchtop), pressing into the PDMS can fracture the slide. Use a soft surface, such as a self-healing mat, for this step.
Use microcrystalline wax to draw barriers between the inlets and outlets of adjacent flow cells (Fig. 4C). This prevents cross-contamination between adjacent flow cells and prevents any accumulated liquid at the inlet or outlet from creeping along the sides of the flow cells.
- Place the slide on the base of the mounted toggle clamp. Place the clamp’s acrylic spacer on the slide. Clamp down on the spacer with gentle pressure (Fig. 4D). The slide should be sufficiently clamped to immobilize it but not so strongly clamped that the flow cells are compressed.Because the flow cells are designed to have weakly sealed edges (see Step 2), physical pressure, delivered by a clamp, is used to prevent the flow cells from leaking. When flow cell disassembly occurs in Basic Protocol 5, the clamp is released.
- Prime the flow cell with Priming Buffer. Pipet 100–200 µL and slowly introduce it into the flow cell, tilting the flow cell vertically such that the flow is assisted by gravity. Try not to introduce air bubbles into the flow cell. Repeat for three exchanges of Priming Buffer.Inspect the flow cells for air bubbles after priming. If air bubbles are present, release the clamp, remove the flow cells from the slide, dry the slide under an air or nitrogen nozzle, and return to step 5. Minimize the probability of air bubble formation by ensuring the PDMS flow cell is dry before priming. Do not inadvertently pipet air into the flow cell.The Priming Buffer has two purposes: first, to prime the flow cell and second, to "activate" the DNA on the surface. We have observed that oligonucleotides on very hydrophobic slides (>90° sessile drop contact angle with water) are not amenable to hybridization unless first treated with certain solutions, such as 100 mM acetic acid. Although we do not understand the mechanism by which this process improves DNA hybridization, we hypothesize that the hydrophobic surface may contribute to a single-stranded DNA conformation unsuitable for hybridization (Monserud and Schwartz, 2012), and the acid may protonate the nitrogenous bases, solvating them and rendering them more soluble.
- Equilibrate each flow cell with 300 µL ice-cold Flow Buffer. Add a small volume (10–30 µL) of Flow Buffer to the inlet and outlet to prevent evaporation from the inside of the flow cell and store, covered, at 4 °C, until ready to attach cells. Flow cells can be stored in this manner for up to one day as long as evaporation is prevented.From this point, the number of steps will vary. You should have already determined a synthetic scheme for the tissue, one that takes into account what DNA is on the template, what DNA is on the cells, and what tissue structure is desired.For each step in the synthetic scheme:
- Working in a laminar flow hood, introduce 20 µL of high-concentration, DNA-labeled, ice-cold cells from Basic Protocol 3 into each flow cell. Let the cells settle for 3 minutes at 4 °C.For most cell types, 2×107 cells/mL, i.e. one million cells resuspended into 50 µL, is a sufficiently high cell concentration. Higher concentrations are better for smaller cells - inspecting the flow cell under a microscope, you ideally see a confluent layer of suspended cells. If it is not feasible to reach this density, several options are available. First, cells can be resuspended into a smaller volume. To compensate for this reduced volume, either fewer flow cells can be used in an experiment, flow cells can be cut to a shorter length (and lower internal volume), or a single bolus of cells can be introduced to the flow cells sequentially. Second, in Step 9, additional iterations can be performed - up to thirty instead of ten. Third, the cells recovered in Step 10 can be centrifuged, resuspended in a small volume, and reused to attach a "second coat" of cells.
- Add 5 µL cells to the inlet and "cycle" the flow cell by pipetting 3 µL up at the outlet and back down at the inlet, moving the cells through the flow cell without dilution. Cycle ten times.If your flow cells contain three-or-more-step assemblies, or if your pattern contains closely spaced (<10 µm separation) cells, alternate the direction of flow every three cyclings by pipetting up at the inlet and back down at the outlet. This will mitigate flow shadow effects.Some cell types do not require cycling for high attachment efficiency, such as cells highly incorporating surface-DNA (i.e., Support Protocol 2 shows a median fluorescence increase >100) or specific cell types such as leukocytes. For such cells, Step 9 can be skipped.
Carefully and slowly introduce 100 µL ice-cold Flow Buffer into each flow cell. Repeat 2–4 times until flow cells are mostly devoid of cells. Recover the cell-dense eluate if you wish to use these cells later on in the synthetic scheme.
- Wash with 100–500 µL ice-cold Flow Buffer to remove any remaining cells aberrantly stuck to the surface.The flow rate of the wash strongly affects the stringency of the wash. Since flow is driven by pipet, it is not feasible to maintain perfectly smooth flow, but an approximate flow rate of 5–10 µL/second is appropriate for washes. If you experience difficulty washing the flow cell, tilting the slide or more vigorously pipetting can increase the flow rate.If your flow cells contain three-or-more-step assemblies, or if your pattern contains closely spaced (<10 µm separation) cells, alternate the direction of flow every two washes by pipetting at the outlet and collecting at the inlet. This will mitigate flow shadow effects.
- For each additional step in the synthetic scheme, return to step 8 using additional, appropriately labeled cells. Otherwise, the Protocol is complete.At any point in this protocol, the cells within the flow cell can be imaged with an inverted microscope with phase contrast illumination. This is useful to verify the extent of attachment of cells to the DNA pattern as well as verify that there is no non-specific background attachment. Image quickly to minimize temperature changes. Imaging can be performed while the flow cells remain in the mounted toggle clamp as long as the objective's working distance exceeds the thickness of the 3/16" acrylic baseplate and as long as the condenser does not hit the clamp handle. In our experience, a variety of standard microscopes such as the Zeiss Axio Vert.A1 and the Nikon Eclipse TE2000 are compatible using 4×, 10×, and some 20× objectives.
Figure 4. Preparation of PDMS flow cells.

(A) PDMS flow cells are cut out individually from a mold, shown here. (B) Prior to placement on the slide, flow cells are flipped upside-down and microcrystalline wax is applied to the underside of the side walls. (C) A 25×75 mm slide (top) can fit four flow cells (middle), each flow cell positioned over a pattern of DNA spots. Wax barriers are drawn between flow cells (bottom), preventing cross-contamination. (D) The slide and flow cells are secured within a mounted toggle clamp.
SUPPORT PROTOCOL 2
Quantification of Cell-DNA Functionalization
In Basic Protocol 4, when handling a new cell type or troubleshooting a failed experiment, it can be useful to measure how much DNA has been incorporated to the cell surfaces. This protocol provides a straightforward approach using flow cytometry.
This protocol compares the median fluorescence increase between cells of interest that have been functionalized with DNA and, as a negative control, otherwise identical cells that have not been functionalized. By comparing fluorescence to a standard reference curve, the number of DNA strands on the cells can be quantified. Results are best compared to a positive control, defined as a DNA-functionalized cell type that has reliably been demonstrated to work well with DPAC, such as Jurkats (e.g., ATCC TIB-152) or MCF-10As (e.g., ATCC CRL-10317).
Additional Materials
5'-FITC-DNA with sequence complementary to that on the cells (Eurofins MWG Operon or similar vendor) (resuspend in dH2O for a 300 µM stock and store at −20 °C in a foil-wrapped tube)
LIVE/DEAD Fixable Far Red Dead Cell Stain Kit (Thermo Fisher, cat. #L10120)
Fluorescent microsphere standards, Quantum™ FITC-5 MESF (Bangs Laboratories, cat. #555)
Steps
Using Basic Protocol 3 or a related method, functionalize cells of interest with DNA. Set aside a portion of unfunctionalized cells as a negative control.
Portion 1×106 cells into an Eppendorf tube. Wash by centrifugation three times, resuspending into 1 mL ice-cold PBS for each wash.
- Resuspend the cells into 50 µL ice-cold PBS and add FITC-DNA (with sequence complementary to that on the cells) to a final concentration of 150 nM.FITC-DNA is light sensitive and will photobleach if exposed to benchtop light. Keep this reagent in the dark.
Incubate the cells on ice, in the dark, for 30 minutes.
Wash the cells by centrifugation three times, resuspending into 1 mL ice-cold PBS for each wash.
Resuspend the cells into 1 mL ice-cold PBS plus 1 µL dissolved Fixable Far Red dye (as per the Thermo Fisher product manual) in a FACS tube and run cells on flow cytometer. Collect at least 30,000 events per sample, collecting data on both 488 nm and 635 nm channels.
- Create a standard curve of FITC fluorescence by running the fluorescent microsphere standards on the flow cytometer. Follow the instructions available online from Bangs Laboratories Product Data Sheet 821.When comparing the labeling of a cell population from day-to-day, Step 7 and the standard curve can be omitted for a faster but non-quantitative method. The median fluorescence increase is still calculated between the experimental and control conditions and gives an approximation of relative labeling efficiency.
Analyze flow cytometry data and compute the median fluorescence increase of 488 nm signal against the negative control, gating out dead cells stained on the 635 nm channel.
SUPPORT PROTOCOL 3
Measurement of Surface-DNA Functionalization
In Basic Protocol 4, when troubleshooting a failed experiment, it can be useful to measure how much DNA on the surface is competent for hybridization. This protocol provides a straightforward approach using microscopy.
This protocol compares the fluorescence intensity of patterned and unpatterned regions of a slide. As with any microscopy-based intensity measurement, care should be taken to ensure identical exposure time, source power, and camera between observations. Care should be taken to avoid bleaching the fluorophore. Results are most meaningful when compared to a positive control, such as a DNA-patterned surface that has reliably been demonstrated to work well with DPAC. Although there is no applicable commercial standard, a standard can be created, the details outlined below.
Additional Materials
Nexterion AL slide (Applied Microarrays, cat. #1064874)
2 mM 20mer 5'-amine-DNA in dH2O (e.g., available from Eurofins MWG Operon)
20× saline sodium citrate (Sigma, cat. #S6639-1L)
5'-Alexa Fluor 488-DNA with sequence complementary to that on the slide (Eurofins MWG Operon or similar vendor) (resuspend in dH2O for a 300 µM stock and store at −20 °C)
18 mm coverslips (Fisher Scientific, cat. #12-542A)
Steps
Prepare a Standard
If a standard has previously been prepared, these steps can be skipped.
-
1.
Bring 4 µL of amine-DNA to a concentration of 1 mM in 3× saline sodium citrate.
-
2.
Prepare tenfold serial dilutions of this solution from 1 mM to 1 µM in 3× saline sodium citrate.
-
3.
On a fresh Nexterion aldehyde slide, spot 0.5 µL droplets as in Fig. 5.
-
4.
Fold a damp paper towel and place it in a Petri dish. Place the slide on top of the paper towel. Seal the dish with parafilm. Incubate for 30 minutes at room temperature.
-
5.
Place the slide in a clean Petri dish and perform reductive amination as per steps 1–6 in Basic Protocol 2. Store the standard slide in a vacuum desiccator unless in use.
Figure 5. Standard slide for surface-DNA quantification.

0.5 µL droplets of DNA solution are pipetted according to this pattern of dotted circles. Each of four DNA concentrations is represented in duplicate across a thousand-fold concentration range. The droplets are spaced such that all eight droplets can fit under an 18×18 mm coverslip. At right, image showing fluorescent complementary DNA and an appropriate spot region (red) and background region (cyan) for fluorescence quantitation.
Probe with Fluorescent Complementary Strand
-
6.
Using Basic Protocol 1 or a related method, functionalize a glass slide with DNA.
-
7.
Place the slide in a Petri dish and wash by immersion in distilled water, adding sufficient distilled water to fully immerse the slide. Dry the slide under an air or nitrogen nozzle.
-
8.
Wet the surface with a 50 µL droplet of PBS containing 150 nM FITC-DNA with sequence complementary to that on the slide.
-
9.
Incubate the surface in the refrigerator for 30 minutes.
-
10.Place the slide in a Petri dish and wash by immersion in PBS, adding sufficient PBS to fully immerse the slide. Dry the slide under an air or nitrogen nozzle.It is important to use PBS instead of dH2O because dH2O decreases the melting temperature of DNA, stripping the fluorescent probe off the surface.
-
11.
Add 20 µL PBS to the slide and cover with an 18×18 mm coverslip.
-
12.
Use a microscope to image the slide with green fluorescence settings.
-
13.
Compute the difference in average fluorescence intensity between a DNA spot and the area around a DNA spot, choosing regions as in Fig. 5.
SUPPORT PROTOCOL 4
Regeneration of DNA-patterned Surfaces
Support Protocol 4 describes a means to purge a flow cell of all cells and cell debris, regenerating the patterned surface for a fresh tissue assembly via Basic Protocol 4. This might be desirable if something has gone wrong - for instance, if the wrong cells were introduced at the wrong time, or if cell viability seems low and the experiment may be more likely to succeed if repeated another day. This protocol provides a means to restart Basic Protocol 4.
Additional Materials
0.05% Trypsin
0.1% SDS in dH2O
Steps
Flow 100 µL 0.05% trypsin through each flow cell. Incubate at room temperature 5 minutes.
Flow 100 µL 0.1% SDS through each flow cell.
Flow 100 µL dH2O through each flow cell.
Flow 100 µL 0.05% trypsin through each flow cell. Incubate at room temperature 5 minutes.
Flow 100 µL dH2O through each flow cell.
If restarting Basic Protocol 4 immediately, skip this step. Otherwise, blow air or nitrogen through each flow cell and store on the benchtop until use.
Proceed with Basic Protocol 4 from Step 6.
SUPPORT PROTOCOL 5
Preparation of PDMS Flow Cells
Basic Protocol 4 requires the use of flow cells, and this is a simple strategy for fabricating flow cells that is accessible to labs without ready access to a cleanroom or lithography equipment. Flow cells of higher precision than those described below can be fabricated using standard lithographic approaches.
Additional Materials
Diamond scribe (Fisher, cat. #17-467-634)
18×18 mm No. 1 coverslips (Fisher, cat. #12-542A)
Permanent double-sided Scotch tape (3M, Tape 665)
Sylgard 184 (Fisher, cat. #NC9644388)
Steps
Using a diamond scribe, cut a coverslip in half twice, lengthwise, yielding a 4.5×18mm fragment.
Press a strip of double-sided tape into the coverslip fragment. Use a razor blade to trim away the excess.
Press the tape and coverslip fragment, tape-side-down, into a 15-cm Petri dish.
Return to step 1, repeating until the 15-cm dish is full. Leave gaps of at least 1 cm between the long edges of the fragments, and leave gaps of at least 3 mm between the short edges of the fragments.
- Weigh out 22 grams of Sylgard 184 (20 grams base, 2 grams curing agent) into a weigh boat and stir, such as with a disposable transfer pipet, for at least one minute and until thoroughly mixed. Pour into the 15-cm dish and wait at least 60 minutes for the Sylgard to degas and level itself.Sylgard 184, especially the base component, is viscous and sticky. Expect to lose ~10% volume on the surface of the weigh boat. Uncured Sylgard 184 can be cleaned with isopropanol.
- Cook the 15-cm dish overnight at 60–70 °C to cure the Sylgard into PDMS.WARNING: Standard polypropylene 15-cm dishes will start to deform above 70 °C or melt at even higher temperatures. Be certain your oven is not too hot.
Peel the PDMS out of the dish (use a razor blade to cut away a small portion of the edge so that you can get underneath it for leverage).
Using a razor blade, cut out individual PDMS flow cells as needed (Fig. 4A). Ensure the flow cells have sidewalls 2–4 mm wide by cutting an appropriate distance away from the flow channel. Flow cells are stable indefinitely on the benchtop.
SUPPORT PROTOCOL 6
Construction of mounted toggle clamp
A mounted toggle clamp is used to secure the PDMS flow cells onto the patterned substrate in Basic Protocol 4. This protocol provides instructions for construction of this device. The acrylic sheets are prone to fractures stemming from the screw holes, and the user should expect to replace these clamps at least once a year, if used frequently. This protocol also describes the construction of an acrylic lattice spacer to accompany the mounted toggle clamp.
Additional Materials
3/16" acrylic sheet (McMaster-Carr, cat. #8560K163)
Laser cutter (e.g., Universal Laser Systems, cat. #VLS 3.50)
Hold-down toggle clamp (McMaster-Carr, cat. #5126A45)
3/8” 8–32 machine screws (McMaster Carr, cat. #91735A192)
36-grit sandpaper pad (McMaster-Carr, cat. #8221A11)
Sigmacote (Sigma, cat. #SL2-25ML)
Steps
Using a laser cutter and Fig. 6, cut a base plate, spacer plate, and lattice from the acrylic sheet.
Stack the toggle clamp on top of the spacer plate, and stack the spacer plate on top of the base plate, aligning the four holes in the toggle clamp to the four holes in the spacer plate to the four holes in the base plate. Use a screwdriver to drive a screw into each of the four holes. The screw threads should dig into the acrylic, embedding the screws.
Use the sandpaper to thoroughly abrade one face of the acrylic lattice. Rub the sandpaper in varied directions to create a non-uniform, rough texture on the acrylic.
- Pipet 200 µL of Sigmacote into a 15 mL conical tube. Position the lattice within the tube such that it is above, but not touching, the Sigmacote. Heat the bottom of this tube to 65 °C for 15 minutes, using a hot plate or water bath.The Sigmacote is deposited on the acrylic by chemical vapor deposition. For this process to work best, the liquid Sigmacote needs to be heated to a higher temperature than the acrylic itself. Under these conditions, the liquid Sigmacote vaporizes then condenses onto the acrylic.
Remove the lattice from the tube, disposing the Sigmacote in a compatible waste stream. Wash the lattice twice with ethanol. Air dry. The lattice is now ready to use.
Figure 6. Vector file for mounted toggle clamp components.

The online version of this figure is an EPS vector file suitable for laser cutting on a Universal Laser Systems VLS laser cutter or similar device. Adjust power settings such that the red lines cut cleanly through 3/16" acrylic without excessive melting. The power is set correctly if all the features of the acrylic lattice spacer can be resolved.
BASIC PROTOCOL 5
3D Transfer into Matrigel
This Protocol transfers the DPAC-assembled tissues into hydrogel to create fully embedded or "on-top" cultures. The authors have tested a variety of hydrogels using this Protocol, including Matrigel, collagen, agarose, fibrin, polyethylene glycol gels, and mixtures thereof. In principle, any sufficiently durable matrix that can exist in both liquid and gelled forms without killing cells is compatible with this Protocol. This Protocol assumes the use of Matrigel. The ensuing Alternate Protocols detail the use of other hydrogels. If altering the Protocol, it is essential to follow this doctrine: the hydrogel must be more adherent to the final vessel (here, a chambered slide) than to the flow cell, and the hydrogel must be more adherent to the flow cell than to the template slide. Complete 3D transfer is otherwise challenging or inefficient.
After completion of this protocol, cell patterns prepared by DPAC can be grown under tissue culture conditions for as long as feasible. However, care should be taken to maintain tissues within 200 µm of the media overlay to ensure suitable oxygen and media transfer. The depth of the tissue in the gel, and the distance of the tissue from the underlying rigid substrate will affect their overall behavior. Depending on factors such as the cells used and the media conditions, cells retain varying viability and conform to the initial pattern to varying degrees. Certain gels, especially cell-laden collagen-rich gels, tend to contract and curl off the culture vessel, becoming free-floating after days to weeks, which typically prevents further microscopy. Gels are amenable to endpoint analysis by fixation and immunofluorescence, including CLARITY processing (Chung and Deisseroth, 2013).
From this point on, it is recommended to perform all manipulations with the most sterile technique feasible, preferably inside a cell culture hood.
Materials
Toggle clamp assembly with flow cells (from Basic Protocol 4)
Cell culture media
Matrigel (Corning, cat. #356231)
Turbo DNase (Life Technologies, cat. #AM2238)
an aluminum tube rack (or other sterilizable, chillable, high heat capacity object) (e.g., Labconco, cat. #4026402)
Autoclaved razor blades
Self-closing curved-tip tweezers (Fisher cat. #50-242-89)
Lab-Tek II Chambered Coverglass, 2-well (Fisher cat. #12565336)
Steps
Invert the aluminum tube rack and place it on ice. Give it enough time to reach 4 °C or lower.
- Prepare a solution of ~9 mg/mL Matrigel supplemented with 1% v/v Turbo DNase. Pipet gently to mix, taking care to not introduce air bubbles. Prepare 160 µL for each flow cell.Aliquots of Matrigel should be stored in the freezer and thawed on ice prior to use. Keep Matrigel on ice at all times to prevent premature gelation. If feasible, use chilled pipet tips. Matrigel is subject to lot-to-lot variability (Debnath et al., 2003), and it is advisable to screen lots for relevant tissue behaviors prior to experiments.
- Swab the chilled aluminum tube rack with 70% ethanol. Place the toggle clamp assembly on the rack. Flow 80 µL Matrigel+DNase into each flow cell.Matrigel is viscous, and this flow-through will be slow. Tilt the slide, even as much as 90°, to assist flow-through. Be aware that tissue features within two to three millimeters of the inlet and outlet are especially prone to damage from viscous flow.
- With a light touch, unclamp the slide, being careful to not lift the flow cells from the slide or introduce air bubbles into the gels. Remove the acrylic lattice spacer.Be especially careful if moisture has accumulated between the flow cells and the spacer. Any adhesion to the roof of the flow cells can cause premature detachment of the flow cells from the slide, damaging 3D transfer. Furthermore, be careful not to jostle the slide once the flow cells have been released from the clamp - the flow cells are only loosely attached to the slide and may inadvertently slip.
Let slide incubate in a 37 °C cell culture incubator for 15 minutes to set the Matrigel into a gel.
- Remove the slide from the incubator. For each flow cell, pipet 6 µL culture media around the edges of the flow cell. Using a razor blade, press against the edge of the flow cell and slide the flow cell towards the edge of the slide. Carefully invert the slide and use the razor blade to push the flow cell off the edge, transferring the flow cell onto the face of the blade. Get a two-well chambered slide and pipet 30 µL cold Matrigel into a chamber. Pluck the flow cell from the face of the razor blade and deposit the flow cell onto the cold Matrigel. Let the dilute Matrigel find its level, on ice, for at least one minute. Repeat this step for each flow cell.See Fig. 7 for a photographic breakdown of the manipulations in this Step.The principle in Step 6 is to float the flow cell on a thin cushion of culture media. Adding too little culture media can create friction between the hydrogel and the slide, causing a variety of 3D transfer defects. Adding too much culture media can allow the hydrogel to detach from the flow cell and float freely, rendering it impossible to manipulate further. The flow cell acts as a rigid backing for the hydrogel, preventing it from folding or curling on itself. The flow cell also acts as a handle for the hydrogel, allowing its manipulation with razor blade and tweezers.
Let chambered slides incubate in 37 °C cell culture incubator for 15 minutes to allow the Matrigel to set.
- Remove the chambered slides from the incubator and add 2 mL prewarmed tissue culture media to each chamber, enough to submerge the flow cells. It is best to slowly drip the media directly on top of the PDMS to thoroughly wet its surface. Otherwise, the hydrophobic nature of the PDMS may prevent its complete immersion. Once the PDMS is immersed, use sharp curved tweezers to slide the PDMS up and off of the gel, exposing it to the media. Remove the PDMS and discard. The patterned 3D organoids are now ready for any downstream growth, observation, or manipulation.Typically, the most challenging part of this step is not sliding the PDMS off the gel but rather removing the PDMS from the chambered slide. One approach is to push the PDMS to the edge of the chambered slide then use the tweezers to tilt the PDMS 90° upward, such that it is standing on-end and sticking out of the media. At this point it becomes easy to grasp the PDMS with the tweezers and remove it. Using this or any other approach, your most important consideration should be to not damage the gel. Avoid fumbling the PDMS back onto the gel and avoid stabbing the gel with the tweezers.Step 8 is necessary to provide media access to the organoids. Even though before Step 8 the organoids are already fully embedded in Matrigel, the PDMS flow cells permit only slight media diffusion and must be removed. Without means of perfusing the 3D gel, cell death typically occurs within a day.
Figure 7. 3D transfer manipulations.

(A) 6 µL of media, here dyed blue, is dispersed around the corners of a flow cell. (B) A razor blade is inserted under the side walls of the flow cell, allowing the media to infiltrate between the PDMS and the glass slide. (C) The razor blade is brought perpendicular to the face of the slide and pushes the flow cell towards the edge of the slide. (D) The slide is inverted, and the razor blade is brought in at an angle such that, as the razor blade pushes the flow cell off the slide, the flow cell falls onto the razor blade. (E) The underlay gel, which is 30 µL of cold Matrigel, is deposited in the center of a two-well chambered coverglass. (F) Self-closing tweezers are used to grip the flow cell on the razor blade. Note the position of the razor blade's hole relative to the flow cell. (G) The flow cell is brought into position above the underlay gel with the tweezers. The flow cell and tweezers are angled such that opening the tweezers drops the flow cell onto the underlay. (H) The flow cell is deposited onto the underlay, spreading out the gel droplet.
ALTERNATE PROTOCOL 1
3D Transfer into Agarose
Although agarose is not a matrix naturally found in mammals, it is useful for some studies due to its non-adhesiveness and its ability to suspend mammalian cells (Whipple et al., 2007). Ordinary agarose has a melting temperature too high to be useful for DPAC, but low-melting-point agarose works well.
Additional Materials
Agarose Type IX-A (Sigma, cat. #A2576)
an aluminum tube rack (or other sterilizable, warmable, high heat capacity object) (e.g., Labconco, cat. #4026402)
Steps
Prepare 5 mL of 2% Type IX-Agarose in PBS in a 50 mL flask. Autoclave to sterilize and dissolve the agarose. Allow the solution to cool to 37 °C in a warm room or water bath. Pipet 200 µL for each flow cell into an Eppendorf tube. Add 1% v/v Turbo DNase. Pipet gently to mix, taking care to not introduce air bubbles.
Place the slide on a 37 °C surface such as a prewarmed aluminum block. Flow 100 µL agarose+DNase into each flow cell.
Step 3 is unchanged from Basic Protocol 5.
Let slide incubate on ice for 5 minutes to gel agarose.
Use 37 °C agarose solution instead of ice-cold Matrigel solution. Otherwise, Step 5 is unchanged from Basic Protocol 5.
Let slide incubate on ice for 5 minutes to gel agarose.
Step 7 is unchanged from Basic Protocol 5.
ALTERNATE PROTOCOL 2
3D Transfer into Collagen/Matrigel Mixtures
Collagen is the most abundant protein in mammals and an important component of the extracellular matrix. Combining collagen and Matrigel provides a matrix supportive of both fibroblasts and epithelial cells. This Protocol provides the formulae necessary to produce matrices with a variety of Matrigel and collagen I concentrations. Gel toughness tends to be unworkably low below 5.0 mg/mL Matrigel, and gel viscosity tends to be unworkably high above 4.0 mg/mL collagen I. We recommend 6.0 mg/mL Matrigel and 2.0 mg/mL collagen I as a starting point for culturing both fibroblasts and epithelial cells.
Note that the properties of collagen vary on the preparation method and the source animal (Dhimolea et al., 2012). Consider comparing different types of collagen, such as rat tail collagen I versus bovine collagen I.
Additional Materials
High-concentration rat-tail collagen I (Corning, cat. #354249)
Sterile 20× PBS (VWR, cat. #101076-202)
Sodium hydroxide (Sigma, cat. #S5881-500G)
pH paper (Fisher, cat. #13-640-502)
Vacuum desiccator (e.g., 150 mm Nalgene desiccator, Thermo, cat. #24987-106)
Steps
Prepare 10 mL of 3 M sodium hydroxide. Sterile filter and store indefinitely at room temperature.
Based on your stock concentrations and desired concentrations of Matrigel and collagen I, calculate the necessary volumes of the various matrix components (Fig. 8).
- Working on ice, create a premix by combining the requisite volumes of 20× PBS, 3 M NaOH, sterile water, and collagen in an Eppendorf tube. Mix by gently stirring and pipetting up and down, taking care not to introduce air bubbles.Consistent timing from Step 3 onward is essential between experiments. Although collagen gelation only occurs at elevated temperature, once neutralized, collagen exhibits certain structural changes over time even on ice, e.g. at 2.0 mg/mL it will set into a stiffer and translucent gel if left on ice for several hours prior to gelation and will otherwise set into a transparent gel if gelled immediately. These structural changes affect the outcome of downstream experiments.
- Use pH paper to verify the premix is neutralized. If not, add additional 3 M NaOH one microliter at a time until neutralization is achieved.Do not dip the pH paper into premix - aside from wasting premix, this can introduce chemicals from the pH paper. Spotting 1µL of premix onto each colored portion of the paper is enough to determine the pH.
- Working on ice, in a new Eppendorf tube, combine the requisite volumes of Matrigel, Turbo DNase, and all but 100 µL of the premix from Step 3. Mix by gently stirring and pipetting up and down, taking care not to introduce air bubbles.100 µL of the premix is left unused because (1) air bubbles tend to form in this solution and (2) collagen tends to be pipetted inaccurately, presenting the possibility of pipetting air if trying to remove every trace of liquid from the Eppendorf tube.It may take a minute or more, depending on your target gel concentrations, to thoroughly mix these components due to the high viscosities of Matrigel and collagen. The components are mixed when optical inhomogeneities, i.e. mixing lines, are no longer visible in the tube.
Fill a sterilized 100 mL beaker with sterile ice, place the Eppendorf tube from Step 5 in the beaker, open the tube, and place the beaker in a sterilized vacuum chamber. Apply vacuum (low strength house/medical vacuum is sufficient) for 5 minutes or until satisfied that the mixture is sufficiently degassed.
Perform Step 2 in Basic Protocol 5, using ice-cold Matrigel+collagen solution instead of ice-cold Matrigel solution.
Perform Step 3 in Basic Protocol 5.
- Perform Step 4 in Basic Protocol 5, incubating for 45 minutes instead of 15 minutes.Depending on gel concentration, it may be feasible to shorten the incubation to as brief as 15 minutes. A longer incubation ensures complete gelation and successful 3D transfer. Be advised that prolonged incubations - an hour or longer - can negatively affect cell viability.
Perform Step 5 in Basic Protocol 5, using ice-cold Matrigel+collagen solution instead of ice-cold Matrigel solution.
- Perform Step 6 in Basic Protocol 5, incubating for 45 minutes instead of 15 minutes.Same considerations as for Step 9.
Perform Step 7 in Basic Protocol 5.
Figure 8. Matrigel/collagen calculations.

The volumes of reagents required for Alternate Protocol 2 depend on the available stocks of Matrigel and collagen I, as well as the desired target concentrations for these gels. See text for associated protocol.
REAGENTS AND SOLUTIONS
4× spotting buffer
90 mM sodium citrate
900 mM sodium chloride
0.4 mg/mL n-octylglucoside (Sigma, cat. #O8001-1G)
20% (w/v) trehalose (Sigma, cat. #T5251-10G)
Dissolve components in Milli-Q water and adjust to pH 9.5 with NaOH or HCl. Store at room temperature for up to a year.
0.1% SDS solution
0.1% (w/v) sodium dodecyl sulfate
Dissolve into water. Store at room temperature indefinitely.
Flow buffer
2% (w/v) bovine serum albumin (Sigma, cat. #BSAV-RO)
0.4% (w/v) EDTA
1% (w/v) Pluronic-F68 (aka Kolliphor P 188) (Sigma, cat. #15759-1KG)
Dissolve components into calcium/magnesium-free phosphate-buffered saline, filter sterilize, and store at 4 °C for up to a month.
Priming buffer
100 mM acetic acid
0.1 mg/mL MgCl2
0.1 mg/mL CaCl2
0.1% (v/v) Tween-20
Adjust to pH 2.5 with HCl and NaOH
Dissolve components in Milli-Q water. Store at 4 °C because Tween-20 degrades under warm acidic conditions (Bates et al., 1973).
COMMENTARY
Background Information
The complex functions of metazoan organisms arise through interacting networks of their constituent cells. These networks exist within three-dimensional tissue structures with specific size, shape, composition, and ECM components. Reconstituting cellular networks in vitro is key to understanding their collective functions and is at the heart of tissue engineering approaches to regenerative medicine. Current methods for reconstituting cell-cell interactions within particular tissue architectures have strengths and weaknesses outlined in Table 1.
Table 1.
Comparison of tissue engineering techniques.
| Direct Patterning of Cell- adhesion Proteins |
Microwell Molding |
Inkjet/Laser Printing |
Dielectrophoresis | Microfluidic Molding |
3D Printing by Extrusion |
Droplet Microfluidics |
|
|---|---|---|---|---|---|---|---|
| Citation | Li et al, 2014 | Nelson et al, 2006 | Matsusaki et al, 2013 | Albrecht et al, 2006 | Hsu et al, 2013 | Murphy et Atala, 2014 | Chan et al, 2013. |
| specification of multiple cell types | yes | no* | yes | up to two | yes | yes | yes |
| cell types independent of cell properties | no | no | yes | no | yes | yes | yes |
| precise (<1um) positioning of single cells | yes | yes | no | yes | no | no | no |
| 3D (cells are in/on hydrogel) | no | yes | yes | yes | yes | yes | yes |
| high viability | yes | yes | no | yes | yes | no | yes |
| rapid pattern development | no | no | yes | no | no | yes | no |
| production of dense tissues | no | yes | no | yes | yes | no | no |
| arbitrary patterns | yes | no | yes | no | no | yes | no |
| arbitrary hydrogel | n/a | must support wells | yes | low ionic strength | yes | cannot be viscous | yes |
for an exception wherein two cell types are specified, see Stevens et al, 2013.
DPAC offers a fundamentally different approach to reconstituting complex tissue architecture in vitro. Rather than physically positioning cells or relying on cells' natural adhesive properties, DPAC uses an orthogonal adhesive mechanism: Watson-Crick base pairing of complementary DNA sequences. DNA-based adhesion is fast, strong, sequence-specific, and reversible. Recent developments in chemical biology have provided several means to functionalize cells with oligonucleotides. DPAC builds off these technologies to provide a flexible platform for controlling the structure of tissues in vitro.
Critical Parameters
It is essential to perform all manipulations on suspension-phase cells as quickly as possible and as cold as possible (without freezing). The viability of suspension-phase cells continuously drops over time. The rate of cell death varies with cell type. It is also undesirable for cell temperature to vary between manipulations.
It is essential that the cell suspension is monodisperse. Clumping cells are less likely to attach to DNA spots correctly. Clumping cells are more likely to non-specifically adhere to the substrate and can also dislodge properly assembled structures from the patterning template. Dead cells and insufficiently trypsinized cells both tend to clump.
Although a variety of hydrogels can be used with this protocol, there are several important parameters to consider. First and most importantly, the matrix must possess the physical and chemical properties necessary to sustain the cell functions and behaviors to be studied in downstream processes. Second, the matrix must be able to transition between ungelled and gelled states without killing the cells. Low-melt agarose is acceptable, whereas regular agarose is not. Third, the matrix must have sufficiently low viscosity while ungelled that it can flow through a flow cell without dislodging the tissues. High concentration gels like 8.0 mg/mL collagen are unacceptable. Other high-viscosity materials like methylcellulose are also challenging to use. The matrix must have sufficient structural integrity to be transferred off the substrate in one piece. Standard Matrigel formulations (9–12 mg/mL) are likely to work, while Matrigel below 5.0 mg/mL and uncrosslinked hyaluronan are unlikely to work.
If designing new DNA strands, it is essential that DNA hybridization between library members is minimized, even at 0–4 °C.
Troubleshooting
See Table 2.
Anticipated Results
In a standard 4.5×18 mm flow cell, it is reasonable to produce an array of small organoids, such as with 200 µm pitch, 17 rows, 70 columns, for 1190 organoids total. Under the same constraints, it is also reasonable to produce an array of large, complex organoids with 1000 µm pitch, 3 rows, 14 columns, for 42 organoids total. With four flow cells per slide, this yields 4760 simple or 168 complex organoids spread across up to four experimental conditions. The fraction of organoids with the correct structure will vary by experiment, but it is reasonable to expect 95% yield from each synthetic step, assuming good technique.
Time Considerations
Excluding time for culturing feedstock cells and preparing reagents, the design and synthesis of a DPAC organoid array can be completed within one day. Four flow cells can fit on a single 25×75mm patterned slide, and it is quite reasonable to process all four flow cells simultaneously in a day's work.
Basic Protocol 1 takes anywhere from one to twelve hours, depending on the size and complexity of the desired pattern. The Nano eNabler prints features at a rate of 2500–3500/hr. When printing multiple sequences, allocate 10–30 minutes to load and align the new SPT and to bring the humidity back to target. Once complete, work can immediately proceed with the Basic Protocol 2 or pause for up to one month with the slide stored in a vacuum desiccator.
Basic Protocol 2 takes approximately 50 minutes. Once complete, work can immediately proceed with Basic Protocol 3 or pause for up to one month with the slide stored in a vacuum desiccator.
Basic Protocol 3 takes approximately one hour. Work must immediately proceed with Basic Protocol 4.
Basic Protocol 4 takes approximately 15 minutes per synthetic step, but this time can be shortened for cells that label especially efficiently, as described in the Protocol. The authors have synthesized organoids with complexity ranging from one to twelve synthetic steps. Ending the workflow at Basic Protocol 4 yields DNA-tethered organoids on the slide, which are stable for up to one day and may be useful for certain applications. Otherwise, work must immediately proceed with Basic Protocol 5.
Basic Protocol 5 takes approximately 30 minutes in addition to the time required to gel the embedding and underlay layers. For Matrigel, this is 15 minutes per layer for 60 minutes total. For collagen, this is 30 minutes per layer for 90 minutes total.
The authors have successfully cultured DPAC organoid arrays for as long as two weeks. The long-term culture of DPAC organoid arrays is limited primarily by the properties of the cells used.
Table 3.
Troubleshooting table.
| Problem | Possible Causes | Differential Diagnosis | Solution |
|---|---|---|---|
| Cells insufficiently attach | Insufficient cell density | Check flow cell under microscope during attachment. Are cells less than confluent? | Use more cells or resuspend cells into a smaller volume. |
| Cell detachment secondary to attachment | Check flow cell under microscope during attachment. Search for attached cells. Apply flow. Do attached cells subsequently detach? | Decrease flow rate. | |
| Insufficient DNA on cells | Use Support Protocol 2. Is MFI below 10? | Increase concentration of DNA, decrease number of cells, use alternative lifting, or use alternative DNA. | |
| Cells are clumping | Spot 5uL cell suspension on glass slide and inspect under microscope. Are cell clumps evident? | Increase trypsinization time. Increase pipetting when resuspending cells. Increase % BSA and/or EDTA in flow buffer. Handle cells more gently. | |
| Insufficient accessible DNA on surface | Use Support Protocol 3. Are fluorescent spots visible under microscope? | Use new aldehyde slides and new sodium borohydride. Increase hybridization time. Prepare new activation buffer. | |
| Surface occluded by cell debris | Use Support Protocol 4, then try again. Did the cell attachment improve? | Use Support Protocol 4 before starting synthesis. If this is a multistep synthesis, decrease attachment time for previous steps. | |
| Cells attach to incorrect DNA spots | Insufficient flow rate | Check flow cell under microscope after attachment. Apply forceful flow. Did some of the inappropriate cells detach? | Increase flow rate. Alternatively, wash with PBS as warm as 37 °C to increase wash stringency. |
| DNA strands improperly designed | Use DINAmelt and/or melting curve analysis to assess cross-hybridization between DNA strands. | Redesign DNA strands. Alternatively, wash with PBS as warm as 37 °C to increase wash stringency. | |
| Cells attach non-specifically to surface | Surface insufficiently passivated | Measure sessile drop contact angle of water on freshly dried surface. Is contact angle < 90 degrees? | Use fresh silanization reagents. Increase silanization time. |
| Flow cell buckling | Flow food coloring into flow cell. Is the intensity of color fainter in the center of the flow cell? | Decrease clamp pressure. Increase thickness of PDMS flow cells. | |
| Cells are clumping | Spot 5uL cell suspension on glass slide and inspect under microscope. Are cell clumps evident? | Increase trypsinization time. Increase pipetting when resuspending cells. Increase % BSA and/or EDTA in flow buffer. Handle cells more gently. | |
| Flow rate insufficient | Check flow cell under microscope after attachment. Apply forceful flow. Did some of the inappropriate cells detach? | Increase flow rate. | |
| Cells die | Temperature too high | Use infrared thermometer to measure temperature. | Adjust amount of/distance from ice to reach target 1–4C range. |
| Manipulations taking too long | Label cells with DNA and set them aside, on ice, for duration of manipulations. Compare viability (e.g., using Trypan blue) before/after. | Decrease number of synthetic steps, decrease hybridization time per step, optimize your workflow. | |
| Flow rate too high | Repeat experiment, reducing the flow rate as much as possible. Does viability improve? | Decrease flow rate. | |
| Assembles fall apart during synthesis | Flow rate too high | Check flow cell under microscope after assembly. Apply flow. Do assemblies disassociate? | Decrease flow rate. |
| Organoids too tall for the flow cell | Does the synthetic scheme contain any 5-step-or-greater assemblies? | Redesign synthetic scheme. | |
| Organoids anchored to surface at too few points | Are any >=3-step assemblies anchored by a single cell? Are any >=4-step assemblies anchored by <=4 cells? | Redesign synthetic scheme and/or pattern. | |
| Insufficient DNA on cells | Use Support Protocol 2. Is MFI below 10? | Increase concentration of DNA, decrease number of cells, use alternative lifting, or use alternative DNA. | |
| Temperature too low | Use infrared thermometer to measure temperature. | Adjust amount of/distance from ice to reach target 1–4C range. | |
| Assemblies grow at inappropriate rate | Cells are clumping | Spot 5uL cell suspension on glass slide and inspect under microscope. Are cell clumps evident? | Increase trypsinization time. Increase pipetting when resuspending cells. Increase % BSA and/or EDTA in flow buffer. Handle cells more gently. |
| Insufficient DNA on cells | Use Support Protocol 2. Is MFI below 10? | Increase concentration of DNA, decrease number of cells, use alternative lifting, or use alternative DNA. | |
| Gel does not flow into flow cell. | Viscosity too high. | Prepare a solution of 85% glycerol at 20 °C. Is your matrix more viscous than this? | Reformulate matrix. Consider lowering bulk protein concentration. |
| Matrix prematurely gelling. | Once gel ceases flowing, pry the flow cell off the surface. Is gelled material left behind? (this is a destructive assay) | Reformulate matrix and/or adjust temperature/light/speed to prevent premature gelation. | |
| Gel distorts/fractures | Poor manual manipulation | Was the blade flush with the flow cell during 3D transfer? Were your movements smooth and sure? | Practice on empty flow cells. Drink less coffee. |
| Failure to remove gel outside flow cell | Before 3D transfer, are there traces of gel outside the flow cell but still connected to the gel within the flow cell? | Use razor blade to cut off excess gel. | |
| Gel shrinkage/swelling | Are air bubbles evident within the flow cell after 3D transfer? Has gel pulled away from the inlet and outlet? | Reformulate matrix. Consider adding surfactants. | |
| Inadequate gel toughness | If the gel is gently poked with a capillary tube, does it fracture? | Reformulate matrix. Consider increasing bulk protein concentration. | |
| Gel curls on itself | During 3D transfer, once the gel is released from the surface and inverted, does the gel ball up? | Add surfactants such as 1% BSA to gel, or increase gel density. | |
| Gel remains on surface during 3D transfer | Surface insufficiently passivated | Measure sessile drop contact angle of water on freshly dried surface. Is contact angle < 90 degrees? | Use fresh silanization reagents. Increase silanization time. |
| Gel detaches from bottom of culture vessel | Insufficient adhesion between gel layers | Add a distinct dye, such as food coloring, to each gel layer. Do the dyes indicate that only certain gel layers detached? | Reformulate matrix. Consider increasing bulk protein concentration or adding adhesive proteins. |
Acknowledgments
The authors thank C. Mosher for technical help with the Nano eNabler. This work was supported by the Department of Defense Breast Cancer Research Program (W81XWH-10-1-1023 and W81XWH-13-1-0221 to ZJG); the National Institutes of Health common fund (DP2 HD080351-01 to ZJG); The Sidney Kimmel Foundation; The National Science Foundation (MCB-1330864 to ZJG); and the University of California San Francisco Program in Breakthrough Biomedical Research. ZJG is supported by the University of California San Francisco Center for Systems and Synthetic Biology (National Institute of General Medical Sciences Systems Biology Center grant P50 GM081879).
Footnotes
Conflict of Interest
A provisional patent application has been filed on the basis of this work. Z.J.G. is a member of the scientific advisory board of Adheren, a company that is commercializing cell-tethering technology.
LITERATURE CITED
- Albrecht DR, Underhill GH, Wassermann TB, Sah RL, Bhatia SN. Probing the role of multicellular organization in three-dimensional microenvironments. Nature Methods. 2006;3:369–375. doi: 10.1038/nmeth873. [DOI] [PubMed] [Google Scholar]
- Application Note 203 -“Speed Printing”; or printing in “No Laser Mode” with the Nano eNabler System™. [Accessed April 5, 2015];BioForce Nanosciences. Available at: http://bioforcenano.com/resources/aapplication-note-203-speed-printing-or-printing-in-no-laser-mode-with-the-nano-enabler-system/
- Bates TR, Nightingale CH, Dixon E. Kinetics of hydrolysis of polyoxyethylene (20) sorbitan fatty acid ester surfactants. Journal of Pharmacy and Pharmacology. 1973;25:470–477. doi: 10.1111/j.2042-7158.1973.tb09135.x. [DOI] [PubMed] [Google Scholar]
- Cerchiari A, Garbe JC, Todhunter ME, Jee NY, Pinney JR, LaBarge MA, Desai TA, Gartner ZJ. Formation of Spatially and Geometrically Controlled Three-Dimensional Tissues in Soft Gels by Sacrificial Micromolding. [Accessed April 8, 2015];Tissue Engineering Part C: Methods. 2014 doi: 10.1089/ten.tec.2014.0450. Available at: http://online.liebertpub.com/doi/abs/10.1089/ten.tec.2014.0450. [DOI] [PMC free article] [PubMed]
- Chan HF, Zhang Y, Ho Y-P, Chiu Y-L, Jung Y, Leong KW. Rapid formation of multicellular spheroids in double-emulsion droplets with controllable microenvironment. [Accessed July 31, 2015];Scientific Reports. 2013 :3. doi: 10.1038/srep03462. Available at: http://www.nature.com/srep/2013/131210/srep03462/full/srep03462.html?message-global=remove&WT.ec_id=SREP-631-20140102. [DOI] [PMC free article] [PubMed]
- Chen S, Bremer AW, Scheideler OJ, Na YS, Todhunter ME, Hsiao S, Bomdica PR, Maharbiz MM, Gartner ZJ, Schaffer DV. Interrogating cellular fate decisions with high-throughput arrays of multiplexed cellular communities. Nat Commun. 2016:7. doi: 10.1038/ncomms10309. Available at: http://dx.doi.org/10.1038/ncomms10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung K, Deisseroth K. CLARITY for mapping the nervous system. Nature Methods. 2013;10:508–513. doi: 10.1038/nmeth.2481. [DOI] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- Dhimolea E, Soto AM, Sonnenschein C. Breast epithelial tissue morphology is affected in 3D cultures by species-specific collagen-based extracellular matrix. Journal of Biomedical Materials Research. Part A. 2012;100:2905–2912. doi: 10.1002/jbm.a.34227. [DOI] [PubMed] [Google Scholar]
- El Muslemany KM, Twite AA, ElSohly AM, Obermeyer AC, Mathies RA, Francis MB. Photoactivated Bioconjugation Between ortho-Azidophenols and Anilines: A Facile Approach to Biomolecular Photopatterning. Journal of the American Chemical Society. 2014;136:12600–12606. doi: 10.1021/ja503056x. [DOI] [PubMed] [Google Scholar]
- Gartner ZJ, Bertozzi CR. Programmed assembly of 3-dimensional microtissues with defined cellular connectivity. Proceedings of the National Academy of Sciences. 2009;106:4606–4610. doi: 10.1073/pnas.0900717106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Y-H, Moya ML, Hughes CCW, George SC, Lee AP. A microfluidic platform for generating large-scale nearly identical human microphysiological vascularized tissue arrays. Lab on a Chip. 2013;13:2990–2998. doi: 10.1039/c3lc50424g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkel JN, Unger KK. Role of solvent and base in the silanization reaction of silicas for reversed-phase high-performance liquid chromatography. Journal of Chromatography A. 1984;316:193–200. [Google Scholar]
- Kumar A, Biebuyck HA, Whitesides GM. Patterning Self-Assembled Monolayers: Applications in Materials Science. Langmuir. 1994;10:1498–1511. [Google Scholar]
- Lange SA, Benes V, Kern DP, Hörber JKH, Bernard A. Microcontact Printing of DNA Molecules. Analytical Chemistry. 2004;76:1641–1647. doi: 10.1021/ac035127w. [DOI] [PubMed] [Google Scholar]
- Li CY, Stevens KR, Schwartz RE, Alejandro BS, Huang JH, Bhatia SN. Micropatterned Cell–Cell Interactions Enable Functional Encapsulation of Primary Hepatocytes in Hydrogel Microtissues. Tissue Engineering Part A. 2014;20:2200–2212. doi: 10.1089/ten.tea.2013.0667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham NR, Zuker M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Research. 2005;33:W577–W581. doi: 10.1093/nar/gki591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsusaki M, Sakaue K, Kadowaki K, Akashi M. Three-Dimensional Human Tissue Chips Fabricated by Rapid and Automatic Inkjet Cell Printing. Advanced Healthcare Materials. 2013;2:534–539. doi: 10.1002/adhm.201200299. [DOI] [PubMed] [Google Scholar]
- Monserud JH, Schwartz DK. Effects of Molecular Size and Surface Hydrophobicity on Oligonucleotide Interfacial Dynamics. Biomacromolecules. 2012;13:4002–4011. doi: 10.1021/bm301289n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SV, Atala A. 3D bioprinting of tissues and organs. Nature Biotechnology. 2014;32:773–785. doi: 10.1038/nbt.2958. [DOI] [PubMed] [Google Scholar]
- Nelson CM, VanDuijn MM, Inman JL, Fletcher DA, Bissell MJ. Tissue Geometry Determines Sites of Mammary Branching Morphogenesis in Organotypic Cultures. Science. 2006;314:298–300. doi: 10.1126/science.1131000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoe H, Hsiao SC, Douglas ES, Gartner ZJ, Bertozzi CR, Francis MB, Mathies RA. Cellular Microfabrication: Observing Intercellular Interactions Using Lithographically-Defined DNA Capture Sequences. Langmuir. 2012;28:8120–8126. doi: 10.1021/la204863s. [DOI] [PubMed] [Google Scholar]
- Selden NS, Todhunter ME, Jee NY, Liu JS, Broaders KE, Gartner ZJ. Chemically Programmed Cell Adhesion with Membrane-Anchored Oligonucleotides. Journal of the American Chemical Society. 2012;134:765–768. doi: 10.1021/ja2080949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens KR, Ungrin MD, Schwartz RE, Ng S, Carvalho B, Christine KS, Chaturvedi RR, Li CY, Zandstra PW, Chen CS, et al. InVERT molding for scalable control of tissue microarchitecture. Nature Communications. 2013;4:1847. doi: 10.1038/ncomms2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twite AA, Hsiao SC, Onoe H, Mathies RA, Francis MB. Direct Attachment of Microbial Organisms to Material Surfaces Through Sequence-Specific DNA Hybridization. Advanced Materials. 2012;24:2380–2385. doi: 10.1002/adma.201104336. [DOI] [PubMed] [Google Scholar]
- Whipple RA, Cheung AM, Martin SS. Detyrosinated microtubule protrusions in suspended mammary epithelial cells promote reattachment. Experimental Cell Research. 2007;313:1326–1336. doi: 10.1016/j.yexcr.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
KEY REFERENCE
- Todhunter ME, Jee NY, Hughes AJ, Coyle MC, Cerchiari A, Farlow J, Garbe JC, LaBarge MA, Desai TA, Gartner ZJ. Programmed synthesis of 3D tissues. Nature Methods. 2015 doi: 10.1038/nmeth.3553. [DOI] [PMC free article] [PubMed] [Google Scholar]