

Abstract
Recent evidence has suggested that IL‐10‐producing effector CD8+ T cells play an important role in regulating excessive inflammation during acute viral infections. However, the cellular and molecular cues regulating the development of IL‐10‐producing effector CD8+ T cells are not completely defined. Here, we show that type I interferons (IFNs) are required for the development of IL‐10‐producing effector CD8+ T cells during influenza virus infection in mice. We find that type I IFNs can enhance IL‐27 production by lung APCs, thereby facilitating IL‐10‐producing CD8+ T‐cell development through a CD8+ T‐cell‐nonautonomous way. Surprisingly, we also demonstrate that direct type I IFN signaling in CD8+ T cells is required for the maximal generation of IL‐10‐producing CD8+ T cells. Type I IFN signaling in CD8+ T cells, in cooperation with IL‐27 and IL‐2 signaling, promotes and sustains the expression of IFN regulatory factor 4 (IRF4) and B‐lymphocyte‐induced maturation protein‐1 (Blimp‐1), two transcription factors required for the production of IL‐10 by effector CD8+ T cells. Our data reveal a critical role of the innate antiviral effector cytokines in regulating the production of a regulatory cytokine by effector CD8+ T cells during respiratory virus infection.
Keywords: CD8+ T cells, IL‐10, IL‐27, Influenza, Type I IFN
Type I IFNs have both cell‐nonautonomous and cell‐autonomous effects in promoting IL‐10 production by CD8+ T cells during influenza infection. The effects of type I IFNs in promoting IL‐10 production by CD8+ T cells require the function of the transcription factors IFN regulatory factor 4 (IRF4) and B‐lymphocyte‐induced maturation protein‐1 (Blimp‐1).
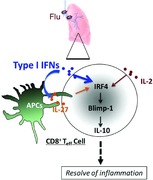
.
Introduction
CD8+ T cells are the key to clear infectious virus, intracellular bacteria, and transformed cells. Upon antigenic encountering in the secondary lymphoid organs, naïve CD8+ T cells undergo stepwise stages of responses including activation, expansion, and differentiation into effector CD8+ T cells (i.e., cytotoxic T lymphocytes) 1, 2. Effector CD8+ T cells acquire tissue tropic chemotactic receptors such as CCR5 and CXCR3 while simultaneously downregulate lymphoid tissue homing receptors such as CCR7 1, 2, which allow them to infiltrate peripheral tissues to clear pathogen‐infected or transformed cells. In the tissue, effector CD8+ T cells use multiple mechanisms to combat invading enemies. Most prominently, effector CD8+ T cells express key cytolytic molecules including granzymes, perforin, and death receptor ligands and are able to clear virus‐infected cells or transformed cells through contact‐dependent mechanisms. In addition, effector CD8+ T cells also express multiple antiviral and antitumor proinflammatory cytokines, such as IFN‐γ and TNF‐α, to inhibit viral replication and kill tumor cells. Notably, these direct and indirect antiviral and tumor activities of effector CD8+ T cells, if unchecked, also can cause severe inflammation and tissue destruction 1, 3, 4, 5.
Recently, we and others have demonstrated that effector CD8+ T cells are able to produce the regulatory cytokine IL‐10 in the lung and brain following acute viral infections 6, 7, 8, 9, 10, 11. The production of IL‐10 by effector CD8+ T cells is important in counterbalancing exuberant inflammation as the blockade of IL‐10 following infection has resulted in exaggerated inflammation and severe host diseases during influenza, RSV, and coronavirus infections 7, 8, 9, 10, 11, 12. Furthermore, we showed that IL‐2 cooperates with innate cell‐derived IL‐27 to upregulate IL‐10 production by CD8+ T cells specifically in the lung through the transcription factor B‐lymphocyte‐induced maturation protein‐1 (Blimp‐1) dependent mechanisms 6. However, the current understanding on the cellular and molecular mechanisms regulating the development of IL‐10‐producing effector CD8+ T cells remains incompletely defined.
Type I IFNs are major antiviral effector cytokines that have critical roles in shaping both innate and adaptive defense against viral infections. Initially discovered as the proinflammatory “signal 3” cytokines, type I IFNs were shown to promote CD8+ T‐cell expansion and effector differentiation during bacterial and viral infections 13, 14. Moreover, type I IFN signaling in CD8+ T cells can protect CD8+ T cell from NK‐cell‐mediated deletion during chronic virus infection 15, 16. Besides their role in enhancing antiviral immune responses, type I IFNs signals also promote IL‐10 and PD‐L1 production/expression and suppress effective CTL responses during chronic LCMV infection 17, 18, suggesting that type I IFN signaling possesses immune‐regulatory functions. Similarly, recent evidence has demonstrated that type I IFNs are required for IL‐10 production during influenza infection 19, 20. Arimori et al. showed that IFN‐α receptor 1 (IFNAR1) deficient mice exhibited diminished IL‐10 levels in the lung, enhanced proinflammatory cytokine production, and increased host mortality following influenza virus infection 20. However, how type I IFN signaling promotes IL‐10 production during influenza infection is currently unknown. Furthermore, given that T cells, particularly effector CD8+ T cells, are a major cellular source of IL‐10 during influenza infection, it is important to determine whether and how type I IFN signaling affects IL‐10 production of T cells.
In this report, we have examined the role of type I IFN signaling in the induction of IL‐10‐producing T cells during influenza infection. We found that type I IFN signaling is critically important in driving IL‐10 production by effector CD8+ T cells, but only plays modest role in promoting IL‐10 production by CD4+ T cells during influenza infection. We found that type I IFN signaling promotes IL‐27 production by APCs to indirectly facilitate CD8+ T‐cell IL‐10 production in a CD8+ T‐cell‐nonautonomous fashion. Interestingly, we found that direct type I IFN signaling in CD8+ T cells is also critical for the maximal generation of IL‐10‐producing effector CD8+ T cells during influenza infection. Mechanistically, type I IFN signaling, in cooperation with IL‐27 and IL‐2 signaling, promotes and sustains the expression of Blimp‐1 and IFN regulatory factor 4 (IRF4), two transcription factors required for the production of IL‐10 by effector CD8+ T cells. Our data have revealed the underlying molecular mechanisms by which innate antiviral effector cytokines regulate the production of a regulatory cytokine by effector CD8+ T cells during respiratory virus infection.
Results
IFNAR1‐ and STAT2‐dependent development of IL‐10‐producing effector CD8+ T cells
To determine the role of type I IFNs in the development of IL‐10‐producing effector CD8+ T cells, we infected WT and IFNAR1‐deficient mice with influenza. At day 7 post infection (at the time when maximal IL‐10 was made in vivo) 6, 8, we stimulated lung T cells with influenza‐infected WT BM‐derived DCs (BMDCs) and measured IL‐10 and IFN‐γ production by T cells following stimulation as we previously reported 6, 8. We found that IFNAR1 deficiency modestly affected IFN‐γ production by CD8+ T cells, but dramatically diminished IL‐10 production by CD8+ T cells (Fig. 1A). We then normalized IL‐10+ CD8+ T cells to IFN‐γ+ CD8+ T cells to determine the relative abundance of IL‐10‐producing cells in IFN‐γ‐producing antigen‐specific CD8 T cells. We found that IFNAR1 deficiency significantly abrogated IL‐10‐producing CD8+ T cells within the antigen‐specific CD8+ T‐cell population (Fig. 1B). IFNAR1 deficiency also decreased the per‐cell production of IL‐10 by effector CD8+ T cells as the mean fluorescence intensity (MFI) of IL‐10 in IL‐10+ cells diminished in the absence of IFNAR1 (Supporting Information Fig. 1A). In addition, IFNAR1 deficiency diminished IL‐10‐producing CD4+ T cells (Fig. 1C), although the relative abundance of IL‐10‐producing CD4+ T cells in antigen‐specific IFN‐γ+ CD4+ T cells only modestly diminished in the absence of IFNAR1 (Fig. 1D). Consistent with their diminished levels of IL‐10‐producing CD8+ and CD4+ T cells, IFNAR1‐deficient mice exhibited diminished airway IL‐10 levels and increased airway IFN‐γ levels in vivo during influenza infection (Figs. 1E and F), consistent with the recent findings that type I IFN signaling is required for IL‐10 but not IFN‐γ production in vivo during influenza infection 19, 20. Following type I IFNs engagement, IFNAR initiates downstream events to form a signaling complex consisting of STAT1/STAT2/IRF9, which mediate the major functions of type I IFNs 23, 24. We therefore examined whether STAT2 is required for the generation of IL‐10‐producing CD8+ T cells in vivo during influenza infection. We found that STAT2 deficiency significantly diminished IL‐10‐producing CD8+ T cells in the lung (Fig. 1G and H). STAT2 deficiency also significantly decreased IL‐10 MFI in IL‐10+ CD8+ effector T cells (Supporting Information Fig. 1B). Taken together, these data suggested that STAT2‐dependent IFNAR1 signaling is required for the generation of IL‐10‐producing effector CD8+ T cells during influenza infection.
Figure 1.
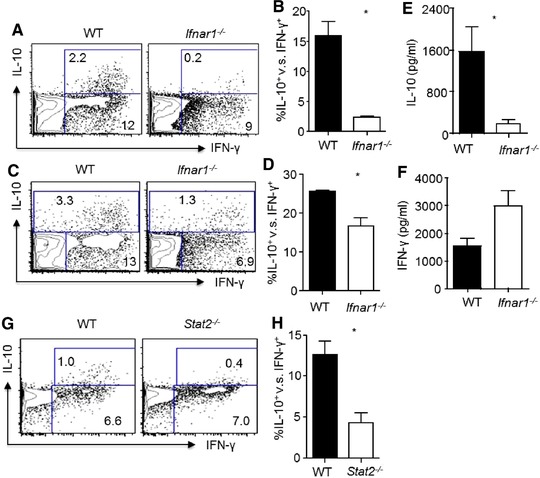
Type I IFN signaling is required for the optimal induction of IL‐10‐producing effector CD8+ T cells. WT or Ifnar1−/− mice were infected with influenza. IL‐10 and IFN‐γ production by lung T cells and IL‐10 levels in the airway were determined at day 7 post infection by flow cytometry (A–D) and ELISA (E–F). (A) Production of IL‐10 and IFN‐γ by lung CD8+ T cells from WT or Ifnar1−/− mice following in vitro antigenic stimulation with influenza‐infected WT BMDCs. (B). Normalized percentages of IL‐10+ cells in influenza‐specific lung CD8+ T cells (IFN‐γ+) from infected WT or Ifnar1−/− mice (C). Production of IL‐10 and IFN‐γ by lung CD4+ T cells following in vitro antigenic stimulation with influenza‐infected WT BMDCs. (D) Normalized percentages of IL‐10+ cells in influenza‐specific lung CD4+ T cells (IFN‐γ+) from infected WT or Ifnar1−/− mice. (E) IL‐10 levels in the BALF from WT or Ifnar1−/− mice were determined through ELISA. (F) IFN‐γ levels in the BALF from WT or Ifnar1−/− mice were determined through ELISA. (G, H) Production of IL‐10 and IFN‐γ by lung CD8+ T cells from WT or Stat2−/− mice was determined by flow cytometry following in vitro antigenic stimulation with influenza‐infected WT BMDCs. (G) Representative density plots and (H) normalized percentages of IL‐10+ cells in influenza‐specific lung CD8+ T cells (IFN‐γ+) from infected WT or Stat2−/− mice. Data in density plots are from a single experiment representative of two to four independent experiments with two to three mice per experiment. Data in graphs are shown as mean + SEM and are representative of two to four separate experiments with two to three mice per experiment. Statistics were determined by unpaired two‐tailed Student's t‐test (*p < 0.05).
To further support this idea, we crossed IFNAR1‐deficient mice to IL‐10‐IRES‐eGFP reporter mice (Vert‐X), as these mice faithfully report IL‐10 expression by T and other cells without the need of in vitro restimulation 25. We infected IFNAR1‐deficient Vert‐X mice with influenza and then examined in vivo IL‐10 production through the reporter eGFP expression on T cells at day 7 post infection. We found that, compared to those of IFNAR1‐sufficient Vert‐X mice, CD8+ T cells from IFNAR1‐deficient mice exhibited diminished percentages of IL‐10/eGFP+ cells (Fig. 2A and B) and a lower per‐cell level of IL‐10/eGFP (Fig. 2 C), consistent with the idea that IFNAR1 signaling is required for the potent induction of IL‐10‐producing CD8+ T cells. We also examined IL‐10/eGFP expression by influenza‐specific PA224 tetramer+ CD8+ T cells and found that the deficiency of IFNAR1 resulted in decreased percentages of IL‐10/eGFP+ cells within PA224+ CD8+ T cell population and diminished IL‐10/eGFP MFI (Fig. 2D–F). The deficiency of IFNAR1 did not significantly decrease IL‐10/eGFP expression by CD4+ T cells (Fig. 2G and H). Taken together, these data suggested that IFNAR1 signaling is preferentially required for the development of IL‐10 expressing CD8+ T cells.
Figure 2.
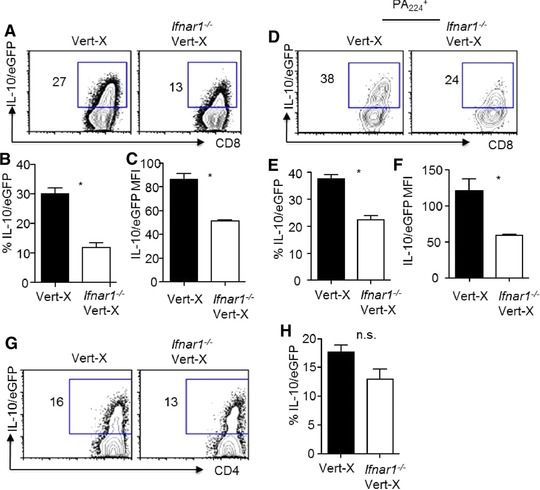
Type I IFN signaling is required for the optimal in vivo expression of IL‐10 by CD8 + T cells. Vert‐X or Ifnar1−/− Vert‐X mice were infected with influenza. IL‐10 expression by T cells in vivo was measured through their eGFP expression by flow cytometry at day 7 p.i. (A). Expression of IL‐10‐eGFP by total lung CD8+ T cells from infected Vert‐X or Ifnar1−/− Vert‐X mice. (B) Percentages IL‐10‐eGFP+ of cells in total lung CD8+ T cells from infected Vert‐X or Ifnar1−/− Vert‐X mice. (C) IL‐10‐eGFP expression levels (MFI) of the IL‐10‐eGFP+ of cells in total lung CD8+ T cells from infected Vert‐X or Ifnar1−/− Vert‐X mice. (D) Expression of IL‐10‐eGFP by influenza‐specific PA224+ lung CD8+ T cells from infected Vert‐X or Ifnar1−/− Vert‐X mice. (E) Percentages IL‐10‐eGFP+ of cells in influenza‐specific PA224+ lung CD8+ T cells from infected Vert‐X or Ifnar1−/− Vert‐X mice. (F) IL‐10‐eGFP expression levels (MFI) of the IL‐10‐eGFP+ of cells in influenza‐specific PA224+ lung CD8+ T cells from infected Vert‐X or Ifnar1−/− Vert‐X mice. (G) Expression of IL‐10‐eGFP by total lung CD4+ T cells from infected Vert‐X or Ifnar1−/− Vert‐X mice. (H) Percentages IL‐10‐eGFP+ of cells in total lung CD4+ T cells from infected Vert‐X or Ifnar1−/− Vert‐X mice. Data in density plots are from a single experiment representative of three independent experiments with three to four mice per experiment. Data in graphs are shown as mean + SEM and are representative of three separate experiments with three to four mice per experiment. Statistics were determined by unpaired two‐tailed Student's t‐test (∗ p < 0.05). n.s., nonsignificant.
Type I IFN signaling is required for the maximal production of IL‐27
We recently have demonstrated that IL‐27 is vital for the development of IL‐10‐producing effector CD8+ T cells during influenza infection. Type I IFNs have been shown to stimulate IL‐27 production by APCs 26. We found that APCs (including macrophages, DCs, and neutrophils) are the main sources of IL‐27 expression (particularly p28 subunit) in the lung at day 5 post influenza infection, at the time when effector T cells begin to migrate to the lungs and upregulate IL‐10 expression there (Fig. 3A). Consistent with the previous findings, we found that type I IFNs were able to induce the expression of IL‐27 in DCs, macrophages, and neutrophils (Supporting Information Fig. 2), the main APC types in the infected lungs when CD8+ T cells start to produce IL‐10 27, 28. Therefore, we reasoned that type I IFNs induce IL‐27 production, thereby supporting IL‐10 production by CD8+ T cells. To address this hypothesis, we examined the expression of IL‐27 subunits in the draining mediastinal LNs (MLN) and the lungs 6, 8. We found that, compared to those of WT mice, EBV‐induced 3 (Ebi3) and p28 expression in the lung of IFNAR1‐deficient mice was significantly decreased (Fig. 3B). Furthermore, airway IL‐27 p28 protein levels were also diminished in IFNAR1‐deficient mice following influenza infection (Fig. 3C). Together, these data suggest that type I IFN signaling is required for the optimal production of the IL‐10 inducing cytokine, IL‐27, during influenza infection.
Figure 3.
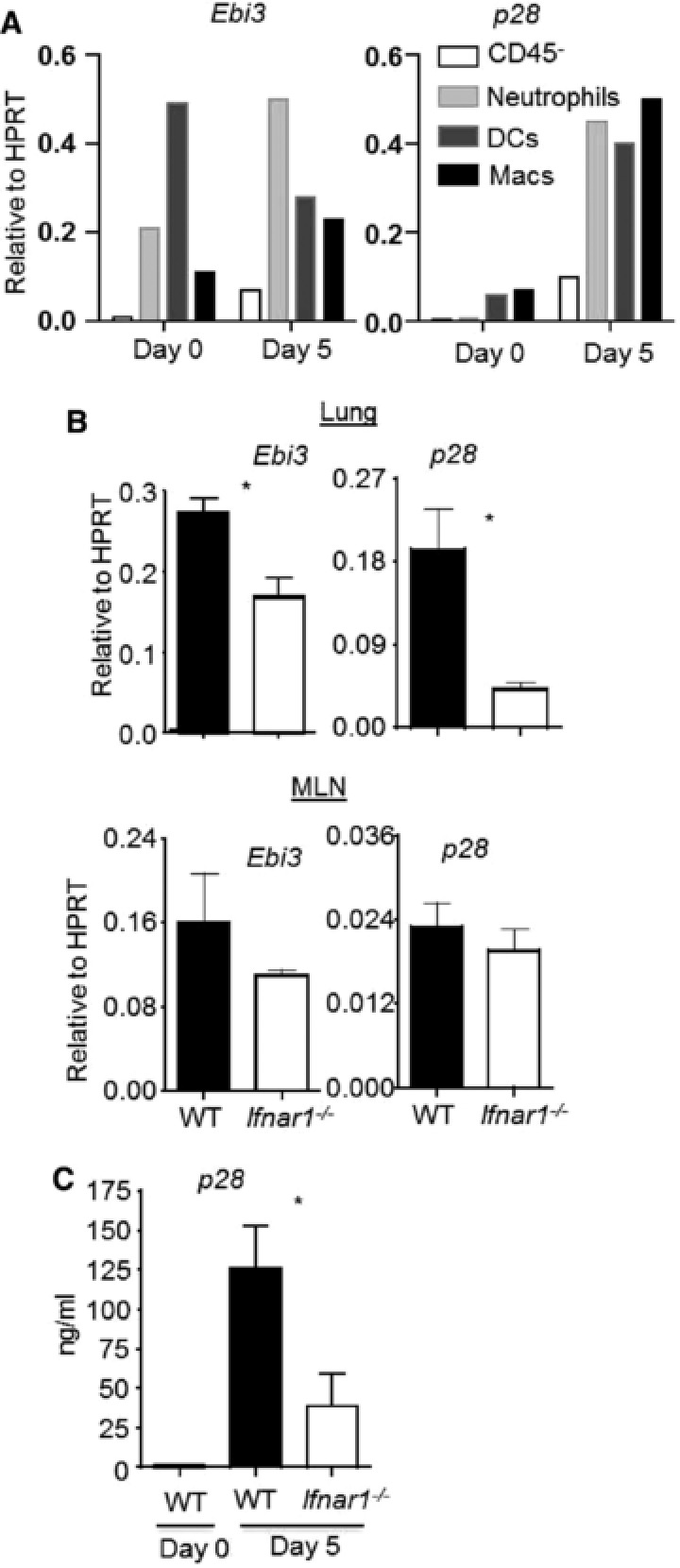
Type I IFN signaling is required for optimal IL‐27 expression during influenza infection. WT mice were infected with influenza. IL‐27 subunit (Ebi3 and p28) gene expression in indicated lung cells’ populations (A), mediastinal LNs (MLN) and lungs (B), and p28 protein production in the airway (C) were determined at day 5 p.i. (A) Ebi3 and p28 gene expression in various cell types sorted from the lungs of WT mice at day 5 p.i. was determined by normalizing the data to HPRT expression through real‐time RT‐PCR. (B) Ebi3 and p28 gene expression in the lung and draining MLN from WT or Ifnar1−/− mice was determined by normalizing the data to HPRT expression through real‐time RT‐PCR. (C) p28 protein levels in the BALF from WT or Ifnar1−/− mice were determined through ELISA. (A) Data are shown as mean pooled from cells isolated from two to three mice and are a single experiment representative of two independent experiments. (B and C) Data in graphs are shown as mean +SEM and are representative of two separate experiments with two to four mice per experiment. Statistics were determined by unpaired two‐tailed Student's t‐test (*p < 0.05).
Cell‐intrinsic IFNAR1 is required for the maximal development of IL‐10‐producing CD8+ T cells
Type I IFNs are important “signal 3” cytokines that can directly drive CD8+ T‐cell differentiation 13, 14. We next want to examine whether direct type I IFN signaling in CD8+ T cells may influence IL‐10 production beyond their cell‐nonautonomous effects in promoting IL‐10 production by CD8+ T cells through the induction of IL‐27 production. To do so, we lethally irradiated WT mice and then reconstituted the mice with 1:1 mixed CD45 mismatched WT and IFNAR1‐deficient BM cells (Fig. 4A). Twelve weeks after reconstitution, we infected the chimeric mice with influenza and found that the T‐cell compartments in the lungs of the infected mice were successfully reconstituted with both WT or Ifnar1−/‐ cells (Supporting Information Fig. 3A). We then checked IL‐10 and IFN‐γ production following in vitro restimulation of T cells. We found that IL‐10 production by IFNAR1‐deficient CD8+ T cells is significantly lower than that of WT CD8+ T cells even within the same hosts (Fig. 4B and C). Furthermore, the IL‐10 MFI is also significantly lower in IFNAR1‐deficient CD8+ T cells than that of WT CD8+ T cells (Fig. 4D). In contrast, IL‐10 production by CD4+ T cells is comparable between WT and IFNAR1‐deficient CD4+ T cells (Fig. 4E and Supporting Information Fig. 3B). Taken together, these data suggest that CD8+ T‐cell‐autonomous type I IFN signaling is also essential for the maximal development of IL‐10‐producing effector CD8+ T cells, but not IL‐10‐producing CD4+ T cells.
Figure 4.
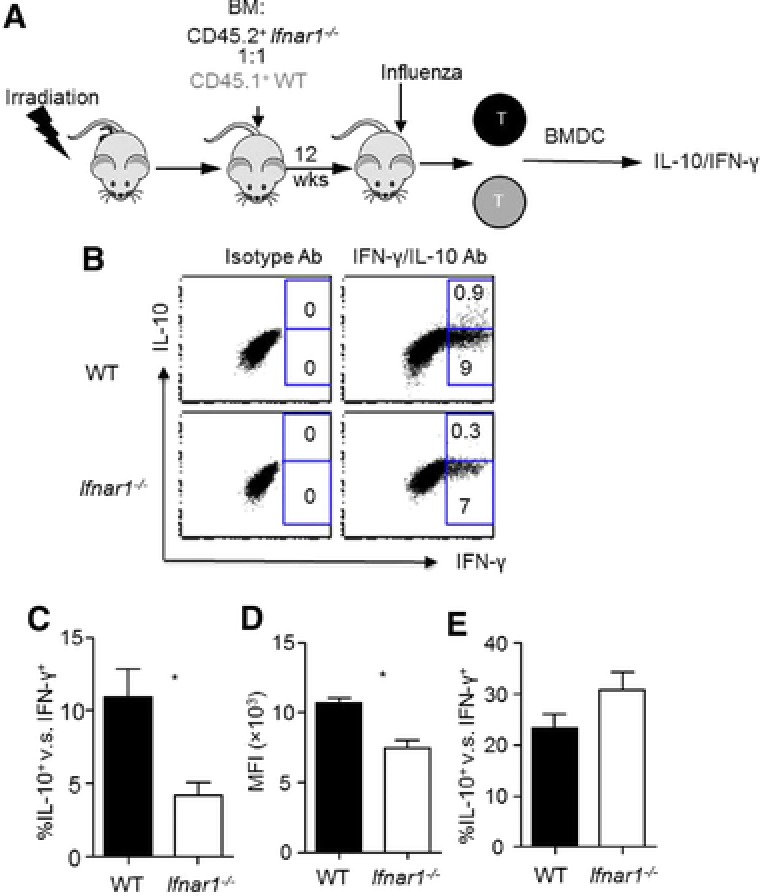
Direct type I IFN signaling in CD8 + T cells is required for optimal IL‐10 production by effector CD8 + T cells. WT and Ifnar1−/− mixed BM chimera mice were infected with influenza. IL‐10 production by lung T cells was determined by flow cytometry at day 7 post infection following stimulation with influenza‐infected WT BMDCs. (A) Schematics of the construction of BM chimeric mice. (B) Production of IL‐10 and IFN‐γ by WT or Ifnar1−/− lung CD8+ T cells following in vitro antigenic stimulation with influenza‐infected WT BMDCs. (C) Normalized percentages of IL‐10+ cells in influenza‐specific WT or Ifnar1−/− lung CD8+ T cells. (D) IL‐10 levels (MFI) produced by WT or Ifnar1−/− lung CD8+ T cells following in vitro antigenic stimulation with influenza‐infected WT BMDCs. (E) Normalized percentages of IL‐10+ cells in influenza‐specific WT or Ifnar1−/− lung CD4+ T cells (IFN‐γ+) from infected WT and Ifnar1−/− mice. Data in density plots are from a single experiment representative of two independent experiments with four to five mice per experiment. Data in graphs are shown as mean + SEM and are representative of two separate experiments with four to five mice per experiment. Statistics were determined by paired two‐tailed Student's t‐test (*p < 0.05).
Type I IFNs further promote IL‐10 production by CD8+ T cells in the presence of IL‐2 and IL‐27
To begin to examine the underlying cellular and molecular mechanisms by which type I IFN signaling promotes IL‐10 production by CD8+ T cells, we employed our previously reported CD8+ T‐cell culture system 6. To this end, we cultured WT CD8+ T cells in the absence or presence of IFN‐α, IL‐2 and IL‐27 (IL‐2/IL‐27), or IFN‐α plus IL‐2/27 (IFN‐α/IL‐2/IL‐27). We found that the inclusion of IFN‐α alone in the culture minimally induced the development of IL‐10‐producing CD8+ T cells in vitro (Fig. 5A). However, IFN‐α helped the development of IL‐10‐producing CD8+ T cells in the presence of IL‐2 and IL‐27 (Fig. 5A and B), which were reported to induce IL‐10‐producing effector CD8+ T cells 6. IFN‐α also promoted IL‐10 mRNA expression in CD8+ T cells in the presence of IL‐2 and IL‐27 (Fig. 5C). In addition, although IFN‐α alone minimally induced IL‐10 secretion by effector CD8+ T cells, it greatly enhanced IL‐10 secretion by CD8+ T cells in the presence of IL‐2 plus IL‐27 (Fig. 5D). Taken together, these data suggested that type I IFNs cooperate with IL‐2 and IL‐27 to promote IL‐10 production by CD8+ T cells.
Figure 5.
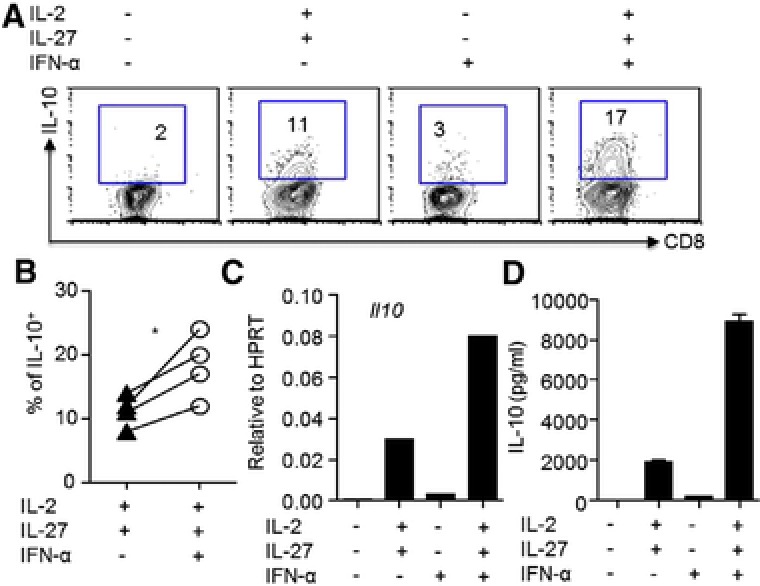
Type I IFNs further promote IL‐10 production by CD8+ T cells in the presence of IL‐2 plus IL‐27. WT CD8+ T cells were cultured under indicated conditions (no cytokines, IFN‐α, IL‐2 plus IL‐27 or IFN‐α plus IL‐2 and IL‐27). (A) Production of IL‐10 by CD8+ T cells and (B) percentages of IL‐10‐producing CD8+ T cells were determined by flow cytometry total of four experiments. (C) IL‐10 gene expression in CD8+ T cells was determined by normalizing the data to HPRT expression through real‐time RT‐PCR. (D) IL‐10 protein production in the supernatants following αCD3 restimulation of CD8+ T cells was determined by ELISA. Data (A, C, and D) are from a single experiment representative of three to four independent experiments with one to two mice per experiment. Data (B) are pooled from total of four experiments with one to two mice per experiment. Statistics were determined by paired two‐tailed Student's t‐test (∗ p < 0.05).
Blimp‐1 and IRF4 are required for the development of IL‐10‐producing effector CD8+ T cells
We next wished to determine the molecular mechanisms by which type I IFNs cooperate with IL‐2 and IL‐27 to promote the development of IL‐10‐producing CD8+ T cells. We have shown previously that Blimp‐1 is required for the development of IL‐10‐producing CD8+ T cells in vitro and in vivo during influenza infection 6. We now observed that type I IFNs cooperated with IL‐2 and IL‐27 to induce the expression of Blimp‐1 in CD8+ T cells (Fig. 6A and B). To determine whether Blimp‐1 is required for the induction of IL‐10‐producing CD8+ T cells in vitro, we cultured WT or Blimp‐1‐deficient CD8+ T cells in the absence or presence of IFN‐α, IL‐2/IL‐27, or IFN‐α/IL‐2/IL‐27. We found that while IFN‐α/IL‐2/IL‐27 induced abundant IL‐10‐producing CD8+ T cells within WT CD8+ T cells, these cytokine failed to stimulate IL‐10 production by Blimp‐1‐deficient CD8+ T cells (Fig. 6C). Furthermore, Blimp‐1 was also required for the secretion of IL‐10 by CD8+ T cells (Fig. 6D). Thus, these data suggest that Blimp‐1 is required for the production of IL‐10 by CD8+ T cells upon type I IFN signaling, consistent with the our previous findings that Blimp‐1 is essential for the development of IL‐10‐producing CD8+ T cells in vivo during influenza infection 6.
Figure 6.
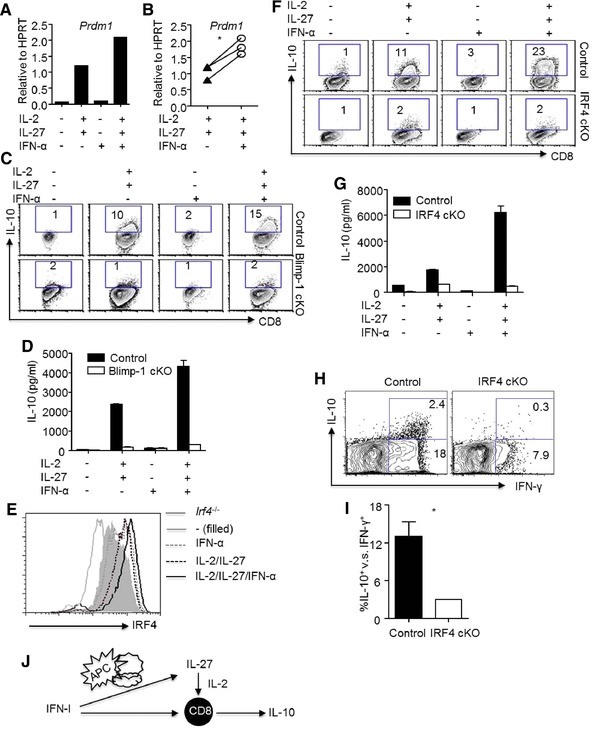
Blimp‐1 and IRF4 are required for the development of IL‐10‐producing effector CD8 + T cells induced by type I IFNs plus IL‐2 and IL‐27. CD8+ T cells were cultured under indicated conditions (no cytokines, IFN‐α, IL‐2 plus IL‐27 or IFN‐α plus IL‐2 and IL‐27). (A) Prdm1 (Blimp‐1 gene) expression in CD8+ T was determined by normalizing the data to HPRT expression through real‐time RT‐PCR. (B) Prdm1 expression in CD8+ T cells was determined by normalizing the data to HPRT expression through real‐time RT‐PCR in total of three experiments. (C) Production of IL‐10 by control or Blimp‐1 cKO CD8+ T cells was determined by flow cytometry. (D) IL‐10 protein production in the supernatants following restimulation of control or Blimp‐1 cKO CD8+ T cells cultured with indicated conditions was determined by ELISA. (E) IRF4 expression in CD8+ T cells cultured with indicated conditions was determined by flow cytometry. (F) Production of IL‐10 by control or IRF4 cKO CD8+ T was determined by flow cytometry. (G) IL‐10 protein production in the supernatants following restimulation of WT or IRF4 cKO CD8+ T was determined by ELISA. (H) Production of IL‐10 and IFN‐γ by lung CD8+ T cells from WT or IRF4 cKO mice was determined by flow cytometry following in vitro antigenic stimulation with influenza‐infected WT BMDCs. (I) Normalized percentages of IL‐10+ cells in influenza‐specific lung CD8+ T cells (IFN‐γ+) from infected control or IRF4 cKO mice were determined by flow cytometry. (J) Model of type‐I‐IFN‐dependent IL‐10 production by CD8+ T cells. Data (A, C, D, E, F, and G) are from a single experiment representative of two to three independent experiments with one to two mice per experiment. Data (B) are pooled from total of three experiments with one to two mice per experiment. Data (H) in density plots are from a single experiment representative of two independent experiments with two to three mice per experiment. Data (I) in graphs are shown as mean + SEM and are representative of two separate experiments with two to three mice per experiment. Statistics were determined by paired (B) or unpaired (I) two‐tailed Student's t‐test (∗ p < 0.05).
We recently reported that IRF4 controls Blimp‐1 expression in CD8+ T cells 29. IRF4 was also shown to regulate IL‐10 production by effector CD4+ T cells and Foxp3+ regulatory CD4+ T cells 30, 31. We therefore examined whether type I IFNs signaling promoted IRF4 expression. We found that IFN‐α was able to cooperate with IL‐2/IL‐27 to promote and sustain IRF4 expression in CD8+ T cells (Fig. 6E and Supporting Information Fig. 4). Furthermore, we found that, following the treatment of IFN‐α/IL‐2/IL‐27, IRF4 expression in CD8+ T cells was required for the production of IL‐10 by CD8+ T cells (Fig. 6F and G). To determine whether IRF4 is required for the induction of IL‐10‐producing CD8+ T cells in vivo, we infected CD8+ T‐cell‐specific IRF4‐deficient mice (IRF4 conditional KO (cKO)) with influenza and examined IL‐10 and IFN‐γ production following in vitro restimulation. Consistent with our previous data 29, selective IRF4‐deficiency in CD8+ T cells diminished IFN‐γ production by CD8+ T cells, suggesting IRF4 is required for the development of antiviral effector CD8+ T cells (Fig. 6H). When IL‐10‐producing CD8+ T cells were normalized to IFN‐γ producing effector CD8+ T cell population, we found that IL‐10 production was diminished in effector CD8+ T cells in the absence of IRF4 (Fig. 6I), suggesting that IRF4 is vital for the production of IL‐10 by effector CD8+ T cells in vivo.
Discussion
In this report, we have investigated the role of type I IFN signaling in regulating the development of IL‐10‐producing effector CD8+ T cells during influenza infection. In accordance with previous observations 20, we found that IFNAR1‐deficient mice exhibited diminished IL‐10 production during influenza virus infection. We further showed that the effect of IFNAR1 deficiency on IL‐10 production was more pronounced on effector CD8+ T cells rather than effector CD4+ T cells. We have also defined the downstream cellular and molecular mechanisms by which type I IFNs control the development of IL‐10‐producing effector CD8+ T cells during influenza virus infection. We found that both indirect and direct effects of type I IFN signaling in CD8+ T cells were required for the optimal induction of IL‐10‐producing CD8+ T cells during influenza infection (Fig. 6I).
Type I IFNs are major antiviral effector cytokines that play important roles in restricting the replication and dissemination of respiratory viruses in vitro and in vivo 24. Besides their direct antiviral functions, type I IFNs are well known to regulate the development of innate and adaptive immunity during respiratory virus infections 32, 33. Early type I IFN signaling was important in the production of inflammatory cytokines during influenza infection 34, 35. Furthermore, type I IFN signaling plays a critical role in regulating the development antiviral CD4+ and CD8+ cytotoxic T lymphocytes during influenza infection 21. Consistent with their role in promoting inflammation and immunity, overproduction of type I IFNs contributed significantly to the pathogenesis of influenza virus and mouse‐adapted coronavirus infection 36, 37, 38. On the other hand, our data here and results from Arimori et al. have also suggested that type I IFNs could have regulatory role by restricting exuberant inflammatory responses through the induction of the antiinflammatory cytokine IL‐10 20. Indeed, IFNAR1‐deficient mice showed an exuberant host inflammatory response and IL‐10 administration could ameliorate excessive lethal inflammation and associated mortality following influenza infection in IFNAR1‐deficient mice 20. Such a regulatory role of type I IFN signaling in inflammation and immunity has been documented during chronic viral infections 17, 18. Thus, type I IFNs could display differing roles in regulating the development of inflammatory responses and immunity following viral infections, and their exact function in regulating inflammation and immunity would be dependent on the infectious organism as well as the timing and levels of the production of these pluripotent antiviral cytokines.
In a similar way, IL‐10 exerts its function in a timing‐dependent fashion during influenza infection. Early production of IL‐10 during influenza infection could hamper the activation of antiviral innate and adaptive immunity as evidenced by the enhanced resistance to influenza virus infection in IL‐10‐deficient mice 39, 40, 41. Conversely, the production of IL‐10 at the time of effector T‐cell infiltration into the lung (i.e., day 5 and thereafter) could suppress the development of lethal pulmonary inflammation during influenza infection 8, 41. Similar beneficial effects of T‐cell‐derived IL‐10 have also been reported during RSV and coronavirus infections 7, 9, 10, 11. Currently, the role of type I IFNs in regulating IL‐10 production by human CD8+ T cells has not been investigated. However, a recent clinical study has established that both type I IFNs and IL‐10 were elevated in patients with severe influenza virus infection 42. These data suggested that type I IFNs might facilitate IL‐10 production by effector T cells during human influenza infection. Whether IL‐10 production is beneficial to the host by keeping inflammation under control or is detrimental to the host by suppressing antiviral immunity in these patients warrants further investigation. However, understanding the timing‐ and dose‐dependent regulating of pulmonary inflammation and immunity by type I IFNs and IL‐10 will be insightful for developing novel therapeutic strategies aiming to dampen excessive respiratory inflammation and diseases during severe influenza infection.
Type I IFNs have been shown to promote macrophage IL‐10 production through the induction of the cytokine IL‐27 26. Similarly, we found that type I IFNs could stimulate IL‐27 expression in major lung APCs during influenza virus infection, including macrophages, DCs, and neutrophils. Furthermore, IL‐27 levels were significantly diminished in the respiratory tract in the absence of type I IFN signaling. Thus, type I IFNs could promote IL‐10 production by CD8+ T cells through its indirect effects on regulating IL‐27 levels in vivo. Surprisingly, through the mixed BM chimera experiments, we have also established that direct type I IFN signaling is important in IL‐10 production by CD8+ T cells during influenza infection. Currently, the relative contribution of the indirect and direct effects of type I IFN signaling in CD8+ T cells in promoting IL‐10 production remains to be determined. Further experiments such as supplementation of influenza‐infected IFNAR‐deficient mice with recombinant IL‐27 would be interesting to evaluate with respect to the induction of IL‐10‐producing CD8 T cells. Nevertheless, type I IFN signaling substantially enhances IL‐10 by CD8+ T cells in vitro in the presence of IL‐2 and IL‐27. At present, the mechanism(s) by which type I IFNs cooperate with IL‐2 and IL‐27 to promote IL‐10 production is unknown. Type I IFN signaling has been shown to sustain the expression of high affinity IL‐2 receptor on CD8+ T‐cell surface 43. So, it is possible that type I IFN signaling promotes IL‐10 production by facilitating IL‐2 and/or IL‐27 signaling in CD8+ T cells. Conversely, it is possible that IL‐2 and IL‐27 treatment could boost IFNAR1 expression and/or its downstream signaling to promote IL‐10 expression.
Mechanistically, we found that type I IFN signaling promotes IL‐10 production through the concerted action of STAT2, IRF4, and Blimp‐1. In a previous report, we identified IRF4 expression as essential for the induction of Blimp‐1 and its downstream antiviral effector cytokine molecules in CD8+ T cells 29. By normalizing IL‐10‐producing cells to IFN‐γ‐producing cells, we showed that IRF4 is also critically important for IL‐10 production by CD8+ T cells. We speculate that IRF4 functions upstream of Blimp‐1 to promote IL‐10 expression in CD8+ T cells. However, it is also possible that IRF4 could directly bind to the IL‐10 locus to promote IL‐10 production in CD8+ T cells since it was shown that the IL‐10 locus contains IRF4‐binding motifs 30. The strength of TCR signaling (i.e., high antigen dose and strong peptide affinity) was also shown to be critical for IRF4 expression in CD8+ T cells. Here we have extended previous findings by demonstrating that IRF4 expression is also modulated by cytokine signaling (i.e., type I IFNs). Of note, high‐dose antigen stimulation has been shown to favor IL‐10 production by CD4+ T cells 44. We have shown previously that IL‐10 production by CD8+ T cells was highly restricted to the early time frame following virus‐specific T‐cell infiltration to the lungs during influenza infection 8, at the time when virus‐derived antigens are plentiful in the infected lungs. Thus, it is possible that both TCR‐derived signaling and type‐I‐IFN‐derived signaling are required for the optimal induction of IRF4 and downstream IL‐10 production in vivo. These possibilities warrant further investigations.
In summary, our data have revealed potential regulatory roles of type I IFN signaling in inflammatory responses during acute viral infections, and may provide the groundwork for manipulating IL‐10 production in effector CD8+ T cells to control excessive host inflammation during acute respiratory virus infection.
Materials and methods
Mouse and infection
WT C57/BL6 mice were purchased from the Jackson Laboratory; Blimp‐1 control (Prdm1fl /fl, where Prdm is PR domain 1), CD4‐Cre transgenic, CD8‐cre transgenic, IRF4 control (Irf4fl /fl), and CD45.1 congenic mice were originally from the Jackson Laboratory and bred inhouse. T‐cell‐specific Blimp‐1 cKO mice were generated through breeding CD4‐cre to Prdm1fl /fl mice. CD8 T‐cell‐specific IRF4 cKO mice were generated through breeding CD8‐cre to Irf4fl /fl mice. IFNAR1‐deficient (Ifnar1 −/−) mice were originally from Dr. U. Deshmukh at the University of Virginia; Vert‐X mice were originally from Dr. C. Karp from Cincinnati Children's Hospital; STAT2‐deficient (Stat2 −/−) mice were originally from Dr. C. Schindler at the Columbia University. All mice were housed in a specific pathogen‐free environment and all animal experiments were performed in accordance with protocols approved by the University of Virginia Animal Care and Use Committee (ACUC) or Indiana University Institutional Animal Care and Use Committee (IACUC). For influenza virus infection, mice were anesthetized first and then intranasally infected with Influenza A/PR8/34 strain (∼150 pfu/mouse in serum‐free IMDM media (Gibco) or PBS) as previously reported 8.
Quantitative RT‐PCR
Lung single‐cell suspensions were prepared as previously described 6, 21. T cells were purified through positive selection via MACS‐beads (Miltenyi Biotech) from pooled two to three lungs per group. mRNA from in vivo purified cells or in vitro cultured cells as indicated was isolated with RNeasy kit (Qiagen) or total RNA isolation kit (Sigma Aldrich) and treated with DNase I (Invitrogen). Random primers (Invitrogen) and Superscript II (Invitrogen) were used to synthesize first‐strand cDNAs from equivalent amounts of RNA from each sample. RT‐PCR was performed with SYBR Green PCR Master Mix (Applied Biosystems). Data were generated with the comparative threshold cycle (Delta CT) method by normalizing to hypoxanthine‐guanine phosphoribosyltransferase (HPRT). Sequences of primers used in the studies are available on request.
In vitro DC/T‐cell coculture
BMDCs were generated as described 22. CD8+ T cells were isolated from spleen and LNs of indicated mice through MACS‐beads (Miltenyi Biotech). BMDC and T‐cell coculture was performed as previously described 6. Briefly, we mixed DC with CD8+ T cells at the ratio of 1 DC: 10 T cells in round‐bottom 96 wells (5 × 104 T cells/well) in the presence of 0.1 μg/mL α‐CD3. The conditions of the culture are indicated in the text. For RNA isolation and RT‐PCR, T cells were harvested at day 3 of culture. For IFN‐γ and IL‐10 staining, T cells were harvested at day 4 of the culture and restimulated with PMA (100 ng/mL, Sigma) or inomycin (1 μg/mL, Sigma) in the presence of Golgi‐Stop (1 μl/mL, BD Biosciences) for 4 h. Intracellular cytokine staining (ICS) was performed as we previously reported 6. To measure T‐cell cytokine production by ELISA, day 4 cultured T cells were restimulated with plate bound α‐CD3 (1 μg/mL) for overnight. Supernatants were then measured by ELISA as previously described 6. The concentrations of the cytokines used in the culture are as following: recombinant human IL‐2 (300 U/mL), mouse IL‐27 (Biolegend, 10 ng/mL), and IFN‐αA (R&D, 250 U/mL), respectively.
Antigenic stimulation of lung effector T cells
On day 6–7 of WT BMDC culture, BMDCs were harvested and infected with influenza virus at approximated 100 MOI for 6 h. Then BMDCs were counted and mixed with total lung cells isolated from WT and transgenic mice as indicated in the text at a 1.5:1 ratio in the presence of Golgi‐Stop (BD Biosciences, 1μL/mL) and hIL‐2 (40 U/mL) for additional 6 h. The surface staining of cell surface markers and intracellular staining of cytokines were performed as described 8.
Generation of mixed BM chimeras
To generate mixed BM chimera, we lethally irradiated (1100 Rads) WT mice and then i.v. injected ∼4 million 1:1 mixed BM cells from WT (CD45.1+) or CD45.2+ IFNAR1‐deficient BM. After 12‐week reconstitution, the chimeric mice were then infected with influenza A/PR8.
Bronchoalveolar lavage (BALF) cytokine determination
BALF was obtained by flushing the airway three to four times with a single use of 600 μL sterile PBS as previous described 8. Cells in BALF were spun down and supernatants were collected for multiplex analysis (Millipore) according to the manufacturer manuals.
Cell sorting
Lung single‐cell suspensions were prepared from uninfected or infected mice (day 5 p.i.). Cells were stained with CD45, F4/80, CD11C, MHCII, CD11b, and Ly6G. Lung eipthelia/stroma cells (CD45−), neutrophils (CD11b+ Ly6G+), DCs (CD11chi MHCIIhi Ly6G−), and macrophages (F4/80+ MHCII+ Ly6G−) were sorted based on their surface marker expression described above.
FACS analysis
All FACS Abs were purchased from Biolegend, BD Biosciences, or eBioscience. Cells were acquired through FACS‐Calibur, FACS‐Canto, or LSR II (BD Biosciences). Data were then analyzed by FlowJo software (Treestar).
Statistical analysis
Data are mean ± SEM. Paired or unpaired two‐tailed Student's t‐test was used. p‐Value <0.05 is considered significant.
Conflicts of interest
The authors declare no financial or commercial conflict of interest.
Abbreviations
- Blimp‐1
B‐lymphocyte‐induced maturation protein‐1
- BMDC
BM‐derived DC
- cKO
conditional KO
- IFNAR1
IFN‐α receptor 1
- IRF4
IFN regulatory factor 4
- Vert‐X
IL‐10‐eGFP reporter mouse (Vert, fr. green; X, roman numeral 10)
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Figure 1: Type I IFN signaling promotes optimal IL‐10 production by CD8+ T cells.
(A) WT and Ifnar1‐/‐ mice were infected with influenza PR8. IL‐10 MFI of lung IL‐10‐producing CD8+ T cells was determined at day 7 post infection.
(B). WT and Stat2‐/‐ mice were infected with influenza PR8. IL‐10 MFI of lung IL‐10‐producing CD8+ T cells was determined at day 7 post infection. (C) WT Vert‐X or Ifnar1‐/‐ Vert‐X mice were infected with influenza. IL‐10‐EGFP MFI in IL‐10‐eGFP+ of lung CD4+ T cells from WT Vert‐X or Ifnar1‐/‐ Vert‐X mice was measured through flow cytometry at day 7 post infection. Data are representative of at least two separate experiments. ∗ P < 0.05.
Figure 2: Type I IFN signaling promotes IL‐27 p28 gene expression by APCs
Purified bone marrow neutrophils (A), bone marrow‐derived macrophages (B) or bone marrow‐derived dendritic cells (C) were treated with IFN‐α and IL‐27 subunit gene expression was determined through real‐time RT‐PCR.
Data are representative of two separate experiments.
Figure 3: Direct type I IFN signaling is modestly required for IL‐10 production by CD4+ T cells during influenza infection.
WT and Ifnar1‐/‐ mixed bone marrow chimera mice (n=5) were infected with influenza. A. Reconstitution rates of WT (CD45.1+) and Ifnar1‐/‐ deficient (CD45.2+) cells in the T cell compartments at day 7 following influenza infection.
B. IL‐10 production by lung CD4+ T cells was determined at day 7 post infection.
Data are representative of two experiments
Figure 4: Type I IFN signaling co‐operates with IL‐2 and IL‐27 to sustain IRF4 expression in CD8+ T cells.
IRF4 MFI at different days post activation in CD8+ T cells cultured with indicated conditions.
Data are representative of two experiments
Acknowledgments
We thank U. Deshmukh and C. Schindler for mice. We thank NIH tetramer facility for tetramer reagents. This work was supported by the U.S. National Institutes of Health (grants AI099753, AI112844, and AG047156 to J.S.; AI083024 to T.J.B), Indiana University Biomedical Research Grant (to J.S.), and American Lung Association Postdoctoral Fellowship Award (RT‐310817 to S.Y.).
Current address: Li Jiang, Su Huang, and Lie Sun; Thoracic Disease Research Unit; Division of Pulmonary and Critical Care Medicine; Mayo Clinic College of Medicine; Rochester; MN55905, USA
References
- 1. Zhang, N. and Bevan, M. J. , CD8(+) T cells: foot soldiers of the immune system. Immunity 2011. 35: 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Braciale, T. J. , Sun, J. and Kim, T. S. , Regulating the adaptive immune response to respiratory virus infection. Nat. Rev. Immunol. 2012. 12: 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim, T. S. , Sun, J. and Braciale, T. J. , T cell responses during influenza infection: getting and keeping control. Trends Immunol. 2011. 32: 225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hufford, M. M. , Kim, T. S. , Sun, J. and Braciale, T. J. , The effector T cell response to influenza infection. Curr. Top Microbiol. Immunol. 2015. 386: 423–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun, J. and Braciale, T. J. , Role of T cell immunity in recovery from influenza virus infection. Curr. Opin. Virol. 2013. 3: 425–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sun, J. , Dodd, H. , Moser, E. K. , Sharma, R. and Braciale, T. J. , CD4+ T cell help and innate‐derived IL‐27 induce Blimp‐1‐dependent IL‐10 production by antiviral CTLs. Nat. Immunol. 2011. 12: 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun, J. , Cardani, A. , Sharma, A. K. , Laubach, V. E. , Jack, R. S. , Muller, W. and Braciale, T. J. , Autocrine regulation of pulmonary inflammation by effector T‐cell derived IL‐10 during infection with respiratory syncytial virus. PLoS Pathog. 2011. 7: e1002173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun, J. , Madan, R. , Karp, C. L. and Braciale, T. J. , Effector T cells control lung inflammation during acute influenza virus infection by producing IL‐10. Nat. Med. 2009. 15: 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trandem, K. , Zhao, J. , Fleming, E. and Perlman, S. , Highly activated cytotoxic CD8 T cells express protective IL‐10 at the peak of coronavirus‐induced encephalitis. J. Immunol. 2011. 186: 3642–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Loebbermann, J. , Schnoeller, C. , Thornton, H. , Durant, L. , Sweeney, N. P. , Schuijs, M. , O'Garra, A. et al., IL‐10 regulates viral lung immunopathology during acute respiratory syncytial virus infection in mice. PLoS One 2012. 7: e32371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Palmer, E. M. , Holbrook, B. C. , Arimilli, S. , Parks, G. D. and Alexander‐Miller, M. A. , IFNgamma‐producing, virus‐specific CD8+ effector cells acquire the ability to produce IL‐10 as a result of entry into the infected lung environment. Virology 2010. 404: 225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stevens, W. W. , Sun, J. , Castillo, J. P. and Braciale, T. J. , Pulmonary eosinophilia is attenuated by early responding CD8(+) memory T cells in a murine model of RSV vaccine‐enhanced disease. Viral Immunol. 2009. 22: 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mescher, M. F. , Curtsinger, J. M. , Agarwal, P. , Casey, K. A. , Gerner, M. , Hammerbeck, C. D. , Popescu, F. et al., Signals required for programming effector and memory development by CD8+ T cells. Immunol. Rev. 2006. 211: 81–92. [DOI] [PubMed] [Google Scholar]
- 14. Kim, M. T. and Harty, J. T. , Impact of inflammatory cytokines on effector and memory CD8+ T cells. Front Immunol. 2014. 5: 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu, H. C. , Grusdat, M. , Pandyra, A. A. , Polz, R. , Huang, J. , Sharma, P. , Deenen, R. et al., Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity 2014. 40: 949–960. [DOI] [PubMed] [Google Scholar]
- 16. Crouse, J. , Bedenikovic, G. , Wiesel, M. , Ibberson, M. , Xenarios, I. , Von Laer, D. , Kalinke, U. Vivier, E. , Jonjic, S. and Oxenius, A. , Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity 2014. 40: 961–973. [DOI] [PubMed] [Google Scholar]
- 17. Wilson, E. B. , Yamada, D. H. , Elsaesser, H. , Herskovitz, J. , Deng, J. , Cheng, G. , Aronow, B. J. et al., Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013. 340: 202–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Teijaro, J. R. , Ng, C. , Lee, A. M. , Sullivan, B. M. , Sheehan, K. C. , Welch, M. , Schreiber, R. D. et al., Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013. 340: 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arimori, Y. , Nakamura, R. , Yamada, H. , Shibata, K. , Maeda, N. , Kase, T. and Yoshikai, Y. , Type I interferon plays opposing roles in cytotoxicity and interferon‐gamma production by natural killer and CD8 T cells after influenza A virus infection in mice. J. Innate Immun. 2014. 6: 456–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arimori, Y. , Nakamura, R. , Yamada, H. , Shibata, K. , Maeda, N. , Kase, T. and Yoshikai, Y. , Type I interferon limits influenza virus‐induced acute lung injury by regulation of excessive inflammation in mice. Antiviral Res. 2013. 99: 230–237. [DOI] [PubMed] [Google Scholar]
- 21. Hua, L. , Yao, S. , Pham, D. , Jiang, L. , Wright, J. , Sawant, D. , Dent, A. L. et al., Cytokine‐dependent induction of CD4+ T cells with cytotoxic potential during influenza virus infection. J. Virol. 2013. 87: 11884–11893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun, J. and Pearce, E. J. , Suppression of early IL‐4 production underlies the failure of CD4 T cells activated by TLR‐stimulated dendritic cells to differentiate into Th2 cells. J. Immunol. 2007. 178: 1635–1644. [DOI] [PubMed] [Google Scholar]
- 23. Park, C. , Li, S. , Cha, E. and Schindler, C. , Immune response in Stat2 knockout mice. Immunity 2000. 13: 795–804. [DOI] [PubMed] [Google Scholar]
- 24. Ivashkiv, L. B. and Donlin, L. T. , Regulation of type I interferon responses. Nat. Rev. Immunol. 2014. 14: 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Madan, R. , Demircik, F. , Surianarayanan, S. , Allen, J. L. , Divanovic, S. , Trompette, A. , Yogev, N. et al., Nonredundant roles for B cell‐derived IL‐10 in immune counter‐regulation. J. Immunol. 2009. 183: 2312–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iyer, S. S. , Ghaffari, A. A. and Cheng, G. , Lipopolysaccharide‐mediated IL‐10 transcriptional regulation requires sequential induction of type I IFNs and IL‐27 in macrophages. J. Immunol. 2010. 185: 6599–6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hufford, M. M. , Richardson, G. , Zhou, H. , Manicassamy, B. , Garcia‐Sastre, A. , Enelow, R. I. and Braciale, T. J. , Influenza‐infected neutrophils within the infected lungs act as antigen presenting cells for anti‐viral CD8(+) T cells. PLoS One 2012. 7: e46581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hufford, M. M. , Kim, T. S. , Sun, J. and Braciale, T. J. , Antiviral CD8+ T cell effector activities in situ are regulated by target cell type. J. Exp. Med. 2011. 208: 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yao, S. , Buzo, B. F. , Pham, D. , Jiang, L. , Taparowsky, E. J. , Kaplan, M. H. and Sun, J. , Interferon regulatory factor 4 sustains CD8(+) T cell expansion and effector differentiation. Immunity 2013. 39: 833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ahyi, A. N. , Chang, H. C. , Dent, A. L. , Nutt, S. L. and Kaplan, M. H. , IFN regulatory factor 4 regulates the expression of a subset of Th2 cytokines. J. Immunol. 2009. 183: 1598–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cretney, E. , Xin, A. , Shi, W. , Minnich, M. , Masson, F. , Miasari, M. , Belz, G. T. et al., The transcription factors Blimp‐1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat. Immunol. 2011. 12: 304–311. [DOI] [PubMed] [Google Scholar]
- 32. McNab, F. , Mayer‐Barber, K. , Sher, A. , Wack, A. and O'Garra, A. , Type I interferons in infectious disease. Nat. Rev. Immunol. 2015. 15: 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Garcia‐Sastre, A. , Induction and evasion of type I interferon responses by influenza viruses. Virus Res. 2011. 162: 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moltedo, B. , Lopez, C. B. , Pazos, M. , Becker, M. I. , Hermesh, T. and Moran, T. M. , Cutting edge: stealth influenza virus replication precedes the initiation of adaptive immunity. J. Immunol. 2009. 183: 3569–3573. [DOI] [PubMed] [Google Scholar]
- 35. Teijaro, J. R. , Walsh, K. B. , Cahalan, S. , Fremgen, D. M. , Roberts, E. , Scott, F. , Martinborough, E. et al., Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011. 146: 980–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kobasa, D. , Jones, S. M. , Shinya, K. , Kash, J. C. , Copps, J. , Ebihara, H. , Hatta, Y. et al., Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 2007. 445: 319–323. [DOI] [PubMed] [Google Scholar]
- 37. Hogner, K. , Wolff, T. , Pleschka, S. , Plog, S. , Gruber, A. D. , Kalinke, U. , Walmrath, H. D. et al., Macrophage‐expressed IFN‐beta contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PLoS Pathog. 2013. 9: e1003188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Channappanavar, R. , Fehr, A. R. , Vijay, R. , Mack, M. , Zhao, J. , Meyerholz, D. K. and Perlman, S. , Dysregulated type i interferon and inflammatory monocyte‐macrophage responses cause lethal pneumonia in SARS‐CoV‐infected mice. Cell Host Microbe 2016. 19: 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun, K. , Torres, L. and Metzger, D. W. , A detrimental effect of interleukin‐10 on protective pulmonary humoral immunity during primary influenza A virus infection. J. Virol. 2010. 84: 5007–5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McKinstry, K. K. , Strutt, T. M. , Buck, A. , Curtis, J. D. , Dibble, J. P. , Huston, G. , Tighe, M. et al., IL‐10 deficiency unleashes an influenza‐specific Th17 response and enhances survival against high‐dose challenge. J. Immunol. 2009. 182: 7353–7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dutta, A. , Huang, C. T. , Chen, T. C. , Lin, C. Y. , Chiu, C. H. , Lin, Y. C. , Chang, C. S. et al., IL‐10 inhibits neuraminidase‐activated TGF‐beta and facilitates Th1 phenotype during early phase of infection. Nat. Commun. 2015. 6: 6374. [DOI] [PubMed] [Google Scholar]
- 42. Oshansky, C. M. , Gartland, A. J. , Wong, S. S. , Jeevan, T. , Wang, D. , Roddam, P. L. , Caniza, M. A. et al., Mucosal immune responses predict clinical outcomes during influenza infection independently of age and viral load. Am. J. Respir. Crit. Care Med. 2014. 189: 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Starbeck‐Miller, G. R. , Xue, H. H. and Harty, J. T. , IL‐12 and type I interferon prolong the division of activated CD8 T cells by maintaining high‐affinity IL‐2 signaling in vivo. J. Exp. Med. 2014. 211: 105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Saraiva, M. , Christensen, J. R. , Veldhoen, M. , Murphy, T. L. , Murphy, K. M. and O'Garra, A. , Interleukin‐10 production by Th1 cells requires interleukin‐12‐induced STAT4 transcription factor and ERK MAP kinase activation by high antigen dose. Immunity 2009. 31: 209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Figure 1: Type I IFN signaling promotes optimal IL‐10 production by CD8+ T cells.
(A) WT and Ifnar1‐/‐ mice were infected with influenza PR8. IL‐10 MFI of lung IL‐10‐producing CD8+ T cells was determined at day 7 post infection.
(B). WT and Stat2‐/‐ mice were infected with influenza PR8. IL‐10 MFI of lung IL‐10‐producing CD8+ T cells was determined at day 7 post infection. (C) WT Vert‐X or Ifnar1‐/‐ Vert‐X mice were infected with influenza. IL‐10‐EGFP MFI in IL‐10‐eGFP+ of lung CD4+ T cells from WT Vert‐X or Ifnar1‐/‐ Vert‐X mice was measured through flow cytometry at day 7 post infection. Data are representative of at least two separate experiments. ∗ P < 0.05.
Figure 2: Type I IFN signaling promotes IL‐27 p28 gene expression by APCs
Purified bone marrow neutrophils (A), bone marrow‐derived macrophages (B) or bone marrow‐derived dendritic cells (C) were treated with IFN‐α and IL‐27 subunit gene expression was determined through real‐time RT‐PCR.
Data are representative of two separate experiments.
Figure 3: Direct type I IFN signaling is modestly required for IL‐10 production by CD4+ T cells during influenza infection.
WT and Ifnar1‐/‐ mixed bone marrow chimera mice (n=5) were infected with influenza. A. Reconstitution rates of WT (CD45.1+) and Ifnar1‐/‐ deficient (CD45.2+) cells in the T cell compartments at day 7 following influenza infection.
B. IL‐10 production by lung CD4+ T cells was determined at day 7 post infection.
Data are representative of two experiments
Figure 4: Type I IFN signaling co‐operates with IL‐2 and IL‐27 to sustain IRF4 expression in CD8+ T cells.
IRF4 MFI at different days post activation in CD8+ T cells cultured with indicated conditions.
Data are representative of two experiments