

Abstract
Orai proteins form highly calcium (Ca2+)-selective channels located in the plasma membrane of both non-excitable and excitable cells, where they make important contributions to many cellular processes. The well-characterized Ca2+ release-activated Ca2+ (CRAC) current is mediated by Orai1 multimers and is activated, upon depletion of inositol 1,4,5-trisphosphate (IP3)-sensitive stores, by direct interaction of Orai1 with the endoplasmic reticulum (ER) Ca2+ sensor, stromal interaction molecule 1 (STIM1). This pathway is known as capacitative Ca2+ entry or store-operated Ca2+ entry (SOCE). While most investigations have focused on STIM1 and Orai1 in their store-dependent mode, emerging evidence suggest that Orai1 and Orai3 heteromultimeric channels can form store-independent Ca2+ selective channels. The role of store-dependent and store-independent channels in excitation-transcription coupling and the pathological remodeling of the cardiovascular system are beginning to come forth. Recent evidence suggests that STIM/Orai-generated Ca2+ signaling couples to gene transcription and subsequent phenotypic changes associated with the processes of cardiac and vascular remodeling. This short review will explore the contributions of native Orai channels to heart and vessel physiology and their role in cardiovascular diseases.
Introduction
Ion channels are membrane proteins with essential functions in a variety of physiological and pathophysiological situations. Defective ion channel function produces clinical syndromes typically termed channelopathies. Mammalian Orai1 proteins (mammals have three Orai genes, Orai1 through 3) are evolutionarily conserved calcium (Ca2+) channels displaying a remarkably high selectivity for Ca2+ [1-4] The channelopathy resulting from Orai1 deficiency is primarily characterized by a severe combined immunodeficiency (SCID)-like phenotype attributed to an impairment in T-cell activation; other notable findings include congenital myopathy and anhidrotic ectodermal dysplasia with defective dental enamel calcification [5]. Mice Homozygous for Orai1 deficiency (Orai1−/−) generated in a mixed ICR genetic background have a phenotype analogous to CRAC-deficient patients bearing the Orai1 homozygous mutation R91W. Orai1−/− mice in a pure inbred C57BL/6 background are characterized by perinatal lethality, presumably from defective skeletal muscle development [6-8]. Despite Orai1 channelopathy being mainly associated with defects of the immune system and skeletal muscle, there is emerging evidence that Orai1 channels (and their homologues) are important to homeostatic cell function as well as disease in almost every cell type, including cardiac and vascular tissues. Thus, the lack of obvious cardiovascular defects in Orai1 channelopathy would suggest that Orai channels likely become more relevant under stressful or pathophysiological conditions. As argued earlier, compensatory mechanisms by Orai2 or Orai3 in Orai1−/− mice are unlikely and all three Orai isoforms probably serve non-redundant functions [9, 10].
Orai channels
Store-operated Ca2+ (SOC) channels
A variety of growth factors, neurohormonal stimuli and inflammatory mediators induce store-operated Ca2+ entry (SOCE) activity. Ligation of agonists to G-protein coupled receptors (GPCR) or receptor tyrosine kinases activates isoforms of phospholipase C (PLC) enzymes, which convert phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). IP3 diffuses into the cytosol and depletes endoplasmic reticulum (ER) Ca2+ stores by opening IP3 receptor (IP3R) Ca2+ release channels. The drop in Ca2+ concentration within the ER lumen activates the single transmembrane ER-resident Ca2+-sensor protein STIM1. When STIM1 loses Ca2+ from its Ca2+-binding EF hand, it undergoes a conformational change that prompts STIM1 oligomerization in areas adjacent to the plasma membrane where its C-terminus achieves an extended conformation to directly gate CRAC channels formed by multimers of Orai1 [1, 3, 4]; this gating involves the binding of a minimal 100 amino acid C-terminal region of STIM1 (called SOAR for STIM/Orai activating region) to both the C- and N-terminus of Orai1 [11, 12]. A homozygous missense mutation in human Orai1, changing amino acid 91 from arginine to tryptophan (R91W) is responsible for defective SOCE observed in a subpopulation of patients with hereditary SCID [1]. Restoration of SOCE in immune cells isolated from these patients was achieved by ectopically expressing wild-type Orai1 [1]. Experiments involving mutagenesis of conserved acidic residues lining the putative pore region further strengthened the notion of Orai1 as an essential pore subunit of CRAC/SOCE [2, 13]. It was also realized that patients with deficient STIM1 function displayed a clinical phenotype similar to that seen in Orai1-deficient patients, including immunodeficiency, nonprogressive myopathy, and malformation of dental enamel [14]. CRAC channels deliver sustained Ca2+ signals essential for a multitude of cellular processes; Ca2+ delivered intracellularly through CRAC channels likely serves to replenish depleted internal stores, thus maintaining a relatively stable concentration of Ca2+ in the lumen of organelles, such as ER and mitochondria, essential for cell homeostasis. A more important function of CRAC is likely the generation of spatial and temporal Ca2+ gradients crucial for activation of specific cell signaling pathways and subsequent gene transcription to coordinate complex long-term cell functions such as cell secretion, migration, growth and proliferation [15-17]. Nuclear factor for activated T cells (NFAT) has been uniformly shown to depend on CRAC channel activity for its nuclear translocation and activity in a number of cell types [18].
There are two human homologues of Drosophila STIM (STIM1 and STIM2). Compared to STIM1, the function of STIM2 remains relatively less understood. STIM2 is a more sensitive Ca2+ sensor and appears to be a weaker activator of CRAC channels compared to STIM1 [19]. STIM2 was proposed to play a role in maintaining ER Ca2+ homeostasis [20]. However, STIM2 is likely endowed with broader signaling functions that remain to be elucidated. Members of mammalian Orai proteins (Orai1, Orai2, Orai3) have been identified based on their homology to the single Drosophila Orai gene [1]. While Orai1 deficiency gives rise to the SCID phenotype described above, channelopathies stemming from defects in either Orai2 or Orai3 have yet to be described. Using ectopic expression in HEK293 cells, all three Orai proteins were found capable of mediating SOCE when co-expressed with STIM1 [21, 22]. However, as argued recently [9], Orai1 consistently and exclusively encodes native CRAC channels in a number of different mammalian cell types despite co-expression of other Orai isoforms by these cells. Ectopic expression of Orai3 in lymphocytes and fibroblasts from SCID patients only weakly reconstituted SOCE while expression of Orai2 had no appreciable effect, suggesting lack of redundancy of Orai proteins, at least in these cell types[18]. There is however at least one reported exception where Orai3 mediates SOCE in a subset of breast cancer cells [23].
Given that Orai proteins lack any obvious homology to other families of ion channels, the structural and functional properties of CRAC channels are inherently unique and many efforts have been directed at understanding such features. CRAC channels are characterized by an extraordinarily high selectivity for Ca2+, paralleled only by voltage-gated Ca2+ channels, and demonstrate poor permeability to Cs+ ions [24]. Further, CRAC channels exhibit an unusually long and narrow pore lined exclusively by residues on the predicted transmembrane domain 1 [25, 26]. These properties are noticeably absent when STIM1 gating of Orai1 channels is circumvented by mutations in Orai1 yielding a constitutive CRAC channel, indicating that STIM1 regulates ion selectivity and the pore architecture of native CRAC channels [27].
Another hallmark of CRAC channels is their rapid Ca2+-dependent inactivation, whereby local elevations of cytosolic Ca2+ quickly diminish the amplitude of CRAC currents [28]. Interestingly, this phenomenon has also been attributed to a direct and specific interaction of Ca2+ and calmodulin with STIM1 [29, 30], further suggesting that STIM1 becomes an intimate component of Orai1 channel during CRAC activity. Lastly, CRAC channels are pharmacologically distinguished by a high sensitivity to blockade by lanthanides mediated by residues glutamine108, aspartate110, and aspartate112 in human Orai1 located in the extracellular TM1-TM2 loop [13, 25]. Previous studies have suggested that functional CRAC channels exist in the plasma membrane as tetrameric complexes [11, 31, 32], with each Orai subunit containing four transmembrane segments and arranged such that both amino and carboxy termini are in the cytosol [1, 3]. Penna et al proposed that in the resting state Orai1 exists as a dimer, and upon channel activation assembles into functional tetramers [32]. All these previous studies are difficult to reconcile with the crystal structure of Drosophila Orai that was recently resolved at 3.35 angstroms, suggesting that drosophila Orai proteins form hexameric structures [33].
Store-independent Ca2+ channels
With native CRAC channels being preferentially composed of Orai1 subunits, it is not surprising that Orai3 (and perhaps Orai2) would contribute to Ca2+ selective currents, either as homomultimeric or heteromutlimeric channels that are activated by mechanisms distinct from store depletion [34, 35]. One particularly well-characterized store-independent Ca2+ channel is the arachidonic acid activated Ca2+ (ARC) channel that is formed by Orai1 and Orai3 [35]. Like CRAC channels, ARC channels are similarly Ca2+ selective and share many of the biophysical properties of CRAC, including dependence on STIM1 for their activation [36, 37]; however, the pool of STIM1 involved was suggested to be the one residing at plasma membrane [37]. Based on experiments using concatenated multimers of Orai, ARC channels were proposed to involve a pentameric assembly of two Orai3 and three Orai1 molecules [34]. We have recently identified a native ARC-like channel in vascular smooth muscle cells (VSMC; discussed below) that is encoded by both Orai1 and Orai3 subunits. This channel is store independent, requires STIM1 for activation and is gated by cytosolic leukotrieneC4 (LTC4) [38]. We have termed this channel LRC (pronounced Lark; for LTC4-regulated Ca2+ channel). To date, the existence of native store-operated or store-independent Ca2+ ionic currents contributed by Orai2 in any cell type remains unknown.
Orai channels in the heart
The mechanical function of the heart is dependent on a continuous alternation between contraction and relaxation of cardiac muscle fibers. Myocardial inotropy is governed by temporally and spatially controlled cytosolic Ca2+ sparks that can be finely tuned to regulate cardiac output and blood pressure [39]. In excitation-contraction coupling, membrane depolarization opens voltage-gated Ca2+ (CaV) channels clustered on plasma membrane invaginations. Due to their spatial proximity with the sarcoplasmic reticulum (SR), these channels direct Ca2+ ions onto nearby ryanodine receptor (RyR) Ca2+-release channels and induce Ca2+ release from the SR into the cytosol to turn on actin-myosin cross bridge cycling [39, 40].
Human patients with Orai1 deficiency do not exhibit any obvious defects in cardiac function, but as mentioned above skeletal myopathy is a prominent clinical feature [41, 42]. SOCE has been shown to be an important regulator of skeletal muscle contractility, although the contribution of SOCE to either cytosolic Ca2+ or Ca2+ store refilling during contractility of skeletal muscle is a contentious issue [43]. CRAC channels can be rapidly activated upon store depletion of skeletal muscle, yet SOCE does not become relevant until Ca2+ stores are substantially reduced [44, 45]. Under physiological conditions, such a state is infrequently achieved, as skeletal muscle is equipped with proteins that minimize store depletion including Ca2+-ATPases, allowing for rapid re-uptake of released Ca2+, as well as highly dynamic Ca2+ buffering proteins within the SR lumen (i.e. calsequestrin) [46]. This would suggest that Ca2+ entry via CRAC channels is likely involved in adaptive signaling required for maintaining muscle fiber homeostasis during development, growth or prolonged muscle work [42]. Supporting this idea, isolated skeletal muscle from STIM1-deficient mice fatigued earlier and generated less force upon high frequency stimulation [45]. Very little is known about how CRAC/SOCE contributes to contraction in other excitable cells including cardiomyocytes. Recently, SOCE was demonstrated in HL-1 cells, a useful in vitro model for investigating Ca2+ handling in unstressed cardiomyocytes [47]. Orai1 knockdown in this cell type not only prevented SOCE, but also lowered baseline Ca2+ levels. These authors proposed that CRAC channels might function in healthy cardiomyocytes to replenish Ca2+ stores and maintain intracellular Ca2+ levels over time [47]. While functional CRAC channels are found in both neonatal and adult hearts, it is unlikely that SOCE is significantly involved in cardiac excitation-contraction coupling. The magnitude of Ca2+ entry in postnatal mouse cardiomyocytes is reportedly half of what has been observed in embryonic cells, and the detection of SOCE in adult mice has been variable [48, 49]. Likewise, the expression of Orai1 dramatically decreases after birth [50]. CaV channels are coincidentally upregulated [51], and it was recently proposed that activated STIM1 impedes CaV channel conductance in VSMC [52]. Indeed, STIM1 knockdown in neonatal rat ventricle cardiomyocytes (NRCM) lowered diastolic Ca2+ levels and also diminished SR Ca2+ content, while silencing Orai1 expression did not alter these parameters [17]. However, as will be discussed below, several studies have proposed a role for STIM1/Orai1 signaling complexes in altered Ca2+ signaling underlying the development of cardiac hypertrophy.
As terminally differentiated cells, cardiomyocytes are largely incapable of cell proliferation. Hypertrophic growth is the primary mechanism by which the heart is able to preserve pump function and maintain adequate cardiovascular support when faced with an increased workload; by increasing wall thickness such growth necessarily decreases stress on the ventricular walls [53]. Cardiac growth during pregnancy or endurance training is transient and occurs without impact on myocardial structure or function [53]. On the other hand, hypertrophy from different pathologic stimuli, including hypertension, coronary insufficiency or valvular defects, is associated with transcription programs that alter myocardial architecture and invariably lead to cardiac dysfunction [54]. The molecular mechanisms underlying the development of physiological and pathological cardiac hypertrophy may be overlapping but are clearly distinct and remain incompletely understood.
Several agonists that converge on common intracellular signaling pathways have been implicated in the activation of hypertrophic gene programs. For example, endothlin-1, angiotensin II and catecholamines were proposed to activate the calcineurin-NFAT and Ca2+/calmodulin-dependent Kinase II (CaMKII) pathways to induce the molecular stress fetal gene program linked to pathologic growth and remodeling. Calcineurin is a Ca2+-dependent serine/threonine phosphatase activated by calmodulin as well as structural proteins localized in the z-disc [55]. Prolonged increases in cystosolic Ca2+ levels maintain calcineurin activation, and in turn keep NFAT transcription factors localized to the nucleus where they induce pro-hypertrophic gene expression [56]. Similarly, CaMKII contributes to hypertrophic gene expression downstream of spatial Ca2+ signals by phosphorylating type II histone deacetylases (HDAC), prompting HDAC exportation from the nucleus and relieving MEF-2-mediated gene expression [57] (Figure 1). However, the sources of Ca2+ responsible for the activation of these Ca2+-dependent signaling circuits are not clear, remain the subject of intensive investigations, with multiple channels being implicated in this process (Figure 1). Ca2+ elevations required for transcriptional activation may be mediated by Ca2+ release from internal stores or influx across the plasma membrane, with evidence supporting involvement of Ca2+ release through cardiac RyR [58] and IP3R [57, 59], and Ca2+ influx through CRAC channels [17], TRPC channels [60] and CaV channels [61].
Figure 1. Ca2+-dependent induction of pathologic cardiac gene expression.
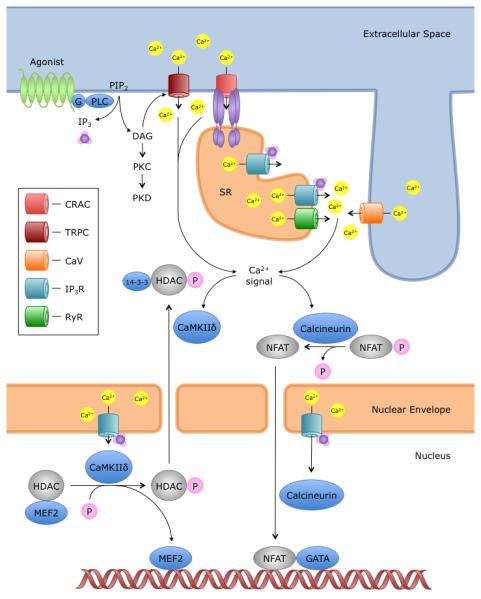
In cardiomyocytes, Ca2+ elevations are achieved by a variety of channels to cause Ca2+-dependent transcriptional activation. On the plasma membrane, agonists binding to G-protein coupled receptors cause IP3 and DAG production. DAG is a direct activator of TRPC3/6/7 channels. IP3, on the other hand, diffuses within the cell and binds to calcium release channels, the IP3 receptors; Ca2+ from SR stores can activate Ca2+ entry via CRAC channels by causing STIM1 clustering, as well as alter Ca2+ flux through RyR and CaV channels in the dyadic junctions [59]. These cytoplasmic Ca2+ signaling events may initiate calcineurin- and/or CaMKII-dependent transcription. Similarly, IP3 has been shown to bind IP3 receptors on the nuclear envelope, inducing Ca2+ transients that activate nearby calcineurin and CaMKII [57]. The dual compartmentalization of Ca2+ signals serves to initiate and maintain activation of prohypertrophic transcription regulators, including nuclear factor of activated T cells (NFAT) and histone deacetylase (HDAC). Abbreviations of Ca2+ channels: CRAC, Ca2+ release-activated Ca2+ channel; TRPC, Canonical transient receptor potential channel; CaV, voltage-activated L-type Ca2+ channel; IP3R, Inositol 1,4,5-trisphosphate receptor; RyR, Ryanodine Receptor.
In NRCM, SOCE activity has been detected upon treating cells with either the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) inhibitor thapsigargin or IP3-generating agonists such as angiotensin II and endothelin-1 [62]. Further, these hypertrophic stimuli were shown to promote NFAT nuclear translocation [62]. Agonist-induced Ca2+ elevations and hypertrophic gene expression were more strongly inhibited when pretreating cells with either glucosamine or SKF96365, both nonselective inhibitors of SOCE, than with the CaV channel antagonist verapamil [62]. Orai1 and STIM1 have since been demonstrated as essential elements of SOCE in cardiac cells [17, 63]. In an in vitro model of cardiac hypertrophy, silencing either STIM1 or Orai1 in cultured NRCM attenuated the phenylephrine-induced hypertrophic response [17, 63]. While protein expression of either Orai2 or Orai3 was not observed in NRCM, Orai2 mRNA level was significantly increased in Orai1 siRNA-treated cells [17]. Hulot et al showed enhanced SOCE in hypertrophic cardiomyocytes [64]. STIM1 expression is elevated in pressure-overloaded hearts, and silencing STIM1 in vivo using cardiotropic viruses prevented hypertrophic responses following abdominal aortic banding [64]. Consistently, overexpressing STIM1 in NRCM enhanced NFAT activity and cell size. However, in the presence of SKF96365, STIM1 overexpression did not induce cellular hypertrophy [64], compatible with the idea that CRAC channel function mediates pathologic cardiac growth. Not surprising, pressure overload in mice induces upregulation of Orai1 in cardiomyocytes [50]. Recently, Volkers et al examined Orai1 in zebrafish development [50]. Injection of zebrafish zygotes with Orai1 antisense oligonucleotides resulted in severe impairments in both skeletal and cardiac muscle function. The cardiac phenotype in this model was characterized by severe heart failure, though no effect on initial myocyte differentiation was reported. The histological findings of cardiomyocytes from Orai1-deficient zebrafish included dramatic alterations in sarcomeric structure, with fewer myofilaments and variable z-disks, suggesting that Orai1 channels are necessary for proper sarcomeric growth and function in cardiac cells [50]. The absence of a comparable cardiac deficit in human patients and mice lacking Orai1 may be attributed to the presence of Orai3 in mammals [65].
Orai channels in vessels
The principle cellular constituents of blood vessel walls are VSMC and endothelial cells (EC). VSMC are most abundant in the tunica media, particularly in the case of muscular arteries and arterioles. While VSMC are maintained in a partially constricted state, moment by moment regulation of intracellular Ca2+ concentrations enables rapid adjustments to vascular tone essential for hemodynamic stability [66]. Although VSMC infrequently divide, they lose their homeostatic quiescence with arterial injury or inflammatory activation [67-69]. Proliferation and migration of VSMC is a key step in the pathogenesis of vascular occlusive diseases such as arterial stenosis in atherosclerosis, neointimal hyperplasia following angioplasty or stent placement, and arteriolar remodeling in hypertension. Similarly, VSMC in the pulmonary circulation adopt this pathologic or “synthetic” phenotype to contribute to vascular remodeling in pulmonary hypertension [70]. EC are the main constituents of the intimal layer of blood vessels, and participate in a wide variety of functions. In addition to forming a selective barrier for cellular and nutrient trafficking, the endothelium also modulates local blood flow and vascular tone, and offers resistance to oxidative stress, inflammation, thrombosis, hemostasis, and VSMC proliferation [71]. Importantly, the EC phenotype is remarkably diverse. Not only do EC display significant structural and functional heterogeneity throughout the cardiovascular system [72], but like VSMC, with the right environmental cues, EC will adopt a pathological phenotype distinguished by the ability to migrate and proliferate [73]. Understanding the regulation of signaling pathways that govern phenotype switching in VSMC and EC is important to the development of targeted therapies against cardiovascular disease.
Endothelial cells
The growth of new vessels or angiogenesis occurs either from preexisting blood vessels or form endothelial precursor cells (EPC) originating in the bone marrow, and is necessary for replacing damaged tissues resulting, for instance, from tissue hypoxia [73]. However, angiogenesis also contributes to the development of vascular networks that support tumor growth and metastasis [73]. EC proliferation and migration is induced by activation of signaling cascades in response to various growth factors, most notably vascular endothelial growth factor (VEGF) [73]. Early studies showed that activation of VEGF receptor triggers Ca2+ release from IP3-sensitive stores and induces a low Ca2+ conductance across the plasma membrane necessary for EC proliferation [74, 75]. Electrophysiological characterization of this conductance was initially problematic given its relatively low and often variable amplitude in endothelial cells. Fasolato and Nilius were first to provide evidence that EC activate a small inward rectifying CRAC-like current in response to store depletion by either IP3, thapsigargin or Ca2+ ionophores [76]. Subsequently, Abdullaev et al showed a requirement for Orai1 and STIM1 in mediating CRAC currents in human umbilical vein EC (HUVEC) after passive store depletion, and that CRAC channels contributed to EC proliferation [77]. Inhibition of Orai1 function by either siRNA, expression of a dominant negative Orai1 mutant or the CRAC channel blocker S66 prevented endothelial tube formation in vitro [78]. However, these results have been challenged by experiments using primary HUVEC, which showed involvement of STIM1 and transient receptor potential canonical channel isoforms, TRPC1 and TRPC4, but not Orai1, in endothelial tube formation [79], suggesting a dissociation between STIM1 and Orai1 in tubulogenesis. Furthermore, knockdown of either STIM1 or STIM2 had only a modest inhibition of EC proliferation relative to that observed with Orai1 knockdown [77]. Furthermore, normal endothelial migration and vasculogenesis was reported in EC-specific STIM1-knockout mice [80].
EPC are present in postnatal circulation where they can proliferate, migrate, and adopt a mature endothelial phenotype [81]. They incorporate into vascular networks associated with tumors, help in the formation of de novo blood vessels and mediate improvements in blood flow to ischemic regions [82, 83]. Sanchez-Hernandez et al provided the first evidence of SOCE expression in EPC [84]. Further characterization of SOCE in this cell type revealed that stimulation with hepatocyte growth factor enhanced STIM1-dependent SOCE activity [85], with STIM1 being required for EPC migration and proliferation [86]. VEGF-treated EPC displayed IP3-dependent Ca2+ oscillations that were mediated through SOCE, and these signals were necessary for the recruitment of NF-κB [87]. These data suggest CRAC channel involvement in EPC Ca2+ signaling pathways governing cell proliferation and tubulogenesis. Importantly, Orai1 channel function has recently been linked to EPC-mediated angiogenesis in patients with renal cell carcinoma [88]. The majority of these tumors arise from functional inactivation of von Hippel-Lindau tumor suppressor, which leads to elevated levels of pro-angiogenic hypoxia-inducible factors (HIF) [89]. EPC isolated form the peripheral blood of patients with renal cell carcinoma were found to have increased expression in both Orai1 and STIM1 in comparison to control EPC, with concordant enhancement of SOCE activity [88]. Pharmacological inhibition of CRAC channel activation using either lanthanides or BTP2 impaired proliferation and in vitro tubulogenesis [88]. It is important to note that in EPC from renal cell carcinoma, VEGF failed to elicit Ca2+ oscillations despite normal expression levels of VEGF receptor; the authors proposed CRAC channel activation may be dependent on alternative physiological agonists in this cell type [88].
EC activation is an adaptive response employed in a variety of physiological and pathological situations. However, when the activation of EC is exaggerated and generalized, as is the case in sepsis and the systemic inflammatory response syndrome (SIRS), endothelial dysfunction follows leading to organ failure and potentially death [90]. An important stimulus of endothelial dysfunction is the bacterial endotoxin, lipopolysaccharide (LPS), which modifies EC phenotype in an organ-specific manner [90]. LPS binding to EC surface receptors, including toll-like receptor 4, activates signaling cascades that can lead to enhanced coagulation, increased vessel permeability, recruitment of inflammatory cells, secretion of vasoactive substances and apoptosis [90]. Ca2+ influx through CRAC channels is suggested to be an important step in the LPS-signaling cascade. The mechanism proposed involves production of reactive oxygen species (ROS) by EC exposed to LPS; ROS would then activate PLC to trigger release of IP3-sensitive Ca2+ stores causing subsequent activation of SOCE [80, 91]. SOCE would mediate its effects by providing sustained Ca2+ entry necessary for sustaining Ca2+-dependent transcriptional activation, namely NFAT transcriptional programs [80]. Additionally, ROS can engage STIM1 independently of Ca2+ store-depletion [92]. Through this pathway, ROS was proposed to cause S-glutathionylation of an evolutionary conserved cysteine residue (cysteine 56) on STIM1, resulting in constitutive STIM1 and CRAC channel activation. While the significance of this redundancy remains unclear, these studies highlight a critical role for STIM1 in EC activation. Indeed, LPS do not elicit a significant inflammatory response in EC-specific STIM1-knockout mice in comparison to wild-type mice. EC isolated from STIM1-knockout mice failed to trigger Ca2+ oscillations or activate NFAT-mediated transcription in response to LPS [80].
Vascular smooth muscle cells
The contribution of SOCE to VSMC contractility is a contentious issue. A number of studies have proposed a role for SOCE in VSMC contractility based on the use of non-specific SOCE inhibitors such as 2-Aminoethoxydiphenyl borate (2-APB) and lanthanides [93-95]. We and others have reported minimal SOCE/CRAC activity and STIM1/Orai1 protein expression in contractile freshly isolated VSMC by comparison to cultured proliferative synthetic VSMC, consistent with a minimal role for SOCE in VSMC contractility [16, 38, 96, 97]. However, the contribution of CRAC channels to vascular tone may be accomplished through their role in EC-mediated vasorelaxation by delivery of Ca2+ signals important for nitric oxide (NO) production in vascular EC [98]. Vasoactive agonists initiate NO-dependent vasodilation by elevating cytosolic endothelial Ca2+ levels and causing activation of endothelial NO synthase [99]. In fact, STIM1 suppression in porcine aortic EC inhibited SOCE, and was associated with a partial reduction in NO production induced by thrombin [100]. Additionally, a reduction in SOCE activity in mouse aortic EC was associated with impaired acetylcholine-induced vessel relaxation [101]. In diabetic mice, hyperglycemia leads to down regulation of STIM1 in EC with attenuation in coronary vascular relaxation, and restoration of STIM1 levels can recover endothelium-dependent relaxation [102].
Dysfunction of the endothelial monolayer is a common inciting event in the development of vascular occlusive diseases, activating inflammatory cells and platelets that result in the local release of a plethora of chemokines, growth factors, and vasoactive molecules [103]. Importantly, these agonists contribute to the phenotypic modulation of VSMC by activating signaling cascades that decrease expression of contractile proteins while upregulating extracellular matrix macromolecule secretion characteristic of VSMC in stenosed vessels [103]. A similar phenotypic modulation is observed when VSMC are cultured in vitro in the presence of serum [104], providing a useful model for the study of proliferating mitogenic synthetic VSMC. In culture, VSMC display enhanced SOCE in comparison to freshly isolated cells [16, 38, 96, 97]. Synthetic VSMC increase expression of STIM1 and Orai1, and knockdown experiments confirmed a requirement for STIM1/Orai1 in mediating SOCE in these cells [96, 97]. Higher expression of Orai1 was demonstrated in aortas of stroke prone hypertensive rats [105] and in neointimal VSMC from either balloon-injured rat carotids or ligation-injured mouse carotids [16]. Inhibition of either STIM1 or Orai1 expression in vivo using specific shRNA encoded by viral vectors lead to impaired CRAC channel function and significantly hindered VSMC proliferation and neointima formation [16, 106-108], implicating Orai1 as a modulator of VSMC phenotype. Orai1 channels appear to be important in sustaining Ca2+ entry required for NFAT nuclear translocation and its continued activation in VSMC proliferation and migration [16].
Interestingly, while Orai1 was detectable in cultured human aortic VSMC, it was relatively low in comparison to abundantly expressed Orai2 and Orai3 when normalized to expression levels in lymphocytes [97]. Orai2 and Orai3 expression was also increased in aortic synthetic VSMC in culture compared with contractile cells [38, 95]. However, silencing either Orai2 or Orai3 in vitro in synthetic VSMC did not affect the amplitude of SOCE, suggesting that CRAC channels contain only Orai1 subunits in these cells [96]. The expression of Orai3 was enhanced in VSMC following carotid injury and in vivo knockdown of Orai3 significantly inhibited neointima formation [38], indicating that Orai3 contributes to additional channels (discussed in detail below) or signaling pathways that promote VSMC synthetic phenotypes.
As noted above, multiple agonists have been implicated in driving VSMC proliferation and migration during vascular remodeling. Paracrine release of platelet-derived growth factor (PDGF) by EC, macrophages and VSMC induces VSMC migration, whereas promotion of intimal proliferation appears to be a secondary phenomenon [109]. Indeed, multiple reports have shown that treatment with PDGF receptor inhibitors significantly attenuates neointima formation in animal models of restenosis [110-112], though the clinical utility of such therapeutics has yet to be achieved. Ligation of PDGF to its receptor induces cytosolic Ca2+ transients [113]; Bisaillon et al showed PDGF-induced Ca2+ entry is specifically contributed by SOCE through STIM1 and Orai1 [106] (Figure 2A). STIM1 and Orai1 are essential for migration of VSMC and airway smooth muscle in response to in vitro stimulation with PDGF [106, 114].
Figure 2. Orai channel activation in VSMC.
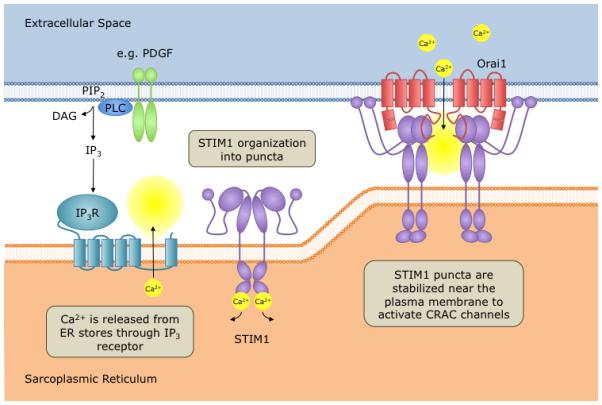
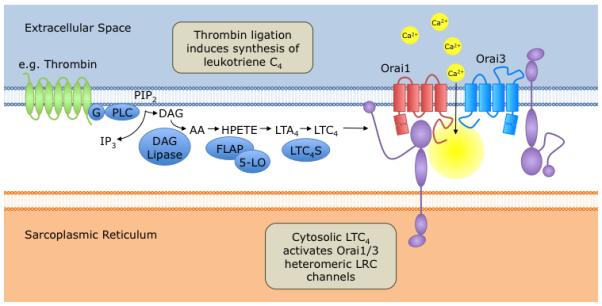
The promigratory and mitogenic agonists (A) PDGF and (B) thrombin activate Orai-dependent store-operated (PDGF) [106] or store-independent (thrombin) [38] Ca2+ entry in VSMC. In B, the involvement of either ER or plasma membrane STIM1 pools, the oligomeric state of functional STIM1 proteins and the nature of the interactions between STIM1 and Orai1/Orai3 LRC channels are unknown. Abbreviations of enzymes: DAG lipase, Diacylgercerol lipase; 5-LO, 5-lipoxygenase; FLAP, 5-lipoxygenase activating protein; LTC4S, LeukotrieneC4 synthase
Thrombin serves as a physiological agonist for proteinase-activated receptors (PAR) on both EC and VSMC. Although the expression of these receptors on VSMC in healthy arteries is limited, in vascular lesions there is an upregulation of PAR [115]. Thrombin simulation is important in mediating VSMC contraction, migration, proliferation, hypertrophy and production of extracellular matrix [116]. As such, thrombin signaling in VSMC is considered an important contributor to the pathogenesis of vascular occlusive disease. In vitro stimulation of synthetic VSMC with thrombin activated a novel Ca2+ entry pathway distinct from SOCE/CRAC. Surprisingly, thrombin-activated Ca2+ channels were highly Ca2+ selective and required STIM1, Orai1 and Orai3 [38] in manner similar to the arachidonic acid (AA)-regulated Ca2+ (ARC) channels [35, 37]. Indeed, despite the requirement for STIM1, this novel thrombin-activated channel was store-independent and was instead, gated by leukotrieneC4 (LTC4) produced downstream of receptor activation and acting from the cytosolic side (Figure 2B) [38]. Whether these channels functionally couple to STIM1 residing on the ER or STIM1 on plasma membrane remains to be determined. Thus, thrombin-activated LTC4-regulated Ca2+ (LRC) channels are the first example of a native store-independent Orai channel in the vasculature activated by a physiological agonist. As previously mentioned, in EC thrombin has been shown to activate store-dependent CRAC channels [77]. Taken together, these findings highlight the diversity of Ca2+ signaling and suggest that not only, distinct Ca2+ signals can be generated by distinct agonists in the same cell type but that, the same agonist can generate distinct Ca2+ entry routes depending on the cell type in question.
In contrast the systemic circulation, pulmonary vessels are not predisposed to becoming hypertensive. However, pulmonary hypertension can occur in the setting of cardiac or pulmonary diseases. Less commonly, pulmonary hypertension arises from intrinsic defects in the pulmonary bed, a rare disease termed idiopathic pulmonary arterial hypertension (IPAH). Proliferation of pulmonary arterial smooth muscle cells (PASMC) is a key step the pathogenesis of IPAH, contributing to the development of pulmonary vascular remodeling, thrombosis, sustained vasoconstriction and subsequent occlusion of pulmonary flow [117]. Studies have shown that SOCE is elevated in PASMC from IPAH patients, and serves as an essential mediator PASMC proliferation [118]. Characterization of SOCE in cultured mouse PASMC revealed that Orai1 channels were responsible for this STIM1-dependent Ca2+ conductance [119], and treatment of PASMC with PDGF enhanced SOCE by upregulating STIM1 and Orai1 through the Akt/mTOR signaling pathway [120]. However, Song et al found that protein expression of STIM2 was enhanced in PASMC from IPAH patients, whereas STIM1 expression was not significantly changed, suggesting that STIM2 may be important in enhancing SOCE in these cells [121]. STIM2 was shown to be necessary for mediating SOCE and proliferation in IPAH PASMC. Interestingly, Orai2 expression was also enhanced in PASMC from IPAH patients, though the exact significance of Orai2 upregulation is unknown [121].
Conclusion and Perspective
STIM and Orai proteins are important regulators of intracellular Ca2+ homeostasis, making pivotal contributions to the Ca2+ signaling mechanisms that support the development of cardiac and vascular disease. As such, there is a heightened interest in targeting Orai channels to treat diseases such as cardiac hypertrophy, atherosclerosis and restenosis. One obstacle with CRAC channel-directed therapies is the absence of specific channel blockers; understanding the molecular details of STIM/Orai structures and interactions and CRAC channel gating will hopefully help in the development of improved small molecule inhibitors. Additionally, the ubiquity of CRAC channels and its prominence in the immune system naturally impedes cell type-specific targeting of these channels for treatment of cardiovascular disease. The recognition of cell type-specific differences in CRAC channel molecular organization and modulation may help in the specific targeting of Orai-mediated Ca2+ signaling in a given system or organ. Likewise, the identification of important cardiovascular processes that depend on Ca2+ currents activated by alternative mechanisms other than store depletion and utilizing other Orai isoforms, whether as homomultimers or in heteromultimeric assemblies of two or more Orais, might hopefully provide newer and perhaps better targets for treatment of cardiovascular disease.
Acknowledgments
Work in the authors’ laboratory is supported by NIH/NHLBI grant R01HL097111 and by startup funds from the College of Nanoscale Science and Engineering (CNSE) to Mohamed Trebak. Brian Ruhle is supported by a Medical Student Research Fellowship from the American Heart Association.
References
- 1.Feske S, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441(7090):179–85. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 2.Prakriya M, et al. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443(7108):230–3. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 3.Vig M, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312(5777):1220–3. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang SL, et al. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci U S A. 2006;103(24):9357–62. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feske S. Immunodeficiency due to defects in store-operated calcium entry. Ann N Y Acad Sci. 2011;1238:74–90. doi: 10.1111/j.1749-6632.2011.06240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gwack Y, et al. Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol Cell Biol. 2008;28(17):5209–22. doi: 10.1128/MCB.00360-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vig M, et al. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol. 2008;9(1):89–96. doi: 10.1038/ni1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feske S. ORAI1 and STIM1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunol Rev. 2009;231(1):189–209. doi: 10.1111/j.1600-065X.2009.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trebak M. PLC: Johnny-come-lately to ORAI and the ups and downs of calcium signalling. J Physiol. 2011;589(Pt 22):5337–8. doi: 10.1113/jphysiol.2011.220517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trebak M. STIM/Orai signalling complexes in vascular smooth muscle. J Physiol. 2012;590(Pt 17):4201–8. doi: 10.1113/jphysiol.2012.233353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park CY, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136(5):876–90. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan JP, et al. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11(3):337–43. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeromin AV, et al. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443(7108):226–9. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Picard C, et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med. 2009;360(19):1971–80. doi: 10.1056/NEJMoa0900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parekh AB. Store-operated CRAC channels: function in health and disease. Nat Rev Drug Discov. 2010;9(5):399–410. doi: 10.1038/nrd3136. [DOI] [PubMed] [Google Scholar]
- 16.Zhang W, et al. Orai1-mediated I (CRAC) is essential for neointima formation after vascular injury. Circ Res. 2011;109(5):534–42. doi: 10.1161/CIRCRESAHA.111.246777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Voelkers M, et al. Orai1 and Stim1 regulate normal and hypertrophic growth in cardiomyocytes. J Mol Cell Cardiol. 2010;48(6):1329–34. doi: 10.1016/j.yjmcc.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gwack Y, et al. Biochemical and functional characterization of Orai proteins. J Biol Chem. 2007;282(22):16232–43. doi: 10.1074/jbc.M609630200. [DOI] [PubMed] [Google Scholar]
- 19.Soboloff J, et al. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol. 2012;13(9):549–65. doi: 10.1038/nrm3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brandman O, et al. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131(7):1327–39. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mercer JC, et al. Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem. 2006;281(34):24979–90. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lis A, et al. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr Biol. 2007;17(9):794–800. doi: 10.1016/j.cub.2007.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Motiani RK, Abdullaev IF, Trebak M. A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J Biol Chem. 2010;285(25):19173–83. doi: 10.1074/jbc.M110.102582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prakriya M, Lewis RS. Separation and characterization of currents through store-operated CRAC channels and Mg2+-inhibited cation (MIC) channels. J Gen Physiol. 2002;119(5):487–507. doi: 10.1085/jgp.20028551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McNally BA, et al. Structural determinants of ion permeation in CRAC channels. Proc Natl Acad Sci U S A. 2009;106(52):22516–21. doi: 10.1073/pnas.0909574106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou Y, et al. Pore architecture of the ORAI1 store-operated calcium channel. Proc Natl Acad Sci U S A. 2010;107(11):4896–901. doi: 10.1073/pnas.1001169107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McNally BA, et al. Gated regulation of CRAC channel ion selectivity by STIM1. Nature. 2012;482(7384):241–5. doi: 10.1038/nature10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fierro L, Parekh AB. Fast calcium-dependent inactivation of calcium release-activated calcium current (CRAC) in RBL-1 cells. J Membr Biol. 1999;168(1):9–17. doi: 10.1007/s002329900493. [DOI] [PubMed] [Google Scholar]
- 29.Mullins FM, et al. STIM1 and calmodulin interact with Orai1 to induce Ca2+-dependent inactivation of CRAC channels. Proc Natl Acad Sci U S A. 2009;106(36):15495–500. doi: 10.1073/pnas.0906781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee KP, et al. Molecular determinants of fast Ca2+-dependent inactivation and gating of the Orai channels. Proc Natl Acad Sci U S A. 2009;106(34):14687–92. doi: 10.1073/pnas.0904664106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mignen O, Thompson JL, Shuttleworth TJ. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J Physiol. 2008;586(2):419–25. doi: 10.1113/jphysiol.2007.147249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Penna A, et al. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature. 2008;456(7218):116–20. doi: 10.1038/nature07338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hou X, et al. Crystal structure of the calcium release-activated calcium channel Orai. Science. 2012;338(6112):1308–13. doi: 10.1126/science.1228757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mignen O, Thompson JL, Shuttleworth TJ. The molecular architecture of the arachidonate-regulated Ca2+-selective ARC channel is a pentameric assembly of Orai1 and Orai3 subunits. J Physiol. 2009;587(Pt 17):4181–97. doi: 10.1113/jphysiol.2009.174193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mignen O, Thompson JL, Shuttleworth TJ. Both Orai1 and Orai3 are essential components of the arachidonate-regulated Ca2+-selective (ARC) channels. J Physiol. 2008;586(1):185–95. doi: 10.1113/jphysiol.2007.146258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mignen O, Shuttleworth TJ. I(ARC), a novel arachidonate-regulated, noncapacitative Ca(2+) entry channel. J Biol Chem. 2000;275(13):9114–9. doi: 10.1074/jbc.275.13.9114. [DOI] [PubMed] [Google Scholar]
- 37.Mignen O, Thompson JL, Shuttleworth TJ. STIM1 regulates Ca2+ entry via arachidonate-regulated Ca2+-selective (ARC) channels without store depletion or translocation to the plasma membrane. J Physiol. 2007;579(Pt 3):703–15. doi: 10.1113/jphysiol.2006.122432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonzalez-Cobos JC, et al. Store-Independent Orai1/3 Channels Activated by Intracrine LeukotrieneC4: Role in Neointimal Hyperplasia. Circ Res. 2013 doi: 10.1161/CIRCRESAHA.111.300220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415(6868):198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 40.Eisner DA, et al. Physiological and pathological modulation of ryanodine receptor function in cardiac muscle. Cell Calcium. 2004;35(6):583–9. doi: 10.1016/j.ceca.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 41.McCarl CA, et al. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J Allergy Clin Immunol. 2009;124(6):1311–1318. e7. doi: 10.1016/j.jaci.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trebak M, et al. What Role for Store-Operated Ca(2+) Entry in Muscle? Microcirculation. 2013 doi: 10.1111/micc.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Launikonis BS, Murphy RM, Edwards JN. Toward the roles of store-operated Ca2+ entry in skeletal muscle. Pflugers Arch. 2010;460(5):813–23. doi: 10.1007/s00424-010-0856-7. [DOI] [PubMed] [Google Scholar]
- 44.Kurebayashi N, Ogawa Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. J Physiol. 2001;533(Pt 1):185–99. doi: 10.1111/j.1469-7793.2001.0185b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stiber J, et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol. 2008;10(6):688–97. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Royer L, Rios E. Deconstructing calsequestrin. Complex buffering in the calcium store of skeletal muscle. J Physiol. 2009;587(Pt 13):3101–11. doi: 10.1113/jphysiol.2009.171934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Touchberry CD, et al. Store-operated calcium entry is present in HL-1 cardiomyocytes and contributes to resting calcium. Biochem Biophys Res Commun. 2011;416(1-2):45–50. doi: 10.1016/j.bbrc.2011.10.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uehara A, et al. Store-operated Ca2+ entry uncoupled with ryanodine receptor and junctional membrane complex in heart muscle cells. Cell Calcium. 2002;31(2):89–96. doi: 10.1054/ceca.2001.0257. [DOI] [PubMed] [Google Scholar]
- 49.Hunton DL, et al. Adult rat cardiomyocytes exhibit capacitative calcium entry. Am J Physiol Heart Circ Physiol. 2004;286(3):H1124–32. doi: 10.1152/ajpheart.00162.2003. [DOI] [PubMed] [Google Scholar]
- 50.Volkers M, et al. Orai1 deficiency leads to heart failure and skeletal myopathy in zebrafish. J Cell Sci. 2012;125(Pt 2):287–94. doi: 10.1242/jcs.090464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harrell MD, et al. Large-scale analysis of ion channel gene expression in the mouse heart during perinatal development. Physiol Genomics. 2007;28(3):273–83. doi: 10.1152/physiolgenomics.00163.2006. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, et al. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science. 2010;330(6000):105–9. doi: 10.1126/science.1191086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358(13):1370–80. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 54.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7(8):589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 55.Heineke J, Ritter O. Cardiomyocyte calcineurin signaling in subcellular domains: from the sarcolemma to the nucleus and beyond. J Mol Cell Cardiol. 2012;52(1):62–73. doi: 10.1016/j.yjmcc.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 56.Molkentin JD, et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93(2):215–28. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu X, et al. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 2006;116(3):675–82. doi: 10.1172/JCI27374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamaguchi N, et al. Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca release channel. J Clin Invest. 2007;117(5):1344–53. doi: 10.1172/JCI29515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harzheim D, et al. Increased InsP3Rs in the junctional sarcoplasmic reticulum augment Ca2+ transients and arrhythmias associated with cardiac hypertrophy. Proc Natl Acad Sci U S A. 2009;106(27):11406–11. doi: 10.1073/pnas.0905485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuwahara K, et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116(12):3114–26. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen X, et al. Calcium influx through Cav1.2 is a proximal signal for pathological cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2011;50(3):460–70. doi: 10.1016/j.yjmcc.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hunton DL, et al. Capacitative calcium entry contributes to nuclear factor of activated T-cells nuclear translocation and hypertrophy in cardiomyocytes. J Biol Chem. 2002;277(16):14266–73. doi: 10.1074/jbc.M107167200. [DOI] [PubMed] [Google Scholar]
- 63.Ohba T, et al. Essential role of STIM1 in the development of cardiomyocyte hypertrophy. Biochem Biophys Res Commun. 2009;389(1):172–6. doi: 10.1016/j.bbrc.2009.08.117. [DOI] [PubMed] [Google Scholar]
- 64.Hulot JS, et al. Critical role for stromal interaction molecule 1 in cardiac hypertrophy. Circulation. 2011;124(7):796–805. doi: 10.1161/CIRCULATIONAHA.111.031229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shuttleworth TJ. Orai3--the 'exceptional' Orai? J Physiol. 2012;590(Pt 2):241–57. doi: 10.1113/jphysiol.2011.220574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bolton TB, et al. Smooth muscle cells and interstitial cells of blood vessels. Cell Calcium. 2004;35(6):643–57. doi: 10.1016/j.ceca.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 67.House SJ, et al. The non-excitable smooth muscle: calcium signaling and phenotypic switching during vascular disease. Pflugers Arch. 2008;456(5):769–85. doi: 10.1007/s00424-008-0491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lompre AM, et al. STIM1 and Orai in cardiac hypertrophy and vascular proliferative diseases. Front Biosci (Schol Ed) 2013;5:766–73. doi: 10.2741/s406. [DOI] [PubMed] [Google Scholar]
- 69.Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95(2):156–64. doi: 10.1093/cvr/cvs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kuhr FK, et al. New mechanisms of pulmonary arterial hypertension: role of Ca(2)(+) signaling. Am J Physiol Heart Circ Physiol. 2012;302(8):H1546–62. doi: 10.1152/ajpheart.00944.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aird WC. Spatial and temporal dynamics of the endothelium. J Thromb Haemost. 2005;3(7):1392–406. doi: 10.1111/j.1538-7836.2005.01328.x. [DOI] [PubMed] [Google Scholar]
- 72.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100(2):158–73. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- 73.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garnier-Raveaud S, et al. Identification of membrane calcium channels essential for cytoplasmic and nuclear calcium elevations induced by vascular endothelial growth factor in human endothelial cells. Growth Factors. 2001;19(1):35–48. doi: 10.3109/08977190109001074. [DOI] [PubMed] [Google Scholar]
- 75.Faehling M, et al. Essential role of calcium in vascular endothelial growth factor A-induced signaling: mechanism of the antiangiogenic effect of carboxyamidotriazole. FASEB J. 2002;16(13):1805–7. doi: 10.1096/fj.01-0938fje. [DOI] [PubMed] [Google Scholar]
- 76.Fasolato C, Nilius B. Store depletion triggers the calcium release-activated calcium current (ICRAC) in macrovascular endothelial cells: a comparison with Jurkat and embryonic kidney cell lines. Pflugers Arch. 1998;436(1):69–74. doi: 10.1007/s004240050605. [DOI] [PubMed] [Google Scholar]
- 77.Abdullaev IF, et al. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ Res. 2008;103(11):1289–99. doi: 10.1161/01.RES.0000338496.95579.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li J, et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ Res. 2011;108(10):1190–8. doi: 10.1161/CIRCRESAHA.111.243352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Antigny F, Girardin N, Frieden M. Transient receptor potential canonical channels are required for in vitro endothelial tube formation. J Biol Chem. 2012;287(8):5917–27. doi: 10.1074/jbc.M111.295733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gandhirajan RK, et al. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J Clin Invest. 2013 doi: 10.1172/JCI65647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Asahara T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964–7. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 82.Rafii S, et al. Vascular and haematopoietic stem cells: novel targets for anti-angiogenesis therapy? Nat Rev Cancer. 2002;2(11):826–35. doi: 10.1038/nrc925. [DOI] [PubMed] [Google Scholar]
- 83.Takahashi T, et al. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5(4):434–8. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 84.Sanchez-Hernandez Y, et al. Store-operated Ca(2+) entry is expressed in human endothelial progenitor cells. Stem Cells Dev. 2010;19(12):1967–81. doi: 10.1089/scd.2010.0047. [DOI] [PubMed] [Google Scholar]
- 85.Shi Y, et al. Knockdown of stromal interaction molecule 1 attenuates hepatocyte growth factor-induced endothelial progenitor cell proliferation. Exp Biol Med (Maywood) 2010;235(3):317–25. doi: 10.1258/ebm.2009.009237. [DOI] [PubMed] [Google Scholar]
- 86.Kuang CY, et al. Silencing stromal interaction molecule 1 by RNA interference inhibits the proliferation and migration of endothelial progenitor cells. Biochem Biophys Res Commun. 2010;398(2):315–20. doi: 10.1016/j.bbrc.2010.06.088. [DOI] [PubMed] [Google Scholar]
- 87.Dragoni S, et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells. 2011;29(11):1898–907. doi: 10.1002/stem.734. [DOI] [PubMed] [Google Scholar]
- 88.Lodola F, et al. Store-operated Ca2+ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS One. 2012;7(9):e42541. doi: 10.1371/journal.pone.0042541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Maranchie JK, et al. The contribution of VHL substrate binding and HIF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell. 2002;1(3):247–55. doi: 10.1016/s1535-6108(02)00044-2. [DOI] [PubMed] [Google Scholar]
- 90.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101(10):3765–77. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 91.Madesh M, et al. Selective role for superoxide in InsP3 receptor-mediated mitochondrial dysfunction and endothelial apoptosis. J Cell Biol. 2005;170(7):1079–90. doi: 10.1083/jcb.200505022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hawkins BJ, et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J Cell Biol. 2010;190(3):391–405. doi: 10.1083/jcb.201004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Trepakova ES, et al. Properties of a native cation channel activated by Ca2+ store depletion in vascular smooth muscle cells. J Biol Chem. 2001;276(11):7782–90. doi: 10.1074/jbc.M010104200. [DOI] [PubMed] [Google Scholar]
- 94.Smani T, et al. Role of Ca2+-independent phospholipase A2 and store-operated pathway in urocortin-induced vasodilatation of rat coronary artery. Circ Res. 2007;101(11):1194–203. doi: 10.1161/CIRCRESAHA.107.159053. [DOI] [PubMed] [Google Scholar]
- 95.Hopson KP, et al. S1P activates store-operated calcium entry via receptor- and non-receptor-mediated pathways in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2011;300(4):C919–26. doi: 10.1152/ajpcell.00350.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Potier M, et al. Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. FASEB J. 2009;23(8):2425–37. doi: 10.1096/fj.09-131128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Berra-Romani R, et al. Ca2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in culture. Am J Physiol Cell Physiol. 2008;295(3):C779–90. doi: 10.1152/ajpcell.00173.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Taniguchi H, et al. Possible involvement of Ca2+ entry and its pharmacological characteristics responsible for endothelium-dependent, NO-mediated relaxation induced by thapsigargin in guinea-pig aorta. J Pharm Pharmacol. 1999;51(7):831–40. doi: 10.1211/0022357991773032. [DOI] [PubMed] [Google Scholar]
- 99.Shaul PW. Regulation of endothelial nitric oxide synthase: location, location, location. Annu Rev Physiol. 2002;64:749–74. doi: 10.1146/annurev.physiol.64.081501.155952. [DOI] [PubMed] [Google Scholar]
- 100.Hirano K, Hirano M, Hanada A. Involvement of STIM1 in the proteinase-activated receptor 1-mediated Ca2+ influx in vascular endothelial cells. J Cell Biochem. 2009;108(2):499–507. doi: 10.1002/jcb.22279. [DOI] [PubMed] [Google Scholar]
- 101.Boittin FX, et al. Ca2+-independent PLA2 controls endothelial store-operated Ca2+ entry and vascular tone in intact aorta. Am J Physiol Heart Circ Physiol. 2008;295(6):H2466–74. doi: 10.1152/ajpheart.00639.2008. [DOI] [PubMed] [Google Scholar]
- 102.Estrada IA, et al. STIM1 restores coronary endothelial function in type 1 diabetic mice. Circ Res. 2012;111(9):1166–75. doi: 10.1161/CIRCRESAHA.112.275743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dzau VJ, Braun-Dullaeus RC, Sedding DG. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med. 2002;8(11):1249–56. doi: 10.1038/nm1102-1249. [DOI] [PubMed] [Google Scholar]
- 104.Thyberg J. Differentiated properties and proliferation of arterial smooth muscle cells in culture. Int Rev Cytol. 1996;169:183–265. doi: 10.1016/s0074-7696(08)61987-7. [DOI] [PubMed] [Google Scholar]
- 105.Giachini FR, et al. Increased activation of stromal interaction molecule-1/Orai-1 in aorta from hypertensive rats: a novel insight into vascular dysfunction. Hypertension. 2009;53(2):409–16. doi: 10.1161/HYPERTENSIONAHA.108.124404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bisaillon JM, et al. Essential role for STIM1/Orai1-mediated calcium influx in PDGF-induced smooth muscle migration. Am J Physiol Cell Physiol. 2010;298(5):C993–1005. doi: 10.1152/ajpcell.00325.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Aubart FC, et al. RNA interference targeting STIM1 suppresses vascular smooth muscle cell proliferation and neointima formation in the rat. Mol Ther. 2009;17(3):455–62. doi: 10.1038/mt.2008.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Guo RW, et al. An essential role for stromal interaction molecule 1 in neointima formation following arterial injury. Cardiovasc Res. 2009;81(4):660–8. doi: 10.1093/cvr/cvn338. [DOI] [PubMed] [Google Scholar]
- 109.Myllarniemi M, et al. Inhibition of platelet-derived growth factor receptor tyrosine kinase inhibits vascular smooth muscle cell migration and proliferation. FASEB J. 1997;11(13):1119–26. doi: 10.1096/fasebj.11.13.9367346. [DOI] [PubMed] [Google Scholar]
- 110.Levitzki A. PDGF receptor kinase inhibitors for the treatment of restenosis. Cardiovasc Res. 2005;65(3):581–6. doi: 10.1016/j.cardiores.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 111.Jandt E, et al. Stent-based release of a selective PDGF-receptor blocker from the bis-indolylmethanon class inhibits restenosis in the rabbit animal model. Vascul Pharmacol. 2010;52(1-2):55–62. doi: 10.1016/j.vph.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 112.Makiyama Y, et al. Imatinib mesilate inhibits neointimal hyperplasia via growth inhibition of vascular smooth muscle cells in a rat model of balloon injury. Tohoku J Exp Med. 2008;215(4):299–306. doi: 10.1620/tjem.215.299. [DOI] [PubMed] [Google Scholar]
- 113.Scherberich A, et al. Migration of human vascular smooth muscle cells involves serum-dependent repeated cytosolic calcium transients. J Cell Sci. 2000;113(Pt 4):653–62. doi: 10.1242/jcs.113.4.653. [DOI] [PubMed] [Google Scholar]
- 114.Spinelli AM, et al. Airway smooth muscle STIM1 and Orai1 are upregulated in asthmatic mice and mediate PDGF-activated SOCE, CRAC currents, proliferation, and migration. Pflugers Arch. 2012;464(5):481–92. doi: 10.1007/s00424-012-1160-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nelken NA, et al. Thrombin receptor expression in normal and atherosclerotic human arteries. J Clin Invest. 1992;90(4):1614–21. doi: 10.1172/JCI116031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hirano K. The roles of proteinase-activated receptors in the vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2007;27(1):27–36. doi: 10.1161/01.ATV.0000251995.73307.2d. [DOI] [PubMed] [Google Scholar]
- 117.Michelakis ED, Wilkins MR, Rabinovitch M. Emerging concepts and translational priorities in pulmonary arterial hypertension. Circulation. 2008;118(14):1486–95. doi: 10.1161/CIRCULATIONAHA.106.673988. [DOI] [PubMed] [Google Scholar]
- 118.Wang C, et al. Inhibition of SOC/Ca2+/NFAT pathway is involved in the anti-proliferative effect of sildenafil on pulmonary artery smooth muscle cells. Respir Res. 2009;10:123. doi: 10.1186/1465-9921-10-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ng LC, et al. Orai1 interacts with STIM1 and mediates capacitative Ca2+ entry in mouse pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol. 2010;299(5):C1079–90. doi: 10.1152/ajpcell.00548.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ogawa A, et al. PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol. 2012;302(2):C405–11. doi: 10.1152/ajpcell.00337.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Song MY, Makino A, Yuan JX. STIM2 Contributes to Enhanced Store-operated Ca Entry in Pulmonary Artery Smooth Muscle Cells from Patients with Idiopathic Pulmonary Arterial Hypertension. Pulm Circ. 2011;1(1):84–94. doi: 10.4103/2045-8932.78106. [DOI] [PMC free article] [PubMed] [Google Scholar]