

Abstract
The autoimmune disease type 1 diabetes in humans and NOD mice is determined by multiple genetic factors, among the strongest of which is the inheritance of diabetes-permissive MHC class II alleles associated with susceptibility to disease. Here we examined whether expression of MHC class II alleles associated with resistance to disease could be used to prevent the occurrence of diabetes. Expression of diabetes-resistant MHC class II I-Aβ chain molecules in NOD mice following retroviral transduction of autologous bone marrow hematopoietic stem cells prevented the development of autoreactive T cells by intrathymic deletion and protected the mice from the development of insulitis and diabetes. These data suggest that type 1 diabetes could be prevented in individuals expressing MHC alleles associated with susceptibility to disease by restoration of protective MHC class II expression through genetic engineering of hematopoietic stem cells.
Introduction
Type 1 diabetes is caused by T cell–mediated autoimmune destruction of insulin-producing β cells in the pancreas. Multiple genes contribute to the development of type 1 diabetes; however, the inheritance of MHC alleles associated with susceptibility to disease (at-risk alleles) that lack a charged amino acid at position 57 of the MHC class II β chain is a principal determining genetic component in humans (1–4). Conversely, inheritance of MHC class II alleles containing a charged amino acid at position 57 of the β chain is associated with protection from disease. In some populations, as many as 28% of individuals that inherit particular HLA-DQ alleles such as DQB1*0302 and 34% that inherit DQB1*0201 develop diabetes (5). The MHC class II region in NOD mice, a model of spontaneous type 1 diabetes (6), encodes a single MHC class II molecule, I-Ag7 (the mouse homologue of HLA-DQB1), which contains polymorphisms that are strikingly similar to those in human alleles associated with type 1 diabetes susceptibility (7).
At-risk alleles in humans and NOD mice are structurally distinct from diabetes-resistant alleles, and in both cases the lack of a charged amino acid at position 57 of the β chain prevents the formation of a salt bridge with an arginine 76 of the α chain. This polymorphism leads to structural abnormalities that result in aberrant peptide binding and presentation (8) and has been suggested to affect the ability of these molecules to effectively mediate efficient negative selection of autoreactive T cells during their development (9–14). Alternatively, structural abnormalities present in at-risk alleles could contribute to diabetes development by failing to positively select T cells that can control disease (15).
Transgenic NOD mice that express MHC class II genes other than I-Ag7 do not develop diabetes (16–23). Transplantation of allogeneic bone marrow from strains that do not spontaneously develop diabetes also prevents the occurrence of diabetes in NOD mice (24–28). While these studies support the idea that MHC plays a major role in the development of type 1 diabetes, the mechanism by which diabetes-resistant MHC alleles prevent disease is unknown. In transgenic mice, diabetes-resistant MHC class II molecules are expressed on both thymic epithelium and bone marrow–derived cells. Therefore, protection from diabetes could occur by positive selection of regulatory T cells or negative selection of self-reactive T cells. In bone marrow chimeras, diabetes-resistant MHC class II molecules are expressed only on hematopoietic cells. However, allogeneic bone marrow cells also express non–MHC-encoded genes that influence diabetes progression in NOD mice (29). Therefore, from these studies, it is impossible to determine the mechanism of the protection and whether expression of diabetes-resistant MHC on bone marrow–derived cells alone is sufficient to prevent diabetes.
We hypothesized that if expression of diabetes-resistant MHC class II alleles on bone marrow–derived cells is sufficient to prevent diabetes, genetic engineering of autologous hematopoietic stem cells could be used to restore expression of diabetes-resistant MHC on bone marrow–derived cells. Furthermore, expression of diabetes-resistant MHC class II alleles on bone marrow–derived cells could mediate negative selection of self-reactive T cells that cause diabetes, thereby preventing disease. This approach could have significant advantages over transplantation of allogeneic bone marrow cells, because the possibility of graft-versus-host disease would be avoided. Here we show that expression of diabetes-resistant MHC class II I-Aβ chain molecules on hematopoietic cells following retroviral transduction of bone marrow is sufficient to prevent the occurrence of type 1 diabetes in NOD mice. Our data demonstrate that expression of diabetes-resistant MHC class II molecules on hematopoietic cells leads to negative selection of self-reactive diabetogenic T cells, thereby preventing insulitis and destruction of β cells in the pancreas. Furthermore, these data suggest that genetic engineering of autologous hematopoietic stem cells could be used to prevent diabetes occurrence in high-risk individuals who inherit diabetes-permissive MHC class II alleles.
Results
Construction and characterization of retroviruses carrying MHC class II–GFP fusion genes.
To examine the role of MHC class II in providing protection from diabetes, genetic engineering of bone marrow was used to achieve expression of diabetes-resistant MHC class II molecules on autologous hematopoietic cells of NOD mice. We reasoned that expression of diabetes-resistant MHC class II β chain genes in NOD hematopoietic cells would reconstitute expression of diabetes-resistant MHC class II molecules by pairing with endogenous I-Aα chains, which are structurally normal. Constructs were prepared in which the cytoplasmic tails of I-Aβ chains from the H-2d and H-2k haplotypes were fused to enhanced GFP to facilitate tracking of diabetes-resistant MHC class II chains in hematopoietic cells. I-Aβ chain–GFP fusion genes were then cloned into the pMMP retroviral vector (Figure 1), and vesicular stomatitis virus G–enveloped retroviruses were prepared from each construct, hereafter referred to as MMP-IAβ-d-GFP and MMP-IAβ-k-GFP virus.
Figure 1.

Diagram of MMP-based retroviral vectors encoding I-Aβd (MMP-IAβ-d-GFP) and I-Aβk (MMP-IAβ-k-GFP) fused to GFP; and diagram of the control MMP-GFP retroviral vector. LTR, long-terminal repeat.
Infection of I-Aβ chain–deficient, surface MHC class II–negative, M12.C3 cells (30) with either MMP-IAβ-d-GFP or MMP-IAβ-k-GFP virus restored expression of MHC class II on the cell surface (Figure 2A), indicating that the retrovirally encoded I-Aβ chains were produced and were able to pair with endogenously encoded α chains. To determine whether retrovirally transduced I-Aβ chains could compete with endogenous MHC class II and be expressed on the cell surface, A20 cells (H-2d positive, BALB/c origin) were infected with MMP-IAβ-k-GFP. Cell surface expression of I-Aβk was detectable on these infected cells by cell surface staining with the I-Aβk chain–reactive antibody 10-3.6 (Figure 2B). In all cases, GFP and I-Aβk were concordantly expressed. The levels of endogenous I-Ad expression were unchanged in A20 cells infected with MMP-IAβ-k-GFP virus (Figure 2B), suggesting that the retrovirally encoded I-Aβk molecules and the endogenous I-Aβd chains are coexpressed on the cell surface.
Figure 2.

Expression of retrovirally transduced MHC class II_GFP fusion proteins. (A) MMP-IAβ-d-GFP and MMP-IAβ-k-GFP retroviruses restore surface expression of MHC class II in I-Aβ chain_deficient M12.C3 cells. M12.C3 cells were cultured with media alone (mock) or infected with cell-free MMP-IAβ-k-GFP or MMP-IAβ-d-GFP retroviral supernatants for 3 days. Cells were harvested, and surface expression of MHC class II was analyzed by staining with anti-MHC mAb’s followed by flow cytometry. Shown is 1 representative example of 3 independent experiments. (B) I-Aβ chains encoded by MMP-IAβ-k-GFP are expressed on the surface of cells that express endogenous MHC class II. A20 cells (H-2d) were cultured with either media alone (mock) or cell-free MMP-IAβ-k-GFP viral supernatant for 3 days. Cells were harvested, and surface expression of MHC class II was analyzed by surface staining with anti_I-Aβk (10-3.6) and anti_I-Aβd (39–10–8) antibodies followed by flow cytometry. Shown is 1 representative example of 3 independent experiments.
Long-term expression of retrovirally encoded MHC class II–GFP fusion genes following reconstitution of NOD mice with transduced syngeneic bone marrow.
We next examined the ability of MMP-IAβ-d-GFP and MMP-IAβ-k-GFP viruses to transfer I-Aβ chain gene expression to primary murine bone marrow cells. Bone marrow was harvested from female NOD/MrkTac mice (hereafter referred to as NOD mice) treated 7 days prior with 150 mg/kg 5-fluorouracil, and transduced as previously described (31). Immediately after transduction, approximately 17.2% ± 2.3% of MMP-IAβ-d-GFP–transduced and 14.4% ± 2.1% of MMP-IAβ-k-GFP–transduced bone marrow cells expressed GFP (Figure 3A). The frequency of bone marrow cells expressing GFP following infection with either MMP-IAβ-d-GFP or MMP-IAβ-k-GFP was similar to that observed following infection with MMP-GFP (16.3% ± 1.8%), a control virus that carries only the gene encoding GFP. Mock-transduced bone marrow cells remained negative (Figure 3A).
Figure 3.

Expression of retrovirally transduced MHC class II β chains in vivo. (A) MMP-IAβ-d-GFP and MMP-IAβ-k-GFP viruses effectively infect primary bone marrow cells. Bone marrow cells were harvested from 5-fluorouracil_treated NOD mice and transduced as described. Immediately after transduction, bone marrow cells were harvested and examined for GFP expression by flow cytometry. One representative experiment of 5 independent experiments is shown. (B) Expression of retrovirally transduced I-Aβ chains is stable. Expression of GFP in PBMCs of NOD mice reconstituted with MMP-IAβ-d-GFP_transduced (squares), MMP-IAβ-k-GFP_transduced (triangles), or control MMP-GFP_transduced bone marrow (diamonds), determined by flow cytometry. Shown are the mean values obtained for 5_6 mice per time point. The data are representative of 3 independent experiments. (C) MHC class II expression levels on the surface of PBMCs. Twenty weeks after bone marrow transplantation, PBMCs were harvested from mice reconstituted with bone marrow transduced with either MMP-IAβ-k-GFP (solid line) or control MMP-GFP virus (dashed line). Cells were stained with anti_I-Ad antibody 39-10-8, which recognizes all cell surface MHC class II, and analyzed by flow cytometry. Left panel: Total cells. Right panel: GFP+ transduced cells.
To examine expression of retrovirally transduced I-Aβ chain genes in vivo, female NOD mice were lethally irradiated (10.25 Gy) and reconstituted with 4 × 106 MMP-IAβ-d-GFP virus–, MMP-IAβ-k-GFP virus–, or MMP-GFP virus–transduced syngeneic bone marrow cells. Eight weeks after bone marrow transplantation, 8.0% ± 0.4% (n = 18) of PBMCs from mice reconstituted with MMP-IAβ-d-GFP–transduced bone marrow expressed GFP. Similarly, 5.0% ± 0.7% (n = 18) of PBMCs from mice reconstituted with MMP-IAβ-k-GFP–transduced bone marrow and 10.6% ± 0.6% (n = 18) of PBMCs from mice reconstituted with control MMP-GFP–transduced bone marrow expressed GFP. The frequency of cells expressing GFP remained stable in all mice over the 36-week follow-up period (Figure 3B). Twenty weeks after reconstitution, GFP expression was detected in CD3+ T cells, B220+ B cells, CD11b+ macrophages, and CD11c+ dendritic cells in the spleen, PBMCs, bone marrow, and thymus (Table 1).
Table 1.
Lineage distribution of GFP-expressing cells in reconstituted mice

We next examined whether expression of retrovirally encoded I-Aβ chain affected expression levels of MHC class II. Twenty weeks after bone marrow transplantation, PBMCs were harvested from mice reconstituted with bone marrow transduced with either MMP-IAβ-k-GFP or control MMP-GFP virus. Cells were stained with anti–I-Ad antibody 39-10-8, which recognizes both the endogenous and the retrovirally encoded MHC class II heterodimers, and analyzed by flow cytometry (Figure 3C). The level of MHC class II expression on PBMCs infected with MMP-IAβ-k-GFP virus was identical to the level on cells infected with control virus. Since total MHC class II levels are unchanged in cells infected with MMP-IAβ-k-GFP virus, and the retrovirally encoded MHC class II β chains must compete with I-Ag7 β chains for endogenously encoded α chain to reach the cell surface, we assume that the total number of I-Ag7 molecules on cells expressing retrovirally encoded MHC class II β chains is reduced.
Retrovirally encoded MHC class II–GFP fusion proteins reach the cell surface only in MHC class II–committed cells.
To examine cellular distribution of retrovirally transduced MHC class II β chains, confocal microscopy was used to analyze the cellular localization of GFP. PBMCs from NOD mice reconstituted with MMP-IAβ-d-GFP–, MMP-IAβ-k-GFP–, or control MMP-GFP–transduced bone marrow were collected, fixed, and double-stained with the anti–I-Ad antibody 39-10-8 (which cross-reacts with I-Ag7) and the anti-GFP antibody JL-8. Analysis of MHC class II–positive cells in blood revealed that expression of GFP was localized in the cytoplasm of cells from mice reconstituted with MMP-GFP–transduced bone marrow and did not colocalize with endogenous MHC class II on the cell surface (Figure 4A). In contrast, in mice reconstituted with MMP-IAβ-d-GFP–transduced or MMP-IAβ-k-GFP–transduced (not shown) bone marrow, GFP expression was colocalized together with MHC class II on the cell surface (Figure 4A). Thus, in MHC class II–expressing cells, retrovirally encoded I-Aβ chains are processed and exported to the cell surface. Analysis of T cells from mice reconstituted with MMP-IAβ-d-GFP–transduced or MMP-IAβ-k-GFP–transduced (not shown) bone marrow by confocal microscopy revealed expression of GFP within the cytoplasm (Figure 4B). GFP expression in T cells did not colocalize with CD3 on the cell surface (Figure 4B). These data suggest that retrovirally transduced MHC class II β chains are expressed in the cytoplasm of MHC class II–negative cells and do not reach the cell surface.
Figure 4.

Subcellular localization of retrovirally encoded I-Aβ chains. (A) Retrovirally encoded I-Aβ chains are expressed on the surface of MHC class II_positive cells. PBMCs were harvested from NOD mice reconstituted with either MMP-GFP_transduced (top panels) or MMP-IAβ-d-GFP_transduced bone marrow (bottom panels), fixed, and stained with antibodies specific for GFP (left panels, green) and I-Ad (middle panels, red). The anti_I-Ad antibody used cross-reacts with I-Ag7. Right panels: Overlay images showing colocalization of retrovirally encoded I-Ad_GFP fusion proteins with endogenous I-Ag7 on the surface in yellow. (B) Retrovirally encoded I-Aβ chains are expressed in the cytoplasm of surface-MHC class II negative cells. PBMCs from NOD mice reconstituted with MMP-IAβ-d-GFP_transduced bone marrow were fixed and stained with antibodies specific for GFP (left panel, green) and anti-CD3 (middle panel, red). An overlay of the 2 images is shown in the right panel, demonstrating that I-Aβd_GFP does not colocalize with CD3 on the cell surface of MHC class II_negative cells. Shown are representative results of 3 independent experiments.
Expression of retrovirally encoded MHC class II–GFP fusion genes in NOD mouse bone marrow–derived cells prevents type 1 diabetes.
To determine the effect of expressing retrovirally transduced diabetes-resistant MHC class II genes in bone marrow–derived cells on diabetes occurrence, 5- to 6-week-old female NOD mice were irradiated and reconstituted with 4 × 106 syngeneic bone marrow cells transduced with MMP-IAβ-d-GFP, MMP-IAβ-k-GFP, or control MMP-GFP virus. Blood glucose levels were then monitored on a weekly basis. Four of six mice reconstituted with MMP-GFP–transduced bone marrow became diabetic by 22 weeks after bone marrow transplantation and were sacrificed (median time of onset, 18 weeks; Figure 5A). In contrast, mice reconstituted with either MMP-IAβ-d-GFP–transduced (n = 6) or MMP-IAβ-k-GFP–transduced (n = 6) bone marrow were protected from diabetes. These mice remained normoglycemic for 40 weeks after reconstitution (P < 0.01, median time of onset >40 weeks after reconstitution, n = 12). All mice remained in good health for the duration of the experiment, and no leukemias or lymphomas were detected (not shown).
Figure 5.

NOD mice reconstituted with either MMP-IAβ-d-GFP_ or MMP-IAβ-k-GFP_transduced bone marrow are protected from diabetes. (A) NOD mice were reconstituted with bone marrow retrovirally transduced with MMP-IAβ-d-GFP (open circles, n = 6), MMP-IAβ-k-GFP (×, n = 6), or control MMP-GFP (filled squares, n = 6). Shown are the percentages of normoglycemic mice at each time point after bone marrow transplantation. (B) NOD mice reconstituted with bone marrow retrovirally transduced with either MMP-IAβ-d-GFP or MMP-IAβ-k-GFP are protected from cyclophosphamide-induced diabetes. NOD mice were reconstituted with bone marrow transduced with MMP-IAβ-d-GFP (open circles, n = 12), MMP-IAβ-k-GFP (×, n = 12), or control MMP-GFP (filled squares, n = 14). Twenty-one weeks after bone marrow transplantation, mice were injected intraperitoneally with 200 mg/kg cyclophosphamide. Shown are the percentages of normoglycemic mice at each time point after bone marrow transplantation. Data in A and B are the combined results of 2 independent experiments. In all experiments, blood glucose levels were measured weekly.
Treatment with cyclophosphamide accelerates the occurrence of diabetes in NOD mice and can induce diabetes in mice in which spontaneous hyperglycemia does not develop (32), suggesting that resistance to cyclophosphamide-induced diabetes is a rigorous test of the ability of gene therapy to prevent diabetes in NOD mice. To test the ability of retroviral gene therapy to protect mice from cyclophosphamide-induced diabetes, female NOD mice were reconstituted with MMP-IAβ-d-GFP–, MMP-IAβ-k-GFP–, or control MMP-GFP–transduced bone marrow as described above, and, 21 weeks after reconstitution, mice in each group were treated with 200 mg/kg cyclophosphamide. In these experiments, recipient mice also received anti-CD4 and anti-CD8 antibodies at the time of bone marrow transplantation to deplete any preexisting mature diabetogenic T cells. Treatment of mice with anti–T cell antibodies delays T cell recovery after bone marrow transplantation and therefore delays the onset of diabetes (J. Iacomini, unpublished observation). Seventy-nine percent of NOD mice (11 of 14) reconstituted with MMP-GFP–transduced bone marrow developed diabetes by 28 weeks after reconstitution and were sacrificed (median time of onset, 23 weeks after reconstitution; Figure 5B). In contrast, mice reconstituted with either MMP-IAβ-d-GFP– or MMP-IAβ-k-GFP–transduced bone marrow were protected from disease and remained normoglycemic for 46 weeks after bone marrow transplantation (Figure 5B), when the experiment was terminated (P < 0.001 compared with MMP-GFP group, n = 24).
Expression of retrovirally encoded MHC class II–GFP fusion genes in NOD mouse bone marrow–derived cells prevents insulitis.
To examine the effect of gene therapy on insulitis, pancreata were harvested from NOD mice reconstituted with MMP-IAβ-d-GFP–, MMP-IAβ-k-GFP–, or control MMP-GFP–transduced bone marrow 20 weeks after reconstitution. Tissue sections were prepared and stained with anti-insulin antibodies and hematoxylin. Pancreatic islets in mice reconstituted with MMP-GFP–transduced bone marrow exhibited significant cellular infiltration, and decreased storage of insulin (n = 4; Figure 6A). Cellular infiltrates consisted primarily of T cells, consistent with insulitis (n = 4; Figure 6B). In contrast, islets from mice reconstituted with either MMP-IAβ-d-GFP– or MMP-IAβ-k-GFP–transduced bone marrow exhibited normal histology (n = 4; Figure 6A). Islets from these mice did not exhibit significant cellular infiltration, and insulin storage appeared to be similar to that observed in tissue sections of pancreata from normal BALB/c controls. Similar results were observed at 40 weeks after reconstitution (not shown). These data suggest that expression of diabetes-resistant MHC genes from protective MHC haplotypes prevents insulitis and the development of islet antigen–reactive T cells that cause islet destruction.
Figure 6.
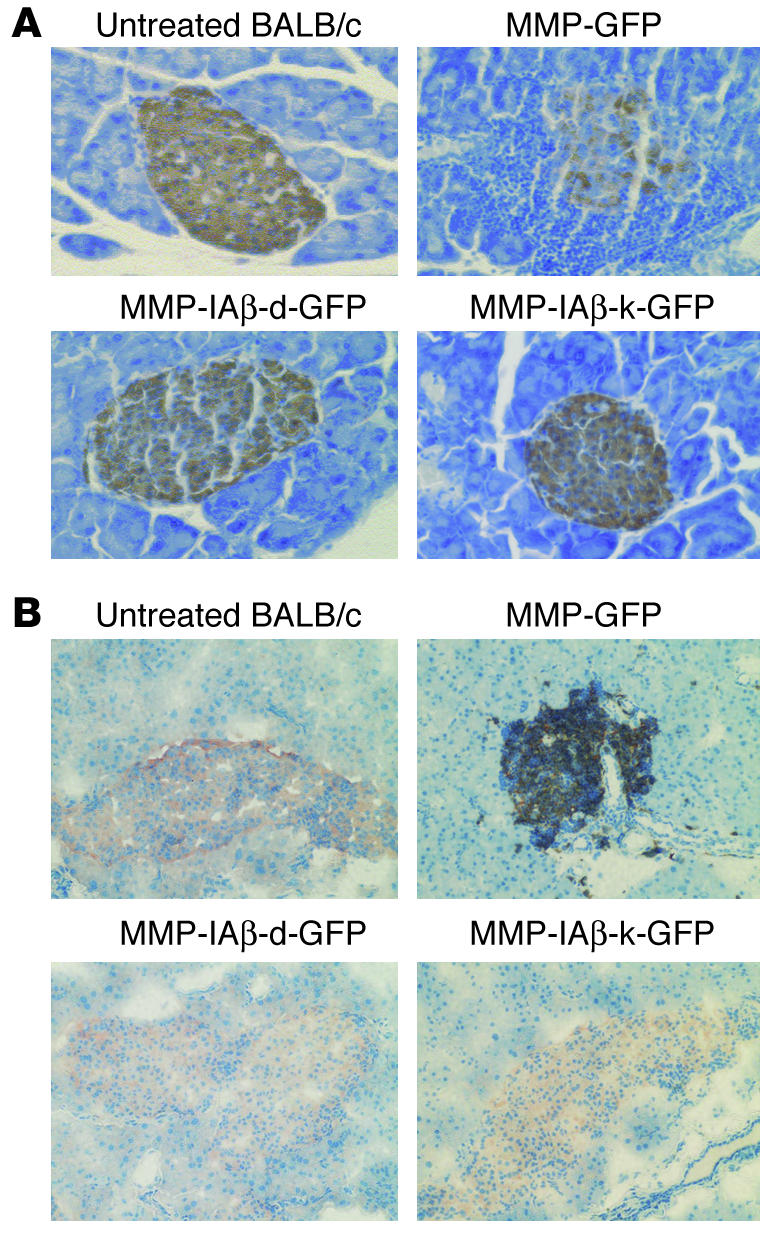
Prevention of insulitis in NOD mice reconstituted with MMP-IAβ-d-GFP_ or MMP-IAβ-k-GFP_transduced bone marrow. (A) NOD mice reconstituted with MMP-IAβ-d-GFP_, MMP-IAβ-k-GFP_, or MMP-GFP_transduced bone marrow were sacrificed 20 weeks after transplantation. Pancreata were then harvested, fixed, paraffin-embedded, sectioned, and stained with anti-insulin antibodies, and counterstained with hematoxylin. Islets from mice reconstituted with MMP-GFP_transduced bone marrow showed significant mononuclear cell infiltration, and decreased storage of insulin (brown staining, top right panel). In contrast, islets from mice reconstituted with either MMP-IAβ-d-GFP_transduced (bottom left) or MMP-IAβ-k-GFP_transduced bone marrow (bottom right) were free of cellular infiltration, and levels of insulin storage were comparable to those observed in tissue sections from control BALB/c pancreas (top left). Shown are representative sections from 3 independent experiments. (B) Cells infiltrating NOD islets are predominantly CD3+. NOD mice reconstituted with MMP-IAβ-d-GFP_, MMP-IAβ-k-GFP_, or MMP-GFP_transduced bone marrow were sacrificed 20 weeks after bone marrow transplantation. Pancreata were snap-frozen, sectioned, fixed, and stained with antibodies specific for insulin (red) and CD3 (black), and then counterstained with hematoxylin (blue). Pancreata from NOD mice reconstituted with MMP-GFP_transduced bone marrow (top right panel) show peri-islet infiltration of CD3+ cells, and little insulin staining. In contrast, pancreata from mice reconstituted with either MMP-IAβ-d-GFP_transduced (bottom left) or MMP-IAβ-k-GFP_transduced bone marrow (bottom right) had no visible cellular infiltrate, and insulin staining was comparable to that observed in control BALB/c pancreata (top left). Shown are representative sections from 3 independent experiments.
Elimination of self-antigen–reactive T cells in NOD mice reconstituted with MHC class II–GFP–transduced bone marrow.
We next examined the frequency of self-antigen–reactive T cells in NOD mice reconstituted with MMP-IAβ-d-GFP–, MMP-IAβ-k-GFP–, or control MMP-GFP–transduced syngeneic bone marrow using cytokine ELISPOT assays. Mice were sacrificed 20–28 weeks after reconstitution, and spleens were harvested. Splenocytes were then restimulated overnight with insulin (whole protein), peptide 206–220 from glutamic acid decarboxylase 65 (GAD 65), or peptide 85–99 from myelin basic protein (MBP) as a control in ELISPOT assays. The GAD 65 peptide was chosen because it is an immunodominant self-antigen peptide in NOD mice (33) and binds with high affinity to I-Ag7 (34). A significant frequency of spleen cells from NOD mice reconstituted with MMP-GFP–transduced bone marrow were able to secrete IFN-γ (34 ± 5 cells per 106), IL-2 (80 ± 12 cells per 106), and IL-4 (88 ± 9 cells per 106) in response to the GAD 65 peptide (n = 7; Figure 7). In contrast, we were unable to detect production of IFN-γ (1 ± 1 cell per 106, P < 0.01), IL-2 (undetectable, P < 0.01), or IL-4 (undetectable, P < 0.01) in response to GAD 65 peptide by spleen cells from NOD mice reconstituted with either MMP-IAβ-d-GFP– or MMP-IAβ-k-GFP–transduced bone marrow (Figure 7). Similarly, a significant frequency of splenocytes from NOD mice reconstituted with control MMP-GFP–transduced bone marrow produced IFN-γ (41 ± 2 cells per 106), IL-2 (226 ± 18 cells per 106), and IL-4 (92 ± 3 cells per 106) in response to the insulin protein, while cells from NOD mice reconstituted with either MMP-IAβ-d-GFP– or MMP-IAβ-k-GFP–transduced bone marrow had significantly lower frequencies of cells producing IFN-γ (2 ± 2 cells per 106, P < 0.01), IL-2 (2 ± 3 cells per 106, P < 0.001), and IL-4 (1 ± 1 cell per 106, P < 0.01) in response to insulin.
Figure 7.

Expression of retrovirally transduced diabetes-resistant MHC class II genes in bone marrow_derived cells prevents the development of functional autoreactive T cells in NOD mice. Twenty to twenty-eight weeks after reconstitution, splenocytes were harvested from NOD mice reconstituted with MMP-IAβ-d-GFP_, MMP-IAβ-k-GFP_, or control MMP-GFP_transduced bone marrow. Single-cell suspensions were prepared and cultured overnight with whole insulin protein, GAD 65 peptide 206_220, or, as a control, peptide 85_99 of MBP. The frequency of cells producing IFN-γ (A), IL-2 (B), and IL-4 (C) was then measured by cytokine ELISPOT assays. Responses to GAD 65 or insulin by splenocytes from NOD mice reconstituted with MMP-GFP_transduced bone marrow (black bars) were compared with responses to GAD 65 or insulin by cells from NOD mice reconstituted with either MMP-IAβ-d-GFP_ or MMP-IAβ-k-GFP_transduced bone marrow (white bars). Shown is 1 representative experiment of 3.
Thymic deletion of islet antigen–specific T cells in NOD mice reconstituted with MHC class II–GFP–transduced bone marrow.
We reasoned that the elimination of functional islet-reactive T cells following expression of a retrovirally encoded diabetes-resistant I-Aβk or I-Aβd allele could result from either central deletion of islet-reactive clones in the thymus (21, 35), or peripheral inactivation of islet-reactive clones through regulatory mechanisms (36). To investigate the mechanism by which functional islet-reactive T cells are eliminated, we used I-Ag7 tetramers loaded with the peptide AAAAVRPLWVRMEAA (BDC-15) (37). This peptide, which was identified through the use of combinatorial peptide libraries (38), is known to stimulate islet-reactive T cell clones that mediate insulitis and cause diabetes. The I-Ag7 tetramer loaded with BDC-15 has been shown to bind a population of T cells in the thymus and pancreatic lymph nodes of NOD mice (37, 39). To determine whether expression of a retrovirally encoded diabetes-resistant I-Aβk or I-Aβd allele on bone marrow–derived cells resulted in thymic deletion, we used I-Ag7 tetramers loaded with the BDC-15 peptide to detect islet-reactive T cells in the thymus of NOD mice. Five- to six-week-old NOD mice were irradiated and reconstituted with 4 × 106 syngeneic bone marrow cells transduced with either MMP-IAβ-d-GFP or control MMP-GFP virus. Eleven weeks after bone marrow transplantation, mice were sacrificed along with age-matched naive NOD and BALB/c control mice, and single-cell thymocyte suspensions were prepared. Thymocytes were stained with anti-CD4 and anti-CD8 antibodies, annexin V, and I-Ag7 tetramers loaded with either the control CLIP peptide or BDC-15 peptide and analyzed by flow cytometry. As expected, we found that I-Ag7/BDC-15 tetramers labeled a population of CD4+ T cells (0.8%; Figure 8A) in the thymus of naive NOD mice, but not in control BALB/c mice (<0.1%). In mice reconstituted with bone marrow transduced with control MMP-GFP virus, CD4+ T cells in the thymus that were labeled with I-Ag7/BDC-15 tetramers were readily detectable at frequencies similar to those observed in naive NOD mice, suggesting that transduction and bone marrow transplantation alone do not affect the frequency of islet-reactive T cells. In contrast, the frequency of CD4+ T cells labeled with I-Ag7/BDC-15 tetramer was substantially reduced (0.1%) in thymocytes from NOD mice reconstituted with bone marrow transduced with MMP-IAβ-d-GFP virus. In all samples, staining with the control I-Ag7/CLIP tetramer resulted in only background labeling (Figure 8A).
Figure 8.

Central deletion of autoreactive T cells is mediated by retrovirally encoded MHC class II β chain. Eleven weeks after bone marrow transplantation, NOD mice reconstituted with either MMP-IAβ-d-GFP_ or control MMP-GFP_transduced bone marrow were sacrificed, and single-cell thymocyte suspensions were prepared. Single-cell suspensions were also prepared from naive age-matched NOD and BALB/c control mice. Thymocytes were stained with anti-CD4 and anti-CD8 antibodies, annexin V, and I-Ag7 tetramers loaded with either the control CLIP peptide or BDC-15 peptide, and analyzed by flow cytometry. (A) Single-positive CD4 T cells from mice reconstituted with bone marrow transduced with MMP-IAβ-d-GFP do not bind I-Ag7/BDC-15 tetramers. Cells were gated on live annexin V_CD4+CD8_ single-positive CD4 T cells. The percentage of these cells that bound to I-Ag7/CLIP (top row) and I-Ag7/BDC-15 tetramers (bottom row) is shown in the upper right quadrant. Shown is 1 representative experiment of 3. (B) Specific deletion of I-Ag7/BDC-15_binding T cells in mice reconstituted with bone marrow transduced with MMP-IAβ-d-GFP. Shown is expression of CD4 and CD8 after gating on I-Ag7/BDC-15 tetramer+, annexin V_ (live) cells. The total number of CD4+ single-positive T cells that bound the I-Ag7/BDC-15 tetramer was calculated by multiplication of the frequency of these cells by the total number of thymocytes recovered and is shown in the upper left quadrant. The total number of CD4+CD8+ double-positive thymocytes that bound the I-Ag7/BDC-15 tetramer was calculated similarly and is shown in the upper right quadrant. One representative experiment of 3 is shown.
To determine at what stage of thymus development elimination of islet-reactive T cells occurred, we gated on I-Ag7/BDC-15 tetramer–labeled cells and examined surface expression of CD4 and CD8. In both naive NOD mice and NOD mice reconstituted with bone marrow transduced with control MMP-GFP virus, the I-Ag7/BDC-15 tetramer labeled both CD4+CD8+ double-positive T cells and CD4+CD8– single-positive T cells (Figure 8B). In contrast, in NOD mice reconstituted with bone marrow transduced with MMP-IAβ-d-GFP virus, the I-Ag7/BDC-15 tetramer labeled CD4+CD8+ double-positive T cells, but not CD4+CD8– single-positive T cells. Naive NOD mice had similar numbers of CD4+CD8+ double-positive T cells (6.4 × 104) compared with NOD mice reconstituted with bone marrow transduced with control MMP-GFP virus (6.0 × 104), while NOD mice reconstituted with bone marrow transduced with MMP-IAβ-d-GFP virus had a reduced number (0.4 × 104). Note that the I-Ag7/BDC-15 tetramer is able to bind cells that express retrovirally encoded MHC class II alleles, since these cells express both I-Ag7 and I-Ad β chains. Together, these data suggest that CD4+ T cells expressing a T cell receptor that binds the I-Ag7/BDC-15 tetramer are negatively selected in the thymus of mice that express retrovirally encoded diabetes-resistant MHC class II alleles.
Discussion
Multiple genes contribute to the development of type 1 diabetes; however, inheritance of particular MHC alleles is the most significant determining factor (40). NOD mice that express MHC class II alleles other than I-Ag7 are protected from development of diabetes (16–18, 21, 41–44), which supports the notion that MHC plays a significant role in type 1 diabetes. However, it has been unclear whether protection results from negative selection of self-antigen–reactive T cells on diabetes-resistant MHC expressed in the thymus, or positive selection of regulatory T cells that can control disease progression. The role of MHC in bone marrow transplantation studies is perhaps less clear, because wild-type bone marrow cells express genes in addition to diabetes-resistant MHC that could influence disease progression (24–28).
Our data suggest that expression of MHC class II genes associated with protection from diabetes in hematopoietic cells of NOD mice alone is sufficient to prevent diabetes occurrence. Expression of diabetes-resistant MHC class II β chains prevented the development of functional T cells that respond to self-antigens that are known to be targets of T cell–mediated destruction of pancreatic β cells. In addition, expression of diabetes-resistant MHC class II β chains prevented T cell infiltration and insulitis. Furthermore, NOD mice expressing I-Ad or I-Ak MHC class II β chains on bone marrow–derived cells were protected from diabetes occurrence following administration of cyclophosphamide, a drug that is known to accelerate the development of diabetes in NOD mice. Together these data suggest that gene therapy approaches provide robust protection from T cell–mediated autoimmunity.
Several T cell clones that recognize islet antigens have been isolated from NOD mice. Transfer of one of these T cell clones, BDC2.5, into NOD/scid mice results in the rapid development of diabetes (45). Although the identity of the autoantigen that these clones recognize is not known, a peptide mimetic that stimulates most of these clones at low peptide concentrations had been identified, and an MHC class II tetramer to which the peptide mimetic is bound will label both these BDC2.5 T cells and a population of CD4+ T cells in the lymph nodes and thymus of prediabetic NOD mice. Expansion of the population of tetramer/peptide mimetic binding autoreactive T cells correlates with increased insulitis and the onset of diabetes (37, 39). Using this reagent, we stained thymocytes from either mice expressing retrovirally transduced diabetes-resistant MHC class II β chains on bone marrow cells, or mice expressing only GFP in bone marrow–derived cells. The thymus of naive NOD and control mice contained a substantial population of CD4+CD8– single-positive T cells able to bind the tetramer/peptide complex. In contrast, tetramer-binding CD4+CD8– single-positive T cells were not detected in the thymus of mice expressing the diabetes-resistant MHC class II β chain on bone marrow–derived cells. These data suggest that the expression of diabetes-resistant MHC class II β chain is sufficient to prevent the development of these autoreactive T cells in the thymus.
Failure to develop autoreactive T cells in these mice may be due to either negative selection of autoreactive clones, or failure to positively select these T cells. Bone marrow–derived cells lack the ability to positively select T cells (46, 47) but are potent mediators of negative selection (48–51), which suggests that the lack of functional self-reactive T cells in NOD mice reconstituted with retrovirally transduced bone marrow is related to the ability of transduced MHC class II genes expressed on bone marrow–derived cells to mediate negative selection of self-reactive T cells in the thymus. Our data demonstrate that CD4+CD8+/lo double-positive as well as CD4+ single-positive T cells in the thymus of control mice receiving MMP-GFP–transduced bone marrow were able to bind the BDC-15 tetramer. The absolute number of CD4+CD8+/lo double-positive T cells able to bind the BDC-15 tetramer was lower in mice reconstituted with MHC class II–transduced bone marrow. Furthermore, we were unable to detect tetramer-binding CD4+ single-positive T cells in these mice. These data suggest that islet antigen–reactive T cells are able to develop to the double-positive stage of T cell development in mice reconstituted with MHC class II β chain–transduced bone marrow and are then negatively selected. Consistent with this idea, we have observed that expression of retrovirally transduced allogeneic MHC antigens on bone marrow–derived cells results in negative selection of alloantigen-reactive T cells in the thymus (52).
Other gene therapy strategies aimed at modifying host islet autoimmunity include the introduction of cytokine genes such as IL-10 (53–58), IL-4 (59–63), TGF-β (64), and IFN-γ receptor (65, 66), and blockade of costimulatory molecules (67, 68). Promising results have been obtained in animal models; however, all of these strategies are geared toward downregulation of existing autoimmune T cells. It is likely therefore, that the more effective of these strategies will cause nonspecific immunosuppression in recipients. However, the disadvantages of long-term immunosuppression are well documented and include increased risk of malignancy and infection. Using genetic engineering to confer expression of diabetes-resistant MHC class II alleles, we show the specific deletion of islet-reactive T cells, and animals are not otherwise immunocompromised.
Several lines of evidence suggest that, in humans, once destructive anti-islet immune responses have developed, an asymptomatic period precedes onset of clinical disease for several months to years (reviewed in ref. 69). In addition, in many patients, even at disease onset there is evidence of residual β cell function as demonstrated by c-peptide secretion. Consequently, the elimination of autoimmunity early after disease onset has the potential to substantially ameliorate type 1 diabetes. One possible intervention is the transplantation of allogeneic bone marrow cells, which can prevent diabetes in NOD mice (1, 24–28). However, transplantation of allogeneic bone marrow carries with it the risk of inducing graft-versus-host disease, which can be fatal, and the conditioning regimen required for allogeneic engraftment makes this therapy unsuitable for treatment of prediabetic patients. Our data suggest that expression of MHC class II genes associated with protection from diabetes through genetic engineering of hematopoietic stem cells could prevent type 1 diabetes in individuals that express MHC alleles associated with susceptibility to disease and exhibit signs of diminishing insulin response. Since this approach involves genetic modification of autologous hematopoietic progenitors to establish molecular rather than cellular chimerism (70), problems such as graft-versus-host disease that are associated with transplantation of allogeneic bone marrow are eliminated. In addition, our data show that diabetes can be prevented with relatively low levels of expression of diabetes-resistant MHC alleles. This suggests that the conditioning regimen required to achieve sufficient engraftment of genetically modified bone marrow progenitors to prevent diabetes may be substantially less rigorous than that required for allogeneic bone marrow transplantation.
For islet transplantation to be successful as a long-term cure for type 1 diabetes (71–73), it is necessary to overcome the underlying problem of autoimmunity that leads to diabetes occurrence and destruction of the transplanted islets (74). Our data demonstrate that the introduction of diabetes-resistant MHC class II β chain on bone marrow–derived cells is sufficient to prevent the development of functional islet-reactive T cells, and to mediate the intrathymic deletion of these cells. This may be of clinical significance in the context of islet transplantation, and this approach could also be used to prevent the recurrence of autoimmunity after islet transplantation.
Methods
Mice.
NOD/MrkTac mice were purchased from Taconic. BALB/c mice were purchased from The Jackson Laboratory. Four- to six-week-old female mice were used in all experiments. All experiments were performed in accordance with institutional guidelines.
Vector construction.
Full-length cDNAs encoding I-Aβd and I-Aβk were cloned by RT-PCR from BALB/c or CBA/Ca spleen cells, respectively. The products were ligated into pEGFP-N2 (BD Biosciences — Clontech) in-frame with eGFP. Genes encoding I-Aβ–GFP fusion proteins were then cloned into the pMMPf2 vector (a kind gift of Richard Mulligan, Children’s Hospital, Boston, Massachusetts, USA) to generate pMMP-IAβ-d-GFP and pMMP-IAβ-k-GFP. The MMP retroviral vector is a derivative of MFG (75).
Production of recombinant retroviruses.
Virus preparations were made at the Harvard Institute of Human Genetics (Boston, Massachusetts, USA) in collaboration with Richard Mulligan (Harvard Gene Therapy Initiative). Virus preparations tested negative for helper virus contamination. The titers of retroviral supernatants used in this study were between 3 × 106 and 5 × 106 infectious particles per milliliter. Control MMP-GFP retrovirus was kindly provided by Richard Mulligan through the Harvard Gene Therapy Initiative.
Transduction of M12.C3 and A20 cell lines.
The M12.C3 cell line was the kind gift of Laurie Glimcher (Harvard Medical School). The A20 cell line was purchased from the American Type Culture Collection. In each case, 106 cells were plated 24 hours prior to transduction, and 1 ml viral supernatant was added to the cells at 24-hour intervals over 3 days. The cells were then washed and cultured for an additional 48 hours prior to analysis by flow cytometry.
Bone marrow transduction and transplantation.
Bone marrow cells were transduced as described previously (31). The multiplicity of infection (MOI) used was 2.2 for MMP-IAβ-d-GFP–transduced, 1.9 for MMP-IAβ-k-GFP–transduced, and 3 for MMP-GFP–transduced cells. After 96 hours in culture, cells were harvested and washed to remove excess virus, and 4 × 106 cells were injected intravenously into preconditioned recipients. Recipients were preconditioned with 10.25 Gy irradiation 1 day prior to bone marrow transplantation. In some experiments, recipients were treated with 0.5 mg of the anti-CD4 antibody GK1.5 (76), and 0.5 mg of the anti-CD8 antibody 116-13.1 (77).
Antibodies.
Monoclonal antibodies specific for CD4 (RM4-5), CD8 (53-6.7), CD3 (145-2C11), Ly-6G (Gr-1, RB6-8C5), B220 (RA3-6B2), CD11b (Mac-1, M1/70), NK cells (DX5), CD11c (HL3), IgG2a (19–15), GFP (JL-8), I-Ad (39–10–8), and I-Aβk (10-3.6) were obtained from BD Biosciences — Pharmingen. Polyclonal guinea pig anti–porcine insulin antibodies were obtained from DAKO Corp. Phycoerythrin-conjugated (PE-conjugated) anti–mouse CD3 (17A2; BD Biosciences — Pharmingen) was used for confocal microscopy studies.
Immunofluorescence and confocal microscopy.
Eighteen weeks after bone marrow transplantation, PBMCs were collected from mice reconstituted with either MMP-IAβ-d-GFP– or MMP-GFP–transduced bone marrow and stained with either biotin-conjugated 39-10-8 or PE-conjugated 17A2 for 30 minutes. Samples stained with 39-10-8 were incubated for an additional 30 minutes with PE-conjugated streptavidin (BD Biosciences — Pharmingen). The cells were then permeabilized with 0.1% Triton X-100 and stained with JL-8 for 30 minutes at 4°C. JL-8 binding was revealed by staining with FITC-conjugated anti-mouse IgG2a. Samples were then analyzed by confocal microscopy.
Blood glucose levels.
Blood glucose levels were monitored weekly using a standard glucometer. Two consecutive readings of greater than 300 mg/dl were considered to be indicative of the development of hyperglycemia.
ELISPOT.
Cytokine ELISPOT assays were performed using HA plates (Millipore Corp.) according to the manufacturer’s specifications. BVD-1D11 (anti–IL-4), JES6-1A12 (anti–IL-2), and R4-6A2 (anti–IFN-γ) (Caltag Laboratories Inc.) were used at 10 μg/ml to coat plates. In ELISPOT assays, 106 (for IL-4), 0.5 × 106 (for IL-2), or 3 × 106 (for IFN-γ) splenocytes were added to each well. Cells were cultured overnight with 0.1 mg/ml insulin (Sigma-Aldrich), MBP peptide (peptide 85–99, ENPVVHFFKNIVTPR), or GAD 65 peptide (peptide 206–220, TYEIAPVFVLLEYVT). Peptides were obtained from Invitrogen Corp. Cytokine production was detected using 10 μg/ml biotinylated BVD-24G2, JES6-5H4, and XMG1.2 (Caltag Laboratories Inc.). Assays were performed in triplicate and averaged. To calculate the specific response to GAD 65 and insulin, the average background response to MBP was subtracted. There was no significant difference in the response to MBP peptide between the groups (not shown).
Histology.
One half of each pancreas analyzed was fixed in 10% buffered formalin and processed for paraffin embedding. The remaining half was snap-frozen in OCT compound (Tissue-Tek; Sakura Finetek USA Inc.). Paraffin-embedded tissue sections (5 μm) were stained with guinea pig anti–porcine insulin antibody for 15 minutes and stained with a biotinylated goat anti–guinea pig secondary antibody (Vector Laboratories Inc.) for 30 minutes. Sections were developed using ABC solution and 3,3′-diaminobenzidine (Vector Laboratories Inc.) and counterstained with hematoxylin. Frozen samples were stained with biotin-conjugated 145-2C11 for 30 minutes and developed with ABC solution (Vector Laboratories Inc.) and 3,3′-diaminobenzidine/nickel. Sections were then stained with anti-insulin antibodies for 15 minutes, followed by biotin-conjugated goat anti–guinea pig secondary antibody (Vector Laboratories Inc.) for 30 minutes. The sections were incubated with streptavidin-HRP (Amersham Biosciences) and developed with AEC substrate–chromogen (DAKO Corp.) according to the manufacturer’s instructions. Coverslips were mounted after counterstaining with hematoxylin.
MHC class II tetramer staining.
Biotinylated, MHC class II monomers were prepared as previously described (37). The tetramers were loaded with the BDC-15 peptide, which differs from the previously used BDC-13 peptide by the addition of an alanine residue at both the N- and the C-terminus (AAAAVRPLWVRMEAA). Both BDC-13 and BDC-15 provide similar levels of labeling. To form tetramers, biotinylated MHC class II monomers and streptavidin-PE (Invitrogen Corp.) were mixed at a molar ratio of 4:1 and incubated for 1 hour. The tetramer was then incubated with 5 × 106 thymocytes at a final concentration of 10 μg/ml for 30 minutes. Cells were additionally stained with anti-CD4 (RM4-5; BD Biosciences — Pharmingen), anti-CD8a (53-6.7; BD Biosciences — Pharmingen), and annexin V (BD Biosciences — Pharmingen) for 20 minutes.
Acknowledgments
We thank Richard C. Mulligan and Jeng-Shin Lee for the MMP retroviral vector and packaging system and for technical advice; and the Harvard Gene Therapy Initiative Vector Core, supported in part by the Association Française contre les Myopathies, for production of retroviruses. In addition, we thank Xandra Breakefield and Gerald L. Waneck for critical review of the manuscript, and members of the Iacomini laboratory for helpful discussions. We thank Denise Malkowski and Lee Bar-Sagi for expert technical assistance. This work was supported in part by NIH grants R01 AI44268 and R01 AI43619 (to J. Iacomini).
Footnotes
See the related Commentary beginning on page 892.
Nonstandard abbreviations used: GAD 65, glutamic acid decarboxylase 65; MBP, myelin basic protein; PE, phycoerythrin.
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Wicker LS, Todd JA, Peterson LB. Genetic control of autoimmune diabetes in the NOD mouse. Annu. Rev. Immunol. 1995;13:179–200. doi: 10.1146/annurev.iy.13.040195.001143. [DOI] [PubMed] [Google Scholar]
- 2.Nepom GT, Kwok WW. Molecular basis for HLA-DQ associations with IDDM. Diabetes. 1998;47:1177–1184. doi: 10.2337/diab.47.8.1177. [DOI] [PubMed] [Google Scholar]
- 3.McDevitt H. The role of MHC class II molecules in susceptibility and resistance to autoimmunity. Curr. Opin. Immunol. 1998;10:677–681. doi: 10.1016/s0952-7915(98)80088-5. [DOI] [PubMed] [Google Scholar]
- 4.Morel PA, Dorman JS, Todd JA, McDevitt HO, Trucco M. Aspartic acid at position 57 of the HLA-DQ beta chain protects against Type I diabetes: a family study. Proc. Natl. Acad. Sci. U. S. A. 1988;85:8111–8115. doi: 10.1073/pnas.85.21.8111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ilonen J, et al. Estimation of genetic risk for type 1 diabetes. Am. J. Med. Genet. 2002;115:30–36. doi: 10.1002/ajmg.10341. [DOI] [PubMed] [Google Scholar]
- 6.Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat. Med. 1999;5:601–604. doi: 10.1038/9442. [DOI] [PubMed] [Google Scholar]
- 7.Hattori M, et al. The NOD mouse: recessive diabetogenic gene in the major histocompatibility complex. Science. 1986;231:733–735. doi: 10.1126/science.3003909. [DOI] [PubMed] [Google Scholar]
- 8.Lee KH, Wucherpfennig KW, Wiley DC. Structure of a human insulin peptide-HLA-DQ8 complex and susceptibility to type 1 diabetes. Nat. Immunol. 2001;2:501–507. doi: 10.1038/88694. [DOI] [PubMed] [Google Scholar]
- 9.Stratmann T, et al. The I-Ag7 MHC class II molecule linked to murine diabetes is a promiscuous peptide binder. J. Immunol. 2000;165:3214–3225. doi: 10.4049/jimmunol.165.6.3214. [DOI] [PubMed] [Google Scholar]
- 10.Ettinger RA, Liu AW, Nepom GT, Kwok WW. Exceptional stability of the HLA-DQA1*0102/DQB1*0602 αβ protein dimer, the class II MHC molecule associated with protection from insulin-dependent diabetes mellitus. J. Immunol. 1998;161:6439–6445. [PubMed] [Google Scholar]
- 11.Corper AL, et al. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science. 2000;288:505–511. doi: 10.1126/science.288.5465.505. [DOI] [PubMed] [Google Scholar]
- 12.Abraham RS, Kudva YC, Wilson SB, Strominger JL, David CS. Co-expression of HLA DR3 and DQ8 results in the development of spontaneous insulitis and loss of tolerance to GAD65 in transgenic mice. Diabetes. 2000;49:548–554. doi: 10.2337/diabetes.49.4.548. [DOI] [PubMed] [Google Scholar]
- 13.Acha-Orbea H, McDevitt HO. The first external domain of the nonobese diabetic mouse class II I-A beta chain is unique. Proc. Natl. Acad. Sci. U. S. A. 1987;84:2435–2439. doi: 10.1073/pnas.84.8.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Todd JA, et al. A molecular basis for MHC class II-associated autoimmunity. Science. 1988;240:1003–1009. doi: 10.1126/science.3368786. [DOI] [PubMed] [Google Scholar]
- 15.Luhder F, Katz J, Benoist C, Mathis D. Major histocompatibility complex class II molecules can protect from diabetes by positively selecting T cells with additional specificities. J. Exp. Med. 1998;187:379–387. doi: 10.1084/jem.187.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uehira M, et al. Development of autoimmune insulitis is prevented in E alpha d but not in A beta k NOD transgenic mice. Int. Immunol. 1989;1:209–213. doi: 10.1093/intimm/1.2.209. [DOI] [PubMed] [Google Scholar]
- 17.Reich EP, Sherwin RS, Kanagawa O, Janeway CAJ. An explanation for the protective effect of the MHC class II I-E molecule in murine diabetes. Nature. 1989;341:326–328. doi: 10.1038/341326a0. [DOI] [PubMed] [Google Scholar]
- 18.Lund T, et al. Prevention of insulin-dependent diabetes mellitus in non-obese diabetic mice by transgenes encoding modified I-A beta-chain or normal I-E alpha-chain. Nature. 1990;345:727–729. doi: 10.1038/345727a0. [DOI] [PubMed] [Google Scholar]
- 19.Slattery RM, et al. Prevention of diabetes in non-obese diabetic I-Ak transgenic mice. Nature. 1990;345:724–726. doi: 10.1038/345724a0. [DOI] [PubMed] [Google Scholar]
- 20.Miyazaki T, et al. Direct evidence for the contribution of the unique I-ANOD to the development of insulitis in non-obese diabetic mice. Nature. 1990;345:722–724. doi: 10.1038/345722a0. [DOI] [PubMed] [Google Scholar]
- 21.Schmidt D, Verdaguer J, Averill N, Santamaria P. A mechanism for the major histocompatibility complex-linked resistance to autoimmunity. J. Exp. Med. 1997;186:1059–1075. doi: 10.1084/jem.186.7.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singer SM, et al. Prevention of diabetes in NOD mice by a mutated I-Ab transgene. Diabetes. 1998;47:1570–1577. doi: 10.2337/diabetes.47.10.1570. [DOI] [PubMed] [Google Scholar]
- 23.Singer SM, Tisch R, Yang XD, McDevitt HO. An Abd transgene prevents diabetes in nonobese diabetic mice by inducing regulatory T cells. Proc. Natl. Acad. Sci. U. S. A. 1993;90:9566–9570. doi: 10.1073/pnas.90.20.9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.LaFace DM, Peck AB. Reciprocal allogeneic bone marrow transplantation between NOD mice and diabetes-nonsusceptible mice associated with transfer and prevention of autoimmune diabetes. Diabetes. 1989;38:894–901. doi: 10.2337/diab.38.7.894. [DOI] [PubMed] [Google Scholar]
- 25.Naji A, Silvers WK, Bellgrau D, Barker CF. Spontaneous diabetes in rats: destruction of islets is prevented by immunological tolerance. Science. 1981;213:1390–1392. doi: 10.1126/science.6791286. [DOI] [PubMed] [Google Scholar]
- 26.Seung E, et al. Allogeneic hematopoietic chimerism in mice treated with sublethal myeloablation and anti-CD154 antibody: absence of graft-versus-host disease, induction of skin allograft tolerance, and prevention of recurrent autoimmunity in islet-allografted NOD/Lt mice. Blood. 2000;95:2175–2182. [PubMed] [Google Scholar]
- 27.Beilhack GF, et al. Purified allogeneic hematopoietic stem cell transplantation blocks diabetes pathogenesis in NOD mice. Diabetes. 2003;52:59–68. doi: 10.2337/diabetes.52.1.59. [DOI] [PubMed] [Google Scholar]
- 28.Li H, et al. Mixed allogeneic chimerism induced by a sublethal approach prevents autoimmune diabetes and reverses insulitis in nonobese diabetic (NOD) mice. J. Immunol. 1996;156:380–388. [PubMed] [Google Scholar]
- 29.Pearson T, et al. Islet cell autoimmunity and transplantation tolerance: two distinct mechanisms? Ann. N. Y. Acad. Sci. 2003;1005:148–156. doi: 10.1196/annals.1288.016. [DOI] [PubMed] [Google Scholar]
- 30.Glimcher LH, et al. IA mutant functional antigen-presenting cell lines. J. Immunol. 1983;130:2287–2294. [PubMed] [Google Scholar]
- 31.Bracy JL, Iacomini J. Induction of B cell tolerance by retroviral gene therapy. Blood. 2000;96:3008–3015. [PubMed] [Google Scholar]
- 32.Andre-Schmutz I, Hindelang C, Benoist C, Mathis D. Cellular and molecular changes accompanying the progression from insulitis to diabetes. Eur. J. Immunol. 1999;29:245–255. doi: 10.1002/(SICI)1521-4141(199901)29:01<245::AID-IMMU245>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 33.Chao CC, Sytwu HK, Chen EL, Toma J, McDevitt H. The role of MHC class II molecules in susceptibility to type I diabetes: identification of peptide epitopes and characterization of the T cell repertoire. Proc. Natl. Acad. Sci. U. S. A. 1999;96:9299–9304. doi: 10.1073/pnas.96.16.9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hausmann DHF, Yu B, Hausmann S, Wucherpfennig KW. pH-dependent peptide binding properties of the type I diabetes-associated I-Ag7 molecule: rapid release of CLIP at an endosomal pH. J. Exp. Med. 1999;189:1723–1733. doi: 10.1084/jem.189.11.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmidt D, Amrani A, Verdaguer J, Bou S, Santamaria P. Autoantigen-independent deletion of diabetogenic CD4+ thymocytes by protective MHC class II molecules. J. Immunol. 1999;162:4627–4636. [PubMed] [Google Scholar]
- 36.Singer SM, Tisch R, Yang XD, McDevitt HO. An Abd transgene prevents diabetes in nonobese diabetic mice by inducing regulatory T cells. Proc. Natl. Acad. Sci. U. S. A. 1993;90:9566–9570. doi: 10.1073/pnas.90.20.9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jang MH, Seth NP, Wucherpfennig KW. Ex vivo analysis of thymic CD4 T cells in nonobese diabetic mice with tetramers generated from I-A(g7)/class II-associated invariant chain peptide precursors. J. Immunol. 2003;171:4175–4186. doi: 10.4049/jimmunol.171.8.4175. [DOI] [PubMed] [Google Scholar]
- 38.Judkowski V, et al. Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J. Immunol. 2001;166:908–917. doi: 10.4049/jimmunol.166.2.908. [DOI] [PubMed] [Google Scholar]
- 39.You S, et al. Detection and characterization of T cells specific for BDC2.5 T cell-stimulating peptides. J. Immunol. 2003;170:4011–4020. doi: 10.4049/jimmunol.170.8.4011. [DOI] [PubMed] [Google Scholar]
- 40.Vyse TJ, Todd JA. Genetic analysis of autoimmune disease. Cell. 1996;85:311–318. doi: 10.1016/s0092-8674(00)81110-1. [DOI] [PubMed] [Google Scholar]
- 41.Slattery RM, et al. Prevention of diabetes in non-obese diabetic I-Ak transgenic mice. Nature. 1990;345:724–726. doi: 10.1038/345724a0. [DOI] [PubMed] [Google Scholar]
- 42.Miyazaki T, et al. Direct evidence for the contribution of the unique I-ANOD to the development of insulitis in non-obese diabetic mice. Nature. 1990;345:722–724. doi: 10.1038/345722a0. [DOI] [PubMed] [Google Scholar]
- 43.Singer SM, et al. Prevention of diabetes in NOD mice by a mutated I-Ab transgene. Diabetes. 1998;47:1570–1577. doi: 10.2337/diabetes.47.10.1570. [DOI] [PubMed] [Google Scholar]
- 44.Singer SM, Tisch R, Yang XD, McDevitt HO. An Abd transgene prevents diabetes in nonobese diabetic mice by inducing regulatory T cells. Proc. Natl. Acad. Sci. U. S. A. 1993;90:9566–9570. doi: 10.1073/pnas.90.20.9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haskins K, McDuffie M. Acceleration of diabetes in young NOD mice with a CD4+ islet-specific T cell clone. Science. 1990;249:1433–1436. doi: 10.1126/science.2205920. [DOI] [PubMed] [Google Scholar]
- 46.Markowitz JS, Auchincloss H, Jr, Grusby MJ, Glimcher LH. Class II-positive hematopoietic cells cannot mediate positive selection of CD4+ T lymphocytes in class II-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 1993;90:2779–2783. doi: 10.1073/pnas.90.7.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Urdahl KB, Sun JC, Bevan MJ. Positive selection of MHC class Ib-restricted CD8(+) T cells on hematopoietic cells. Nat. Immunol. 2002;3:772–779. doi: 10.1038/ni814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jenkinson EJ, Anderson G, Owen JJ. Studies on T cell maturation on defined thymic stromal cell populations in vitro. J. Exp. Med. 1992;176:845–853. doi: 10.1084/jem.176.3.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brocker T, Riedinger M, Karjalainen K. Targeted expression of major histocompatibility complex (MHC) class II molecules demonstrates that dendritic cells can induce negative but not positive selection of thymocytes in vivo. J. Exp. Med. 1997;185:541–550. doi: 10.1084/jem.185.3.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Owen RD. Immunogenetic consequences of vascular anastomoses between bovine twins. Science. 1945;102:400–401. doi: 10.1126/science.102.2651.400. [DOI] [PubMed] [Google Scholar]
- 51.Billingham RE, Brent L, Medawar PB. Actively acquired tolerance to foreign cells. Nature. 1953;172:603–606. doi: 10.1038/172603a0. [DOI] [PubMed] [Google Scholar]
- 52.Kang ES, Iacomini J. Induction of central deletional T cell tolerance by gene therapy. J. Immunol. 2002;169:1930–1935. doi: 10.4049/jimmunol.169.4.1930. [DOI] [PubMed] [Google Scholar]
- 53.Ko KS, Lee M, Koh JJ, Kim SW. Combined administration of plasmids encoding IL-4 and IL-10 prevents the development of autoimmune diabetes in nonobese diabetic mice. Mol. Ther. 2001;4:313–316. doi: 10.1006/mthe.2001.0459. [DOI] [PubMed] [Google Scholar]
- 54.Koh JJ, et al. Degradable polymeric carrier for the delivery of IL-10 plasmid DNA to prevent autoimmune insulitis of NOD mice. Gene Ther. 2000;7:2099–2104. doi: 10.1038/sj.gt.3301334. [DOI] [PubMed] [Google Scholar]
- 55.Goudy K, et al. Adeno-associated virus vector-mediated IL-10 gene delivery prevents type 1 diabetes in NOD mice. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13913–13918. doi: 10.1073/pnas.251532298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang YC, et al. Adeno-associated virus-mediated IL-10 gene therapy inhibits diabetes recurrence in syngeneic islet cell transplantation of NOD mice. Diabetes. 2003;52:708–716. doi: 10.2337/diabetes.52.3.708. [DOI] [PubMed] [Google Scholar]
- 57.Yang Z, et al. Suppression of autoimmune diabetes by viral IL-10 gene transfer. J. Immunol. 2002;168:6479–6485. doi: 10.4049/jimmunol.168.12.6479. [DOI] [PubMed] [Google Scholar]
- 58.Moritani M, et al. Prevention of adoptively transferred diabetes in nonobese diabetic mice with IL-10-transduced islet-specific Th1 lymphocytes. A gene therapy model for autoimmune diabetes. J. Clin. Invest. 1996;98:1851–1859. doi: 10.1172/JCI118986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zipris D, Karnieli E. A single treatment with IL-4 via retrovirally transduced lymphocytes partially protects against diabetes in BioBreeding (BB) rats. JOP. 2002;3:76–82. [PubMed] [Google Scholar]
- 60.Hayashi T, Yasutomi Y, Hasegawa K, Sasaki Y, Onodera T. Interleukin-4-expressing plasmid DNA inhibits reovirus type-2-triggered autoimmune insulitis in DBA/1 J suckling mice. Int. J. Exp. Pathol. 2003;84:101–106. doi: 10.1046/j.1365-2613.2003.00341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Horiki M, et al. High-level expression of interleukin-4 following electroporation-mediated gene transfer accelerates Type 1 diabetes in NOD mice. J. Autoimmun. 2003;20:111–117. doi: 10.1016/s0896-8411(03)00004-0. [DOI] [PubMed] [Google Scholar]
- 62.Feili-Hariri M, et al. Dendritic cells transduced to express interleukin-4 prevent diabetes in nonobese diabetic mice with advanced insulitis. Hum. Gene Ther. 2003;14:13–23. doi: 10.1089/10430340360464679. [DOI] [PubMed] [Google Scholar]
- 63.Lee M, Ko KS, Oh S, Kim SW. Prevention of autoimmune insulitis by delivery of a chimeric plasmid encoding interleukin-4 and interleukin-10. J. Control. Release. 2003;88:333–342. doi: 10.1016/s0168-3659(03)00031-2. [DOI] [PubMed] [Google Scholar]
- 64.Piccirillo CA, Chang Y, Prud’homme GJ. TGF-β1 somatic gene therapy prevents autoimmune disease in nonobese diabetic mice. J. Immunol. 1998;161:3950–3956. [PubMed] [Google Scholar]
- 65.Chang Y, Prud’homme GJ. Intramuscular administration of expression plasmids encoding interferon-gamma receptor/IgG1 or IL-4/IgG1 chimeric proteins protects from autoimmunity. J. Gene Med. 1999;1:415–423. doi: 10.1002/(SICI)1521-2254(199911/12)1:6<415::AID-JGM66>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 66.Prud’homme GJ, Chang Y. Prevention of autoimmune diabetes by intramuscular gene therapy with a nonviral vector encoding an interferon-gamma receptor/IgG1 fusion protein. Gene Ther. 1999;6:771–777. doi: 10.1038/sj.gt.3300879. [DOI] [PubMed] [Google Scholar]
- 67.Lu L, et al. Transduction of dendritic cells with adenoviral vectors encoding CTLA4-Ig markedly reduces their allostimulatory activity [abstract] Transplant. Proc. 1999;31:797. doi: 10.1016/s0041-1345(98)01774-6. [DOI] [PubMed] [Google Scholar]
- 68.Lu L, et al. Genetic engineering of dendritic cells to express immunosuppressive molecules (viral IL-10, TGF-beta, and CTLA4Ig) J. Leukoc. Biol. 1999;66:293–296. doi: 10.1002/jlb.66.2.293. [DOI] [PubMed] [Google Scholar]
- 69.Daaboul J, Schatz D. Overview of prevention and intervention trials for type 1 diabetes. Rev. Endocr. Metab. Disord. 2003;4:317–323. doi: 10.1023/a:1027308310837. [DOI] [PubMed] [Google Scholar]
- 70.Bagley J, Iacomini J. Gene therapy progress and prospects: gene therapy in organ transplantation. Gene Ther. 2003;10:605–611. doi: 10.1038/sj.gt.3302020. [DOI] [PubMed] [Google Scholar]
- 71.White SA, Nicholson ML, Hering BJ. Can islet cell transplantation treat diabetes? BMJ. 2000;321:651–652. doi: 10.1136/bmj.321.7262.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shapiro J, et al. Islet transplantation in seven patients with type-1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 2000;343:230–238. doi: 10.1056/NEJM200007273430401. [DOI] [PubMed] [Google Scholar]
- 73.Ryan EA, et al. Clinical outcomes and insulin secretion after islet transplantation with the Edmonton protocol. Diabetes. 2001;50:710–719. doi: 10.2337/diabetes.50.4.710. [DOI] [PubMed] [Google Scholar]
- 74.Sutherland DE, et al. Twin-to-twin pancreas transplantation: reversal and reenactment of the pathogenesis of type I diabetes. Trans. Assoc. Am. Physicians. 1984;97:80–87. [PubMed] [Google Scholar]
- 75.Riviere I, Brose K, Mulligan RC. Effects of retroviral vector design on expression of human adenosine deaminase in murine marrow transplant recipients engrafted with genetically modified cells. Proc. Natl. Acad. Sci. U. S. A. 1998;92:6733–6737. doi: 10.1073/pnas.92.15.6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dialynas DP, et al. Characterization of the murine T cell surface molecule designated L3T4, identified by monoclonal antibody GK1.5: similarity of L3T4 to the human Leu 3/T4 molecule. J. Immunol. 1984;131:2445–2451. [PubMed] [Google Scholar]
- 77.Shen, F.-W. 1983. Monoclonal antibodies to mouse lymphocyte differentiation alloantigens. In Monoclonal antibodies and T-cell hybridomas. G.J. Hammerling, U. Hammerling, and J.F. Kearney, editors. Elsevier North-Holland. Amsterdam, The Netherlands. 25–31.