

Abstract
Adult bone marrow-derived cells can participate in muscle regeneration after bone marrow transplantation. In recent studies a single hematopoietic stem cell (HSC) was shown to give rise to cells that not only reconstituted all of the lineages of the blood, but also contributed to mature muscle fibers. However, the relevant HSC derivative with this potential has not yet been definitively identified. Here we use fluorescence-activated cell sorter-based protocols to test distinct hematopoietic fractions and show that only fractions containing c-kit+ immature myelomonocytic precursors are capable of contributing to muscle fibers after i.m. injection. Although these cells belong to the myeloid lineage, they do not include mature CD11b+ myelomonocytic cells, such as macrophages. Of the four sources of mature macrophages tested that were derived either from monocytic culture, bone marrow, peripheral blood after granulocyte colony-stimulating factor mobilization, or injured muscle, none contributed to muscle. In addition, after transplantation of bone marrow isolated from CD11b-Cre-transgenic mice into the Cre-reporter strain (Z/EG), no GFP myofibers were detected, demonstrating that macrophages expressing CD11b do not fuse with myofibers. Irrespective of the underlying mechanisms, these data suggest that the HSC derivatives that integrate into regenerating muscle fibers exist in the pool of hematopoietic cells known as myelomonocytic progenitors.
For decades, it has been debated whether cells from the circulation could participate in skeletal muscle regeneration in adult animals (reviewed in ref. 1). Recently, bone marrow (BM)-derived cells (BMDC) have been shown by investigators in several groups to integrate into diverse adult tissues, such as epithelia (2), liver (3), heart (4), brain (5), and skeletal muscle in both mice and humans (reviewed in ref. 6). Moreover, BMDC have the capacity to rescue a genetically lethal metabolic liver disease, highlighting their therapeutic potential (7). Numerous reports clearly demonstrate that BMDC contribute to skeletal muscle fibers (4, 8–14). Although the frequency of these events is generally reported to be low (0.01–0.1%), it increases markedly and can reach 5% of total fibers in some muscles with high contractile activity or in muscles damaged by stress (13, 14). We and others have demonstrated that BMDC en route to muscle can follow a biological progression, first giving rise to muscle-specific stem cells (satellite cells) and then fusing under physiological conditions to form mature myofibers (12, 13, 15). Whether all BMDC follow this progression remains unclear, and it is quite possible that a proportion of cells fuse directly with myofibers and that both mechanisms coexist in the same tissue.
It has now been shown by single-cell transplantation experiments that a hematopoietic stem cell (HSC) can give rise to progeny that reconstitute the blood and integrate into regenerating myofibers (16, 17). However, whether the HSC themselves or only a subset of HSC derivatives are the cells involved in this process remains to be determined. Definitive identification of the BMDC that integrate into muscle fibers is required, because the nature of the precise population of cells involved in this process will be critical in guiding future research in this field.
Here we identify the HSC derivatives that have the capacity to incorporate into muscle fibers. These derivatives were isolated by using fluorescence-activated cell sorter (FACS)-based fractionation of cells from BM, peripheral blood (PB), and damaged muscle, followed by direct i.m. injection. This approach made it possible to test more mature fractions that were not capable of reconstituting the blood. We show that HSC and their progeny that express markers of the premyelomonocytic cells can incorporate into myotoxin-damaged myofibers, whereas, with four distinct paradigms, mature myelomonocytic cells (macrophages) cannot. Knowledge of the subset of BMDC derivatives with the potential to contribute to muscle should aid elucidation of the mechanisms by which they are recruited, incorporated, and reprogrammed during this process.
Materials and Methods
Mice and BM Transplantation (BMT) Procedure. C57BL/6, cytomegalovirus (CMV)-Cre, and C57BL/6-GFP transgenic mice were purchased from The Jackson Laboratory. CD11b-Cre transgenic and Cre-reporter transgenic mice (Z/EG) (18, 19) were generously provided by D. G. Tenen (Harvard Institutes of Medicine, Boston) and C. G. Lobe (College Health Centre, Toronto), respectively. BMT and i.m. injection procedures are described in detail in Supporting Materials and Methods, which is published as supporting information on the PNAS web site. All protocols were approved by the Administrative Panel on Laboratory Animal Care at Stanford University School of Medicine.
Cell Sorting by Flow Cytometry. BM, PB, or skeletal muscle cells (20) from GFP mice were stained with specific antibodies to CD45, c-kit, Sca-1, IL7R, CD31, and CD34 directly conjugated to phycoerythrin, allophycocyanin, or phycoerythrin-Cy7 (all from Pharmingen) and/or a lineage panel (Lin; mixture of biotinylated antibodies to Ter119, B220, CD3, Gr1, and CD11b) (BD Biosciences). Lin– cells were separated by magnetic depletion by using streptavidin coupled to magnetic beads (Miltenyi Biotec, Auburn, CA) and counterstained by using streptavidin Texas red (Pharmingen). Cells were then fractionated twice by flow cytometry (DIVA-Van, Becton Dickinson) before i.m. injection into the tibialis anteriors (TAs) of C57BL/6 mice that had just received a single 10-μl injection of notexin (NTX) (10 μg/ml).
Tissue Analysis. At 4 weeks after injection, TA muscles were harvested, imbedded in OCT compound, snap-frozen in liquid nitrogen-cooled isobutane, and then cut into 10-μm transverse sections. The detailed tissue-staining procedures are described in Supporting Materials and Methods. Briefly, tissue sections were blocked in PBS/normal goat serum/Triton X-100; incubated with primary antibodies against GFP (Molecular Probes), laminin-β2 (Upstate Biotechnology, Lake Placid, NY), or CD11b (Pharmingen); and incubated in secondary antibodies before overnight wash. GFP+ muscle fibers were counted on a Zeiss Axioplan epifluorescent microscope by using a double-bandpass filter to ensure that the GFP signal was authentic (this filter distinguishes between GFP and autofluorescence). CD11b staining was imaged by using a Zeiss laser-scanning confocal microscope (LSM 510) (1-μm optical sections).
Mononuclear Cell Mobilization in PB. C57BL/6 GFP mice were anesthetized and an ALZET osmotic minipump (Durect, Cupertino, CA) containing recombinant mouse granulocyte colony-stimulating factor (G-CSF) dosed to 50 μg/kg·day–1 (R & D Systems) was implanted subcutaneously. PB was harvested directly from the heart 5 days after minipump implantation, and the PB cells were stained as described above.
Myeloid and Myoblast Cell Isolation from Muscle Tissues. Myelomonocytic/macrophage cells of the myeloid lineage and primary myoblasts were isolated from the TA muscle of 6- to 8-week-old mice as described in ref. 21. Cells isolated from muscle extract of GFP or CMV-Cre transgenic mice (22) were labeled with CD45-phycoerythrin- and CD11b-allophycocyanin-conjugated antibodies (Pharmingen), sorted twice by FACS, and injected i.m. into the TA of either C57BL/6 or Z/EG recipient mice. The precise efficiency of the CMV-Cre recombination cannot be determined in such experiments, because only cells that effectively recombined the reporter gene are detected.
BM Cell Culture and FACS Analysis. After two rounds of cell sorting, 5 × 103 Lin– c-kit+ BM cells were seeded in complete methylcellulose medium (MethoCult, StemCell Technologies, Vancouver). After 1 and 2 weeks of culture, cells were harvested, washed twice with PBS, and injected i.m. into TAs previously damaged by NTX. An aliquot of the cells injected was resuspended in FACS buffer and stained with c-kit, CD31, CD13, or CD11b antibodies for FACS analysis.
Results
HSC and Myeloid Precursor Fractions Within BM Contribute to Regenerating Muscle. Previously, we and others reported that HSC within BM had the potential to contribute nuclei to regenerating muscle fibers (16, 17). To further characterize the HSC derivatives with this capacity, we fractionated BM cells by using well documented markers (reviewed in ref. 23). Fractions were isolated from BM of GFP transgenic mice by using Lin-specific markers and flow cytometry before direct injection into regenerating muscle (i.m.). This i.m. assay bypassed the need for BM reconstitution, allowing more specialized derivatives of HSC not capable of sustained hematopoietic engraftment or BM reconstitution to be tested. This method has previously been used to show that total BM is capable of participating in muscle regeneration after i.m. injection by Ferrari et al. (8). To test only the hematopoietic fraction of the BM (excluding stromal cells), we performed i.m. injections of GFP+ CD45+ BM cells into muscles damaged with the myotoxin NTX (24). Of three animals that received 106 CD45+ BM cells in the TA muscles, two had GFP+ myofibers (Table 1). The results do not discern whether nonhematopoietic cells within BM also have this capacity, but they provide evidence that the i.m. assay can be used to assess which hematopoietic fraction is capable of incorporation into regenerating muscles.
Table 1. Fractionation of total BM cells.
| BM fractionation | Cells injected per TA, n | TAs injected,*n | TAs with GFP+ fibers,†n (%) | Total GFP+ fibers observed,‡n | |
|---|---|---|---|---|---|
| Group 1: Total CD45+ BM | CD45+ | 106 | 3 | 2 (67) | 6 |
| PBS | NA | 3 | 0 (0) | 0 | |
| Group 2: Fractions containing hematopoietic stem cells | Lin-c-kit+ | 105 | 8 | 8 (100) | 44 |
| Lin-c-kit+ | 104 | 8 | 6 (75) | 53 | |
| c-kit+Sca1+ | 104 | 6 | 4 (65) | 6 | |
| Lin-c-kit+Sca1+ | 5 × 104 | 2 | 1 (50) | 6 | |
| Lin-c-kit+CD34- | 104 | 4 | 3 (75) | 19 | |
| Lin-c-kit-CD34+ | 104 | 8 | 6 (75) | 38 | |
| Total | NA | 36 | 28 (80) | 166 | |
| Group 3: Fractions containing myeloid progenitors | c-kit+ Sca1- | 104 | 3 | 1 (30) | 12 |
| Lin-c-kit+Sca1- | 105 | 6 | 6 (100) | 17 | |
| Lin-c-kit+CD31+ | 104 | 4 | 3 (75) | 15 | |
| Total | NA | 13 | 10 (75) | 44 | |
| Group 4: Fractions containing lymphoid progenitors§ | Lin-c-kit- | 105 | 5 | 1 (20) | 3 |
| Lin-IL7R+ | 104 | 2 | 0 (0) | 0 | |
| Lin-c-kit-Sca1- | 104 | 8 | 1 (10) | 1 | |
| Lin-c-kit-Sca1+ | 104 | 7 | 0 (0) | 0 | |
| Total | NA | 23 | 2 (9) | 4 | |
| Group 5: Fractions containing committed precursors and mature myeloid/lymphoid cells | Lin+c-kit- | 105 | 5 | 0 (0) | 0 |
| Lin-c-kit+ | 105 | 8 | 0 (0) | 0 | |
| Gr1+ | 105 | 2 | 0 (0) | 0 | |
| Gr1+c-kit+ | 104 | 4 | 0 (0) | 0 | |
| CD11b+ | 105 | 6 | 0 (0) | 0 | |
| CD11b+c-kit+ | 104 | 4 | 0 (0) | 0 | |
| Total | NA | 29 | 0 (0) | 0 |
NA, not applicable. Totals are shown in boldface type.
Total number of TAs injected for each fraction or group of fractions.
Total number of TAs presenting one or multiple GFP+ myofibers when microscopy analyses of transverse cross section of the TA were performed 4 weeks after NTX/cell fraction injections. Percentage represents the proportion of TA with GFP+ fibers to the total number of TA injected.
Total number of fiber counted for all the TAs analyzed. Although 12 cross sections were scanned for GFP+ fibers for each TA sample, only one cross section per TA was counted to avoid counting the same myofiber twice.
The low frequency of GFP+ fibers observed in fractions containing lymphoid progenitors is likely due to the inevitable 2-5% contamination of even 2× FACS-sorted cells.
We further fractionated whole BM cells by FACS, based on the expression of cell-surface markers (25). These markers included a mixture of antibodies to lineage antigens (Ter119 for er ythroid cells, B220 for B-lymphocytes, CD3 for T-lymphocytes, Gr1 for granulocytes, and CD11b for monocytes/macrophages), the stem cell factor receptor (c-kit) (26), and the stem cell surface antigen (Sca-1) (27). Representative results obtained for fractions of Lin+, Lin–c-kit–, Lin–c-kit+Sca-1–, and Lin–c-kit+Sca-1+ cells are shown (Fig. 1). These GFP-labeled fractions were injected in replicate experiments directly into the TA muscles of wild-type C57BL/6 mice that had just received NTX. GFP+ myofibers were detected only with fractions Lin–c-kit+Sca-1– and Lin–c-kit+Sca-1+, whereas fractions Lin+ and Lin–c-kit– did not contribute to muscle regeneration (Fig. 1 A′–D′). Similar experiments were carried out with 18 different FACS fractions tested in a total of 101 NTX-damaged TA muscles (Table 1). The results were pooled into four groups. BM cells that were incorporated into skeletal muscle belonged to the fractions that either contained HSC (Table 1, Group 2) or myeloid progenitors (Table 1, Group 3). On the contrary, fractions containing lymphoid progenitors (Table 1, Group 4) or mature cells of either lymphoid or myeloid lineages (Table 1, Group 5) did not incorporate into muscle fibers.
Fig. 1.

HSC (Lin–c-kit+Sca-1+) and common myeloid progenitors (Lin–c-kit+Sca-1–), but not Lin+ and c-kit– mature hematopoietic cells, contribute to muscle regeneration. (A–D) Example of fractionation from the BM of GFP transgenic mice: Total BM cells were Lin-depleted by using purification with magnetic beads, and the depleted fraction (Lin–) was then fractionated on the basis of c-kit and Sca-1 expression by flow cytometry. All separated fractions were injected i.m. into the TAs of mice that had just received a NTX injection. (A′–D′) Transverse sections of TA muscles 4 weeks after i.m. fraction/NTX injection are shown stained for GFP (green) and laminin (red) expression; nuclei are counterstained with Hoechst 33342 (blue). Representative TA sections of mice injected with 105 Lin+ cells (A′), 105 Lin–c-kit– cells (B′), 105 Lin–c-kit+Sca-1– cells (C′), or 5.104 Lin–c-kit+Sca-1+ cells (D′) are shown. The total number of GFP+ muscle fibers observed 4 weeks after injection in several experiments and the percentage of injected TA presenting GFP+ fibers for each fractionation is provided in Table 1. (Scale bars, 50 μm.)
Each stem cell fraction that contained HSC (Lin–c-kit+; c-kit+Sca-1+; Lin–c-kit+Sca-1+) gave rise to readily detectable GFP+ myofibers, whereas the presence or absence of the marker CD34 did not distinguish cells with or without the potential to integrate in regenerating myofibers (Table 1). The ability of BM cells to incorporate into muscle also did not specifically cosegregate with the expression of the stem cell antigen Sca-1 (Table 1). Myeloid progenitor fractions that expressed high amounts of the tyrosine kinase receptor c-kit but that lacked Sca-1 (c-kit+Sca-1– and Lin–c-kit+Sca-1–) were found to contribute to muscle regeneration with the same efficiency as fractions containing stem cells (≈75% of muscles) (Table 1, Group 3). In contrast, none or a very few GFP+ myofibers were observed when fractions containing either lymphoid progenitors (Lin–c-kit–, Lin–IL7R+, Lin–c-kit–Sca-1–, and Lin–c-kit–Sca-1+) (Table 1, Group 4) or mature lymphoid or myeloid cells (Lin+c-kit–Lin+c-kit+) (Table 1, Group 5) were tested (9% or 0% of muscles, respectively). Distinction between the four populations is evident from this analysis and 98% of all GFP+ fibers derived from fractions containing either HSC or myeloid progenitors, and only 2% were from fractions containing either lymphoid progenitors or committed precursors and mature myeloid/lymphoid cells. Taken together, these data suggest that the cells in the BM that integrate into myofibers during muscle regeneration are within the fractions containing HSC and myelomonocytic progenitors.
Mature Myeloid Cells (Macrophages) Do Not Contribute to Regenerating Muscle. Experiments were designed to determine whether mature macrophages participate in muscle regeneration. These cells have been considered by others to be the prime candidate for an HSC derivative that contributes to muscle, due in part to their innate fusogenic activity and to their abundance in regenerating tissues (28). The participation of GFP+ cells in the inflammatory response was evident in most of our experiments. GFP+ cells in clusters were observed that were similar in size to muscle fibers but could be distinguished by a lack of the basal laminal membrane that always encompasses muscle fibers (Fig. 2 A and A′, arrow heads). Moreover, these GFP+ clusters stained strongly with CD11b/Mac1, a marker of mature myelomonocytic cells and therefore likely represent macrophages and neutrophils engulfing dying myofibers and participating in a process of muscle fiber degeneration, not regeneration (Fig. 2 B and B′). By contrast, GFP+ structures that were determined to be myofibers were always surrounded by a basal lamina (Fig. 2 A and A′, arrows) and did not express CD11b (Fig. 2 C and C′).
Fig. 2.
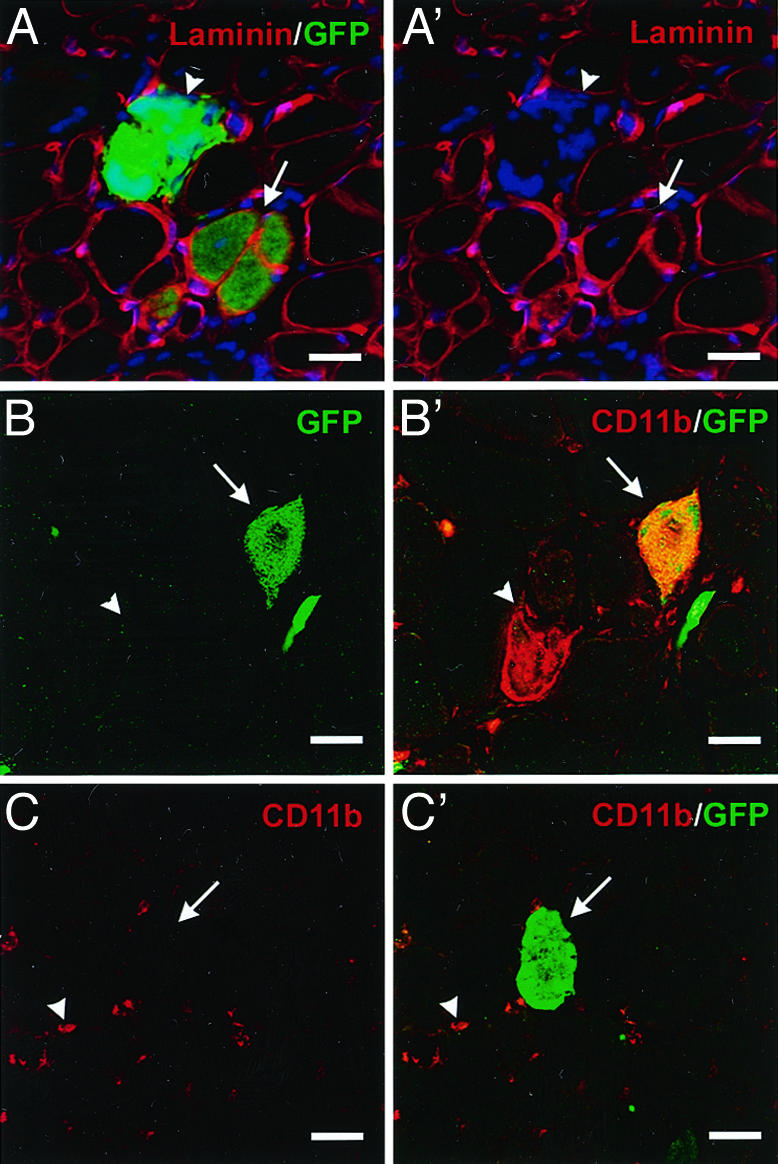
Injected BMDC contribute to intact myofibers or give rise to CD11b/Mac1+ cells that invade degenerating fibers. (A and A′) Representative confocal microscopic analyses of transverse sections of skeletal muscle 4 weeks after i.m. injection of cells fractionated from the BM of GFP transgenic mice detect GFP (green in A only), laminin (A and A′, red) and nuclei (A and A′, blue). Both GFP+ myofibers surrounded by basal lamina (arrow) and GFP+ clusters of cells not surrounded by basal laminal membranes (arrow heads) were observed in TA cross sections (note number of nuclei in GFP+ cluster in A′). (B and B′) GFP+ clusters (arrows) (green) were positive for CD11b (B′, red) and therefore identified as donor-derived macrophages that have invaded degenerating myofibers. Endogenous macrophages (arrowheads) are not GFP+.(C and C′) True GFP+ myofibers (arrows) (C′, green) were found to lack CD11b (red), whereas infiltrating endogenous macrophages CD11b+ (arrowheads) were observed. (Scale bars, 50 μm.)
To further explore the potential of cells from the myelomonocytic lineage to contribute to muscle regeneration, we tested whether mature myelomonocytic cells that express CD11b could contribute to myofibers in several experimental paradigms. First, when CD11b+ or Gr1+ cells isolated directly from BM were injected i.m. into regenerating TA muscles, no GFP+ myofibers were detected after 4 weeks (Fig. 3 A and B and Table 2). Second, when CD11b+ cells isolated from the PB after G-CSF delivery and consequent mobilization in vivo (29) were injected i.m. into regenerating TA muscles, no GFP+ myofibers were detected after 4 weeks (Fig. 3C and Table 2). By contrast, in control mice that had received an i.m. injection of c-kit+ fractions from either BM or mobilized PB, several laminin-ensheathed GFP+ myofibers were consistently observed (Fig. 3 A and C and Table 2).
Fig. 3.

Mature CD11b+ myelomonocytic cells from BM, PB after G-CSF mobilization, NTX-damaged muscle, or monocytic cultures do not participate in myofiber regeneration. (A and B) Flow-cytometric analysis of BM and sorting gates for isolation of Gr1+ (A) or CD11b+ (B) cells. (C) Sorting gates for CD11b+ and CD11b–c-kit+ cells from PB after G-CSF mobilization. (D) Sorting gates for CD45+CD11b+ and CD45–CD11b– cells from muscle damaged by NTX. All cells were analyzed by flow cytometry and isolated from GFP+ transgenic mice before i.m. injection into skeletal muscles that had previously received an NTX injection. The number of TA muscles with GFP+ myofibers and the total number of GFP+ myofibers observed for each fraction analyzed are shown in Table 2. (E) Representative transverse section of TA muscle CD11b-Cre Z/EG BMT 4 weeks after NTX damage. No GFP+ myofibers were detected, whereas GFP+ macrophages (arrows) resulting from fusion between Z/EG endogenous and CD11b-Cre donor macrophages were observed. (F) TA muscle of Z/EG transgenic mice injected with myoblasts from muscle extract of CMV-Cre transgenic mice was used as positive control. (G) Lin–c-kit+ cells expanded in methylcellulose culture for 1 or 2 weeks and the phenotypes of the cells tested for contribution to muscle regeneration are shown. The number of TA muscles with GFP+ myofibers and total number of GFP+ myofibers observed for each culture time point are shown in Table 2.
Table 2. Fractionation of progenitors and mature myelomonocytic cells.
| Fraction | Cells injected per TA, n | TAs injected, n | TAs with GFP+ fibers, n (%) | Total GFP+ fibers observed, n | |
|---|---|---|---|---|---|
| Mature cells from BM | Gr1+ | 105 | 4 | 0 (0) | 0 |
| CD11b+ | 105 | 6 | 0 (0) | 0 | |
| Immature cells from BM | Gr1-c-kit+ | 105 | 4 | 4 (100) | 43 |
| CD11b-c-kit+ | 105 | 6 | 5 (85) | 27 | |
| G-CSF-mobilized PB cells | CD11b+ | 105 | 4 | 0 (0) | 0 |
| CD11b-c-kit+ | 104 | 2 | 2 (100) | 5 | |
| Muscle cells after NTX damage | CD45+CD11b+ | 105 | 10 | 1 (10) | 5 |
| CD45-CD11b- | 3.104 | 8 | 6 (75) | 123 | |
| Lin-c-kit+ cultured cells | 1 week | 105 | 6 | 6 (100) | 20 |
| 2 weeks | 105 | 6 | 0 (0) | 0 |
In a third paradigm, we determined whether mature myelomonocytic cells capable of participating in the inflammatory response in injured muscles were involved in myofiber regeneration. TA muscles of 8-week-old GFP transgenic mice were injured with NTX, and 2 days later the injured muscles were harvested. CD45+CD11b+ cells were isolated from minced muscles and injected into regenerating muscles of C57BL/6 mice (Fig. 3D). Once again, no GFP+ myofibers were detected in 9 of 10 muscles when mature myelomonocytic cells from injured muscle were injected, whereas CD45– CD11b– cells isolated from the same muscle and likely to be myoblasts contributed robustly to myofiber regeneration (Table 2). To further test whether myelomonocytic precursors matured to macrophages before integrating into skeletal muscle, we transplanted Z/EG mice that express GFP upon Cre-mediated excision of lacZ (19) with BM from transgenic donor mice in which Cre recombinase was under the control of CD11b promoter (18). Because in adults CD11b is specifically expressed by mature myelomonocytic cells but not by cells earlier in the lineage (i.e., premyelomonocytic cells), recombination and GFP expression should occur only if CD11b-Cre BMDC currently expressing CD11b fuse with muscle fibers that contained the conditional Crereporter GFP gene. CD11b-Cre BM cells were injected into three lethally irradiated Z/EG mice, and both legs of reconstituted mice were injected with NTX 3 months after BMT. Four weeks after muscle damage, GFP+ fibers were not detected in any of the six NTX-injected TAs and in any of the six non-NTX-injected upper front legs. Although GFP+ myofibers were not detected in these mice, GFP+ endogenous macrophages that fused with other macrophages from the CD11b-Cre transgenic donor mice where observed at the site of regeneration (Fig. 3E), showing that the Cre recombinase was expressed in macrophages. As an additional control to show that the reporter constructs were capable of Cre recombination in myofibers, the TAs of four Z/EG recipient mice were injected with myoblasts isolated from transgenic mice ubiquitously expressing Cre recombinase under the transcriptional control of a CMV minimal promoter (22). Under these conditions, GFP+ myofibers were detected (Fig. 3F). Together, these four experimental paradigms provide evidence that only immature myelomonocytic cells and not mature macrophages can integrate into muscle fibers.
Myelomonocytic Precursor Cells Retain Their Capacity to Contribute to Muscle After in Vitro Culture. To test whether myelomonocytic cells isolated from the BM can be maintained in vitro and yet be capable of participating in muscle regeneration, Lin–c-kit+ cells freshly isolated from the BM of GFP transgenic mice were seeded in methylcellulose supplemented with cytokines (stem cell factor, IL-3, and IL-6) and cultured for 1 and 2 weeks (Fig. 3G). After 1 week in culture, the cell population had proliferated extensively and was primarily of an immature phenotype that expressed c-kit (76%), CD31 (67%), and CD13 (85%), whereas only 33% expressed the mature monocytic/macrophage marker CD11b. Thus, the cells were predominantly in an early stage of myelomonocytic differentiation (30–33). After 2 weeks in culture, the expression patterns of the cells had shifted such that a relatively small proportion now expressed the stem cell receptor c-kit (11%) and the immature monocytic markers CD13 and CD31 (29% and 30%, respectively). By contrast, >60% of the cells expressed the mature monocytic/macrophage marker CD11b (Fig. 3G). This finding is in accordance with published studies demonstrating that in vitro myeloid differentiation is accompanied by down-regulation of immature monocytic markers, such as c-kit, CD13, and CD31, and up-regulation of a marker typical of mature macrophages, CD11b (34).
Both of the cell populations that had been cultured for 1 or 2 weeks were injected into TA muscles treated with NTX and then analyzed 4 weeks later. Only the cells that were grown in culture for 1 week were found to give rise to GFP+ myofibers (Table 2). These data demonstrate that myeloid progenitors within BM can be maintained in culture and still retain the ability to contribute myonuclei to regenerating myofibers. In addition, the fact that the cells cultured for 2 weeks, which primarily comprised mature monocytic cells, did not participate in myofiber regeneration provides another line of evidence that mature macrophages do not fuse with myofibers.
Discussion
The identification of a specific cell type within BM that is capable of integrating into muscle fibers is critical to understanding and potentially enhancing this process. In this report, we identify the relevant HSC derivative by using FACS-based BM fractionation and i.m. injections. This approach allowed us to test cellular fractions for their ability to contribute to muscle irrespectively of their blood reconstitution capabilities. The results were consistent among replicates and clearly indicated that mature progeny of myelomonocytic progenitors (macrophages), although fusogenic by nature (35), do not fuse spontaneously with muscle fibers and do not contribute to muscle regeneration in this assay. By contrast, more primitive HSC derivatives, such as myelomonocytic progenitors, can incorporate into regenerating muscle fibers.
The finding that both HSC and their derivatives yield GFP+ myofibers is in good agreement with recent reports showing that individual transplanted HSC can contribute to muscle regeneration (16, 17). Although myeloid precursors have previously been implicated in this process (16), the methodology used had inherent problems. First, because in this study, bitransgenic LysM-Cre/Rosa flox/STOP mice were analyzed, even transient lysozyme expression during embryogenesis could have led to permanent β-galactosidase expression in all of the progeny of the cell in which the recombination happened. Second, the lysozyme promoter Cre-construct, although expressed in mature macrophages, has recently been shown to be transiently expressed in HSC resulting in β-galactosidase expression in ≈10% of cells in each hematopoietic lineage (36). Thus, expression of β-galactosidase in these animals does not provide definitive evidence that the cells that integrated into muscle fibers were macrophages, because they could have derived from any hematopoietic lineage.
In this report, we segregate BM cells by cell fractionation and demonstrate that the cells capable of incorporating into regenerating myofibers are restricted to the myelomonocytic fraction, which agrees well with findings obtained by others in liver (37, 38). In addition, we show that mature macrophages from three tissue sources are not able to contribute myonuclei during muscle regeneration in contrast to their myelomonocytic progenitors. To further demonstrate that these progenitors do not require maturation to macrophages before incorporating into muscle fibers, we took advantage of the Cre/lox system in a different setting than what has been recently published (16). First, by using BMT rather than mating the mice, we did not allow recombination events to occur before the fusion of donor cells with recipient myofibers. Second, CD11b, although expressed in HSC during embryonic development, is not expressed in adult HSC or their progeny before reaching late monocytic differentiation and macrophage maturation (39, 40). By using positive controls, we were able to show (i) that CD11b-Cre macrophages expressed Cre and were capable of recombination after spontaneous fusion with endogenous macrophages and (ii) that recombination following introduction of myoblasts from CMV-Cre mice into Z/EG mouse muscle was possible in myofiber nuclei in vivo.
The myelomonocytic progenitor cell fraction identified here represents only 5% of the total BM, and, by using FACS fractionation, it is possible to obtain a substantial enrichment of the relevant cell type. Identification of these cells as the major players in the BM-to-muscle transition is important in that it will facilitate elucidation of the mechanisms by which they incorporate into muscle fibers. Although CD11b is expressed on neutrophils and natural killer cells in addition to macrophages, our results show that none of these populations contributes to muscle, whereas the premyelomonocytic cells clearly do. Based on the findings presented here, we propose that, upon muscle damage, immature myelomonocytic cells are recruited to the injured area and incorporate into muscle fibers, contributing to multinucleate myofiber regeneration. Whether these cells assume a muscle stem cell-like state (satellite cells), directly fuse with regenerating myofibers, or employ a combination of these two pathways remains to be determined. In addition, it remains unknown whether there is a relationship between the myelomonocytic cells described here and other recently described endogenous cells that are capable of contributing to muscle, such as the CD45+Sca-1+ cells found in the interstitial regions of muscle (41) or mesoangioblasts within the embryonic aorta (42). Further lineage studies using genetic markers will be required to define the intermediate cell types that may be involved in the blood-to-muscle pathway and the factors that mediate their fusion with muscle fibers. The myelomonocytic progenitors identified here as potential donors of myonuclei could provide a readily accessible cell population for therapeutic applications.
Supplementary Material
Acknowledgments
We thank Kassie Koleckar and Peggy Kraft for excellent technical support, Dr. James Weimann for confocal microscopy expertise, Dr. Jason Pomerantz for useful comments on the manuscript, and Drs. Daniel Tenen and Corrinne Lobe for providing CD11b-Cre and Z/EG reporter mice. M.A.L. was supported by National Institutes of Health Predoctoral Fellowship TG AG00259. H.M.B. was supported by National Institutes of Health Grants AG09521, HL65572, HD18179, and AG20961, Senior Ellison Medical Foundation Award AG-33-0817, the McKnight Endowment Fund, and the Baxter Foundation.
Abbreviations: BM, bone marrow; BMDC, BM-derived cells; NTX, notexin; BMT, BM transplantation; HSC, hematopoietic stem cell; TA, tibialis anterior; PB, peripheral blood; G-CSF, granulocyte colony-stimulating factor; FACS, fluorescence-activated cell sorter; CMV, cytomegalovirus; Lin, lineage panel (mixture of biotinylated antibodies to Ter119, B220, CD3, Gr1, and CD11b).
References
- 1.Grounds, M. D., White, J. D., Rosenthal, N. & Bogoyevitch, M. A. (2002) J. Histochem. Cytochem. 50, 589–610. [DOI] [PubMed] [Google Scholar]
- 2.Krause, D. S., Theise, N. D., Collector, M. I., Henegariu, O., Hwang, S., Gardner, R., Neutzel, S. & Sharkis, S. J. (2001) Cell 105, 369–377. [DOI] [PubMed] [Google Scholar]
- 3.Lagasse, E., Connors, H., Al-Dhalimy, M., Reitsma, M., Dohse, M., Osborne, L., Wang, X., Finegold, M., Weissman, I. L. & Grompe, M. (2000) Nat. Med. 6, 1229–1234. [DOI] [PubMed] [Google Scholar]
- 4.Bittner, R. E., Schofer, C., Weipoltshammer, K., Ivanova, S., Streubel, B., Hauser, E., Freilinger, M., Hoger, H., Elbe-Burger, A. & Wachtler, F. (1999) Anat. Embryol. 199, 391–396. [DOI] [PubMed] [Google Scholar]
- 5.Mezey, E., Chandross, K. J., Harta, G., Maki, R. A. & McKercher, S. R. (2000) Science 290, 1779–1782. [DOI] [PubMed] [Google Scholar]
- 6.Blau, H. M., Brazelton, T. R. & Weimann, J. M. (2001) Cell 105, 829–841. [DOI] [PubMed] [Google Scholar]
- 7.Wang, X., Montini, E., Al-Dhalimy, M., Lagasse, E., Finegold, M. & Grompe, M. (2002) Am. J. Pathol. 161, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrari, G., Cusella-De Angelis, G., Coletta, M., Paolucci, E., Stornaiuolo, A., Cossu, G. & Mavilio, F. (1998) Science 279, 1528–1530. [DOI] [PubMed] [Google Scholar]
- 9.Gussoni, E., Soneoka, Y., Strickland, C. D., Buzney, E. A., Khan, M. K., Flint, A. F., Kunkel, L. M. & Mulligan, R. C. (1999) Nature 401, 390–394. [DOI] [PubMed] [Google Scholar]
- 10.Ferrari, G., Stornaiuolo, A. & Mavilio, F. (2001) Nature 411, 1014–1015. [DOI] [PubMed] [Google Scholar]
- 11.Gussoni, E., Bennett, R. R., Muskiewicz, K. R., Meyerrose, T., Nolta, J. A., Gilgoff, I., Stein, J., Chan, Y. M., Lidov, H. G., Bèonnemann, C. G., et al. (2002) J. Clin. Invest. 110, 807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fukada, S., Miyagoe-Suzuki, Y., Tsukihara, H., Yuasa, K., Higuchi, S., Ono, S., Tsujikawa, K., Takeda, S. & Yamamoto, H. (2002) J. Cell Sci. 115, 1285–1293. [DOI] [PubMed] [Google Scholar]
- 13.LaBarge, M. A. & Blau, H. M. (2002) Cell 111, 589–601. [DOI] [PubMed] [Google Scholar]
- 14.Brazelton, T. R., Nystrom, M. & Blau, H. M. (2003) Dev. Biol. 262, 64–74. [DOI] [PubMed] [Google Scholar]
- 15.Dreyfus, P. A., Chretien, F., Chazaud, B., Kirova, Y., Caramelle, P., Garcia, L., Butler-Browne, G. & Gherardi, R. K. (2004) Am. J. Pathol. 164, 773–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Camargo, F. D., Green, R., Capetenaki, Y., Jackson, K. A. & Goodell, M. A. (2003) Nat. Med. 9, 1520–1527. [DOI] [PubMed] [Google Scholar]
- 17.Corbel, S. Y., Lee, A., Yi, L., Duenas, J., Brazelton, T. R., Blau, H. M. & Rossi, F. M. (2003) Nat. Med. 9, 1528–1532. [DOI] [PubMed] [Google Scholar]
- 18.Dziennis, S., Van Etten, R. A., Pahl, H. L., Morris, D. L., Rothstein, T. L., Blosch, C. M., Perlmutter, R. M. & Tenen, D. G. (1995) Blood 85, 319–329. [PubMed] [Google Scholar]
- 19.Novak, A., Guo, C., Yang, W., Nagy, A. & Lobe, C. G. (2000) Genesis 28, 147–155. [PubMed] [Google Scholar]
- 20.Rando, T. A. & Blau, H. M. (1994) J. Cell Biol. 125, 1275–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Springer, M. L., Rando, T. A. & Blau, H. M. (1997) Gene Delivery to Muscle (Wiley, New York). [DOI] [PubMed]
- 22.Schwenk, F., Baron, U. & Rajewsky, K. (1995) Nucleic Acids Res. 23, 5080–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kondo, M., Wagers, A. J., Manz, M. G., Prohaska, S. S., Scherer, D. C., Beilhack, G. F., Shizuru, J. A. & Weissman, I. L. (2003) Annu. Rev. Immunol. 21, 759–806. [DOI] [PubMed] [Google Scholar]
- 24.Harris, J. B. & Johnson, M. A. (1978) Clin. Exp. Pharmacol. Physiol. 5, 587–600. [DOI] [PubMed] [Google Scholar]
- 25.Morrison, S. J., Wandycz, A. M., Hemmati, H. D., Wright, D. E. & Weissman, I. L. (1997) Development (Cambridge, U.K.) 124, 1929–1939. [DOI] [PubMed] [Google Scholar]
- 26.Ogawa, M., Matsuzaki, Y., Nishikawa, S., Hayashi, S., Kunisada, T., Sudo, T., Kina, T. & Nakauchi, H. (1991) J. Exp. Med. 174, 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van de Rijn, M., Heimfeld, S., Spangrude, G. J. & Weissman, I. L. (1989) Proc. Natl. Acad. Sci. USA 86, 4634–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charge, S. & Rudnicki, M. A. (2003) Cell 113, 422–423. [DOI] [PubMed] [Google Scholar]
- 29.Morrison, S. J., Wright, D. E. & Weissman, I. L. (1997) Proc. Natl. Acad. Sci. USA 94, 1908–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terskikh, A. V., Miyamoto, T., Chang, C., Diatchenko, L. & Weissman, I. L. (2003) Blood 102, 94–101. [DOI] [PubMed] [Google Scholar]
- 31.Newman, P. J. (1997) J. Clin. Invest. 100, Suppl. 11, S25–S29. [PubMed] [Google Scholar]
- 32.Nomdedeu, J. F., Mateu, R., Altes, A., Llorente, A., Rio, C., Estivill, C., Lopez, O., Ubeda, J. & Rubiol, E. (1999) Leuk. Res. 23, 341–347. [DOI] [PubMed] [Google Scholar]
- 33.Nikolic, T., de Bruijn, M. F., Lutz, M. B. & Leenen, P. J. (2003) Int. Immunol. 15, 515–524. [DOI] [PubMed] [Google Scholar]
- 34.Ho, M. K. & Springer, T. A. (1982) J. Immunol. 128, 2281–2286. [PubMed] [Google Scholar]
- 35.McInnes, A. & Rennick, D. M. (1988) J. Exp. Med. 167, 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye, M., Iwasaki, H., Laiosa, C. V., Stadtfeld, M., Xie, H., Heck, S., Clausen, B., Akashi, K. & Graf, T. (2003) Immunity 19, 689–699. [DOI] [PubMed] [Google Scholar]
- 37.Willenbring, H., Bailey, A. S., Foster, M., Akkari, Y., Dorrell, C., Olson, S., Finegold, M., Fleming, W. H. & Grompe, M. (2004) Nat. Med. 10, 744–748. [DOI] [PubMed] [Google Scholar]
- 38.Camargo, F. D., Finegold, M. & Goodell, M. A. (2004) J. Clin. Invest. 113, 1266–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morrison, S. J., Hemmati, H. D., Wandycz, A. M. & Weissman, I. L. (1995) Proc. Natl. Acad. Sci. USA 92, 10302–10306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pahl, H. L., Rosmarin, A. G. & Tenen, D. G. (1992) Blood 79, 865–870. [PubMed] [Google Scholar]
- 41.Polesskaya, A., Seale, P. & Rudnicki, M. A. (2003) Cell 113, 841–852. [DOI] [PubMed] [Google Scholar]
- 42.Sampaolesi, M., Torrente, Y., Innocenzi, A., Tonlorenzi, R., D'Antona, G., Pellegrino, M. A., Barresi, R., Bresolin, N., De Angelis, M. G., Campbell, K. P., et al. (2003) Science 301, 487–492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}