

Abstract
Laboratory mice bearing inactivating mutations in the genes encoding the NPAS1 and NPAS3 transcription factors have been shown to exhibit a spectrum of behavioral and neurochemical abnormalities. Behavioral abnormalities included diminished startle response, as measured by prepulse inhibition, and impaired social recognition. NPAS1/NPAS3-deficient mice also exhibited stereotypic darting behavior at weaning and increased locomotor activity. Immunohistochemical staining assays showed that the NPAS1 and NPAS3 proteins are expressed in inhibitory interneurons and that the viability and anatomical distribution of these neurons are unaffected by the absence of either transcription factor. Adult brain tissues from NPAS3- and NPAS1/NPAS3-deficient mice exhibited a distinct reduction in reelin, a large, secreted protein whose expression has been reported to be attenuated in the postmortem brain tissue of patients with schizophrenia. These observations raise the possibility that a regulatory program controlled in inhibitory interneurons by the NPAS1 and NPAS3 transcription factors may be either substantively or tangentially relevant to psychosis.
Mice, humans, and other vertebrates contain genes encoding two related transcription factors designated neuronal PAS domain protein 1 (NPAS1) and NPAS3 (1, 2). The two proteins are members of the basic helix–loop–helix (bHLH)PAS domain family of transcription factors. Members of this family respond to various environmental stimuli, including light, oxygen, voltage, and redox potential (3–5). NPAS1 and NPAS3 are highly related in primary amino acid sequence and indistinguishable when tested by a variety of functional assays. One notable difference between NPAS1 and NPAS3 is the size of their encoding genes. The 11 exons of the NPAS1 gene are housed within a 20-kb segment of mouse chromosome 7 (human chromosome 19). By contrast, the equivalent exons of the NPAS3 gene span 791 kb of mouse chromosome 12 (human chromosome 14).
A disruption in the human NPAS3 gene has recently been reported in a family suffering from schizophrenia (6). A translocation between chromosomes 9 and 14 results in truncation of the NPAS3 gene between exons 2 and 3. If expressed, the protein product of the disrupted NPAS3 locus can be predicted to contain an intact bHLH DNA-binding domain missing both PAS domains and all amino acids C-terminal to the PAS domains. Affected mother and daughter are reported to carry the t(9;14) (q34;q13) translocation, whereas the unaffected father does not.
NPAS1-, NPAS3-, and doubly deficient mouse strains have been prepared by targeted gene disruption. Behavioral studies of these mice reveal distinctive abnormalities, including impaired startle response, diminished social recognition, stereotypic darting behavior, and enhanced locomotor activity. Immunohistochemical staining assays of CNS tissues further reveal attenuation in the expression of reelin in NPAS3- and NPAS3/NPAS1-deficient mice. We hereby hypothesize that these transcription factors will control regulatory pathways that may possibly be relevant to the etiology of psychosis.
Methods
Approval for the animal experiments described herein was obtained by the University of Texas Southwestern Medical Center Institutional Animal Care and Use Committee. A full description of methods for the targeting of the NPAS1 and NPAS3 genes can be found in Supporting Text, which is published as supporting information on the PNAS web site.
Mice. NPAS1+/–:NPAS3+/– females (F1) mated to NPAS1+/–: NPAS3+/– males (F1) produced 129/SvEv:C57BL/6J mixed strain litters. Healthy pups were ear-tagged and tail-snipped when weaned (21 days). Pups that displayed growth deficiencies were group-housed and received special care (nesting squares, isolation foam under the cage, and 11% soaked food) until viable. Eventually all animals were group-housed until time of testing. Genotypes for the NPAS1 and NPAS3 loci were established by PCR analysis.
Behavioral Test Battery. To enable testing in the active, dark period, animals were housed in reversed light cycle (12 h/12 h) starting 3 weeks before testing. Before each test, the mice were allowed acclimatize for 1 h in the test room. Four groups of animals (age, 17–20 weeks) were tested in either red light or dim light, depending on the test.
The behavioral test battery consisted of the following tests conducted on the days indicated: day 1, social recognition 1; day 2, measurement of general parameters; day 3, hanging wire 1; day 4, social recognition 2; day 5, open field and hanging wire 2; days 8–10, rotarod; day 12, ataxia; day 15, prepulse inhibition; days 17–19, cued and contextual fear; day 22, anosmia; days 23–24, shock threshold. A complete description of each of these tests is available in Supporting Text.
Immunocytochemical Localization. For immunocytochemical experiments, mice were anesthetized with an overdose of ketamine/xylazine and transcardially perfused with saline followed by 4% paraformaldehyde in 0.15 M phosphate buffer. Free-floating vibratome sections of 40 μm were incubated overnight in antibodies to NPAS1 (1:500) or NPAS3 (1:1,000) and were then processed through the standard immunocytochemical protocol by using the Vector Elite kit (Vector Laboratories) with a biotinylated anti-rabbit IgG. The final horseradish peroxidase reaction product was developed with the Vector DAB peroxidase substrate kit. Sections were floated onto subbed slides, allowed to air dry, dipped in Histoclear, and coverslipped with Permount. Sections were viewed with a Nikon Eclipse E1000M microscope equipped with a DMX1200F digital camera. Images were acquired with act-1 software. Contrast adjustments of the digital images were made by using photoshop (Adobe Systems, San Jose, CA). A more complete description of the immunocytochemical protocol, details of antibody generation, sources, and dilutions of other antibodies examined can be found in Supporting Text.
Measurement of Reelin Expression. Mice of the following genotypes were perfused as described above: NPAS1+/+:NPAS3+/+ (n = 6); NPAS1–/–:NPAS3+/+ (n = 6); NPAS1+/+:NPAS3–/– (n = 7); and NPAS1–/–:NPAS3–/– (n = 5). The brains were sectioned from the preoptic area to the midbrain and processed through the immunocytochemical protocol described above, except that the Vector Elite Mouse IgG kit was used. To reduce background staining, the Vector MOM kit was used according to the manufacturer's instructions. Sections were examined on a Zeiss IIIRS microscope equipped with a Polaroid digital camera. Reelin-positive neurons were counted at a final magnification of ×125 from five sections of each brain. The anatomical areas examined for reelin immunoreactivity were as follows: cingulate cortex, a field encompassing the junction of the primary motor cortex and the somatosenory cortex, the dentate gyrus of the hippocampus, piriform cortex, and medial amygdala. Reelinpositive cells were counted on one side of the brain by an experimenter blind to genotype. The cell counts for each area were compared by a matched-pair t test.
Results
Anatomical Expression Profiles of NPAS1 and NPAS3. Two methods were used to define the distribution of NPAS1 expression in the adult mouse brain. The targeting construct used to disrupt the NPAS1 gene placed the β-galactosidase gene of Escherichia coli in frame with the N-terminal 41 aa of the truncated NPAS1 polypeptide. Coronally sectioned material from wild-type (wt) mice, NPAS1 heterozygotes and NPAS1 homozygotes were stained for β-galactosidase according to a protocol contained in Supporting Text. Intensely stained β-galactosidase-positive cells were observed in the dentate gyrus of NPAS1 homozygotes (Fig. 1A). Similarly distributed but less intense staining was observed in NPAS1 heterozygotes and no discernable staining was observed in the brain tissue of wt mice. That this β-galactosidasestaining pattern recapitulates the bona fide expression pattern of the NPAS1 gene was confirmed by use of antibodies prepared against the recombinantly expressed NPAS1 protein. NPAS1-specific antibodies stain the nuclear compartment of the same cell population observed to express β-galactosidase in NPAS1 homozygotes and heterozygotes (Fig. 1B). Given that antibody staining was eliminated in NPAS1 homozygotes and diminished in NPAS1 heterozygotes (relative to tissue derived from wt animals), we conclude that both the antibody- and β-galactosidase-staining patterns accurately reveal the pattern of NPAS1 gene expression in the adult mouse brain.
Fig. 1.

Samples of NPAS1 and NPAS3 expression in the adult mouse brain. (A) Patterns of β-galactosidase enzyme activity in coronal sections of brain tissue derived from wt (Top), NPAS1+/– (Middle), and NPAS1–/– (Bottom) animals. (B) Staining patterns of matching tissue samples with antibodies prepared against recombinant NPAS1 protein. NPAS1-positive cells are observed in wt (Top) and NPAS1+/– (Middle) samples, but absent in the NPAS1–/– sample (Bottom). (C) Coronal sections of the cerebellum from a wt mouse immunohistochemically processed with the antibody to NPAS3. NPAS3-positive neurons are seen in the granular layer of the cerebellum immediately outside of the Purkinje (P) cell layer. (Upper Right) Shown is the high-magnification image of the boxed area (Left), revealing nuclear, antibody-reactive material. (Lower Right) Shown is hematoxylin/eosin staining of an analogous region of the cerebellum prepared from a wt mouse. (D) Coronal section as in C but from an NPAS3–/– mouse. Despite the absence of antibody-reactive material in the cerebellum of the NPAS3–/– sample (Upper Right), the hematoxylin/eosin staining pattern (Lower Right) could not be distinguished from wt.
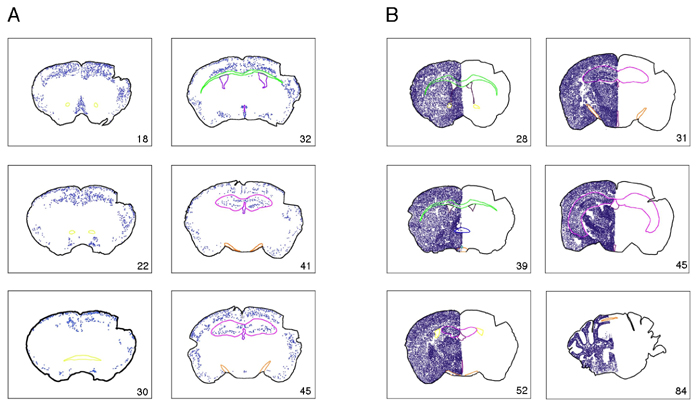
NPAS1 is expressed in a relatively small number of cells distributed throughout the brain (Fig. 5A, which is published as supporting information on the PNAS web site). Sites of expression include the hippocampus, dentate gyrus, piriform cortex, medial septum, and cortical layers 1, 4, and 5. The specific cells expressing NPAS1 were relatively small compared with the large, excitatory pyramidal neurons in the cortex, indicating that the protein might be expressed in inhibitory interneurons. Double-labeling experiments were used to define the types of cells expressing NPAS1. Almost all NPAS1-positive cells costained with γ-aminobutyric acid (GABA) or glutamic acid decarboxylase 67 (GAD-67) (Fig. 2A). Additionally, many NPAS1-positive cells also expressed calretinin (Fig. 6, which is published as supporting information on the PNAS web site). In the hippocampus, the majority of calretinin-expressing neurons are GABAergic inhibitory interneurons (7, 8). Taken together, these observations provide evidence that the NPAS1 gene is expressed primarily in inhibitory interneurons.
Fig. 2.

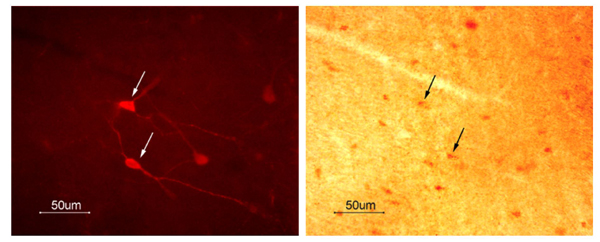
NPAS1 and NPAS3 neurons colocalize with GABA. (A) Photomicrographs of brain sections subjected to double-labeling immunocytochemistry for NPAS1 and GABA (Left) and NPAS1 and GAD-67 (Right). (Left) The 10-μm paraffin section was taken from the hippocampus of a wt mouse. The NPAS1 reaction product in the nucleus was detected with the Vector DAB peroxidase substrate, whereas the blue-gray GABA reaction product in the cytoplasm was detected with the Vector SG peroxidase substrate. (Magnification, ×700.) (Right) This micrograph is a 40-μm vibratome section through the cerebral cortex of an NPAS1–/– mouse stained for β-galactosidase. The blue, nuclear reaction product marks the presence of NPAS1. The section was then exposed to anti-GAD-67 (Chemicon, 1:1,500) followed by the Vector DAB peroxidase substrate to yield a brown, cytoplasmic reaction product. (Calibration bar, 20 μm.) (B) Photomicrographs from the CA1 (Upper) and CA3 (Lower) areas of the hippocampus from a wt mouse taken under fluorescent and bright-field illumination. The nuclear NPAS3 brown reaction product was detected with the Vector DAB peroxidase substrate kit. The fluorescent GAD-67-expressing neurons were detected by using Texas Red Avidin D (Vector Laboratories). Arrows indicate double-labeled neurons. (Calibration bar, 50 μm.)
The targeting construct used to disrupt the NPAS3 gene did not mark the gene with β-galactosidase. We instead relied solely on the use of antibodies prepared against the recombinant NPAS3 protein to define its sites of expression in the adult mouse brain. NPAS3 immunoreactivity was again observed in small cells widely distributed throughout the CNS. Antibody staining was restricted to the nuclear compartment of sectioned material from wt mice (Fig. 1C), yet it was totally absent from NPAS3 homozygotes (Fig. 1D). The number of NPAS3-positive cells exceeded NPAS1-positive cells by at least an order of magnitude, and staining was observed in a fairly uniform manner throughout the brain (Fig. 5B). Costaining assays revealed no overlap with the glial fibrillary acidic protein marker for astroglial cells (Fig. 7, which is published as supporting information on the PNAS web site), yet almost perfect concordance with GABA (Fig. 2B). Some NPAS3-expressing neurons were also calretinin-positive (Fig. 8, which is published as supporting information on the PNAS web site). We therefore conclude that both the NPAS1 and NPAS3 transcription factors are primarily expressed in inhibitory interneurons of the adult mouse brain.
Behavioral Deficits in NPAS1, NPAS3, and NPAS1/NPAS3 Mutant Mice. Among the eight genotypes under study, informative differences in behavioral and physiological parameters could be deduced from analyses of a subset of only four, NPAS1–/–:NPAS3–/– (double homozygote), NPAS1–/–:NPAS3+/+ (NPAS1 homozygote), NPAS1+/+:NPAS3–/– (NPAS3 homozygote), and NPAS1+/+:NPAS3+/+ (wt). Heterozygosity of either locus compounded over homozygosity of the paralogous locus did reveal phenotypic trends consistent with deficits revealed by the four genotypes listed above. However, in no case did such data achieve statistical significance.
Upon weaning into groups of 5 or more it was observed that a small number of animals displayed an abnormal, stereotypic darting behavior. An example can be viewed in Movie 1, which is published as supporting information on the PNAS web site. Of the 89 mice that were systematically observed, only four exhibited this behavior. All four mice were homozygous null at the NPAS3 locus and either homozygous null or heterozygous at the NPAS1 locus. Of the remaining 85 mice, none were homozygous null at the NPAS3 locus. Therefore, this stereotypic locomotor activity was exclusively dependent on the absence of NPAS3.
With respect to the general health and behavioral parameters measured at 4 months of age, no genotype-dependent differences were observed regarding body temperature, eye-blink response, ear-twitch response, whisker orientation, hearing, smell, or grip strength (Table 1, which is published as supporting information on the PNAS web site). Additionally, no differences were observed in longevity among the four genotypes. Both sexes of NPAS1+/+:NPAS3–/– and compound nulls were ≈20% smaller than wt and NPAS1–/–:NPAS3+/+ animals. Animals of the former two genotypes also went limp upon tail suspension, displayed a shorter and irregular stride length indicative of a slightly abnormal gait, and were slightly uncoordinated on early rotarod trails. As with wt and NPAS1 homozygotes, however, NPAS3 homozygotes and double-null animals were able to learn effective motor coordination upon repeated rotarod testing. No genotype-dependent differences were detected in the hanging wire test, the rotarod test, the test for anosmia, or the shock-threshold test (data not shown). A genotype-dependent difference was observed in the cued learning task. Instead of freezing in response to a conditioning stimulus previously coupled with a mild foot shock, NPAS1:NPAS3 double-null animals exhibited an enhanced level of motion in this behavioral assay (Fig. 9, which is published as supporting information on the PNAS web site). Finally, females of all genotypes were fertile, yet the NPAS1+/+:NPAS3–/– and NPAS1–/–:NPAS3–/– genotypes failed to properly nest and mother their litters, resulting in pup mortality 1–2 days postpartum. A video of this poor mothering behavior is available (Movie 2, which is published as supporting information on the PNAS web site).
Impaired Prepulse Inhibition. Initially, mice of all four genotypes were tested for startle responses to sound stimuli ranging from 70 dB (background noise) to 120 dB. No genotype-dependent differences in hearing ability were observed (Fig. 10, which is published as supporting information on the PNAS web site). For the prepulse inhibition test, mice were first assessed for acoustic startle response to 40 msec of a 120-dB pulse in the presence of the 70-dB background. Mice of all genotypes responded equivalently to this baseline startle pulse. Prepulse inhibition was then measured by applying prepulses of 78 or 86 dB followed by the 120-dB startle pulse. A statistically significant reduction in prepulse inhibition was observed in NPAS1–/–:NPAS3–/– animals relative to NPAS1–/–:NPAS3+/+ and wt mice (Fig. 3A). NPAS1+/+:NPAS3–/– mice appeared to be attenuated in prepulse inhibition at both the 78- and 86-dB prepulse sound levels, yet in neither case did such data achieve statistical significance.
Fig. 3.

Aberrant behavioral patterns in mice deficient in NPAS1 and NPAS3. (A) Impaired prepulse inhibition (PPI) in NPAS1–/–:NPAS3–/– adult mice. Animals were assayed for prepulse inhibition at two prepulse sound levels, 78 and 86 dB. Statistically significant impairment for prepulse inhibition was observed at both sound levels only for double-null animals. (B) Impaired social recognition in NPAS3–/– and NPAS1–/–:NPAS3–/– adult mice. Animals were assayed for social recognition by comparing the investigation time of unfamiliar juveniles on day 1 and reinvestigation on day 4. (Left) Double-null animals showed a statistically significant reduction in initial investigation time. (Right) Both NPAS3 homozygotes and double-null animals failed to display statistically significant social recognition when comparing investigation times on days 1 and 4. (C) Enhanced open-field locomotor activity in NPAS3–/– and NPAS1–/–:NPAS3–/– adult mice. Animals were assayed for open-field locomotor activity. NPAS3 homozygotes and double-null animals showed a statistically significant increase in total distance traveled compared with NPAS1 homozygotes and wt littermates. Thigmotactic analysis revealed that extra locomotor activity took place in the outer rim of the open-field cage.
Impaired Social Recognition. Social recognition was investigated by assessing the response of adults of varying genotypes to wt juvenile mice. On day 1, animals were allowed an initial investigation of the juveniles for 2 min. Three days later (day 4) interaction time between the same two individuals was scored. The normal response to this assay is a reduction in social interaction scored on day 4, indicating a process of learned familiarization. Deficits in this paradigm of social recognition were observed in both NPAS3 homozygotes and NPAS1:NPAS3 double-null animals. Only wt animals and NPAS1 homozygotes displayed significant social recognition (Fig. 3B). Double-null animals interacted with juveniles for a substantially shorter amount of time on day 1 (Fig. 3B Left). The level of interaction in the double-null animals actually increased slightly on day 4, leading to an enhanced duration of investigation as integrated between the two test days (Fig. 3B Right). NPAS1+/+:NPAS3–/– animals also exhibited a reduced level of interaction on day 1 and failed to achieve significant social recognition.
Enhanced Open-Field Activity. Animals of each genotype were analyzed for locomotor activity for 12 min, as assayed in an open-field box. Activity was then analyzed for distance traveled, stereotypic counts, and thigmotaxis. Both NPAS1–/–:NPAS3–/– and NPAS1+/+:NPAS3–/– animals exhibited enhanced open-field activity relative to wt and NPAS1–/–:NPAS3+/+ mice (Fig. 3C). Double-null animals were slightly more active in the open-field test than NPAS3 homozygotes, and thigmotactic analyses indicated that the enhanced locomotor activity of double nulls and NPAS3 homozygotes took place in the outer rim of the open-field test box.
Attenuated Reelin Expression in NPAS3 Homozygotes and NPAS1:NPAS3 Double-Null Animals. Inhibitory interneurons expressing the NPAS1 and NPAS3 transcription factors appear to be alive and present in the CNS of adult mice irrespective of the expression of functional protein from either locus. This finding was most clearly established in NPAS1 homozygotes. The distribution of β-galactosidase-expressing neurons was indistinguishable in sectioned brain tissue derived from NPAS1–/– and NPAS1+/– animals. Moreover, this β-galactosidase staining pattern was anatomically indistinguishable from the immunohistochemical staining patterns with antibodies specific to the NPAS1 transcription factor in NPAS1+/+ and NPAS1+/– animals (Fig. 1 A and B). Such data strongly argue that abrogation of NPAS1 function does not eliminate survival of the neurons in which this transcription factor is normally expressed. A similar interpretation can be gleaned from immunohistochemical studies of sectioned brain tissue derived from wt and NPAS3 homozygotes. A high fraction of GAD-67-expressing neurons in the cortex of adult mice costained for the NPAS3 transcription factor (Fig. 2B). The GAD-67 staining pattern was indistinguishable in wt and NPAS3 homozygotes (Fig. 11, which is published as supporting information on the PNAS web site), giving indirect evidence that the neurons normally expressing NPAS3 are not only alive and present in NPAS3 homozygotes, but unimpaired in their ability to express the GAD enzyme critical for production of the inhibitory neurotransmitter, GABA.
To assess whether other regulatory proteins, enzymes, or neurotransmitters might be expressed in an enhanced or attenuated manner in NPAS1, NPAS3, or doubly deficient mice, we performed immunohistochemical staining assays on brain tissue sectioned from wt, NPAS1–/–, NPAS3–/–, and double-null animals. No substantive differences were observed for GAD-67, parvalbumin (see Fig. 11), neuropeptide Y (NPY), calbindin D-28k, calretinin, or GABA (data not shown) among animals of the four genotypes. By contrast, reelin expression was substantially attenuated in numerous brain regions of both NPAS3 homozygotes and NPAS1:NPAS3 double-null animals.
Reelin was observed most prominently in the soma of small inhibitory interneurons of wt animals and was substantially diminished in cortical brain sections of NPAS3 homozygotes and NPAS1:NPAS3 double-null animals (Fig. 4 A–D). The pattern of attenuation of reelin staining in NPAS3 homozygotes and NPAS1:NPAS3 double-null animals was not uniform across the entire CNS. For example, clusters of reelin-positive neurons located in the piriform cortex were unaffected as a function of genotype (Fig. 4 E–H). That changes in reelin expression in various regions of the mouse brain achieved, or failed to achieve, statistical significance was confirmed by quantitative measurements scored in a blinded fashion with respect to sample genotype (Fig. 4I). Moreover, double-labeling immunocytochemical experiments revealed that reelin extensively colocalized with NPAS1- and NPAS3-expressing neurons (Fig. 12, which is published as supporting information on the PNAS web site).
Fig. 4.

Attenuation in reelin expression in brain tissue derived from NPAS1+/+: NPAS3–/– and NPAS1–/–:NPAS3–/– adult mice. Coronal sections of adult mouse brain tissue were prepared from wt (A and E), NPAS1 homozygotes (B and F), NPAS3 homozygotes (C and G), and double-null (D and H) animals. Sections were stained with antibodies specific to reelin. Shown are antibody staining patterns on matching coronal sections of the frontal (A–D) and piriform (E–H) cortex. Irrespective of brain region, reelin-positive antibody staining was uniformly restricted to the cytoplasm of inhibitory interneurons. Significant attenuation in reelin antibody staining was observed in the frontal cortex of NPAS3 homozygotes and double-null animals. The roman numerals in A designate cortical layers I–VI. (I) Quantitative assessment of reelin expression in adult brain tissue derived from wt, NPAS1+/+:NPAS31–/–, NPAS1–/–: NPAS3+/+, and NPAS1–/–:NPAS3–/– mice. Coronal sections were prepared from matching regions of cerebral and piriform cortex of at least five animals of the four genotypes under study. Sections were stained with antibodies specific to reelin, photographed, and quantitated over equivalent 1.6-mm2 regions for reelin-positive neurons. Observers were blinded to genotype.
Discussion
The observations reported in this study tentatively suggest that the NPAS3 and NPAS1 transcription factors may control regulatory pathways relevant to schizophrenia. First, behavioral studies of NPAS3 homozygotes and NPAS1:NPAS3 double-null animals gave statistically significant evidence of deficits in prepulse inhibition and social recognition, as well as substantially increased open-field locomotor activity. These behavioral abnormalities in laboratory mice, as best possible for a rodent model, mimic psychosis (9–11). As such, they may be viewed in support of the conclusions of Kamnasaran et al. (6) who discovered a mutation in the NPAS3 gene in a family suffering from schizophrenia.
It is difficult to draw direct parallels between the behavioral abnormalities observed in NPAS1- and NPAS3-deficient mice and the complex, delusional, and cognitive defects that characterize human schizophrenia. In the human family with the NPAS3 translocation, schizophrenia was transmitted as a dominant trait, and the affected patients carried one normal allele at the NPAS3 locus. One important possibility is that the human brain is more sensitive to NPAS3 haploinsufficiency than the mouse brain, particularly regarding the relatively subtle, thought-based defects in schizophrenia. It is also possible that the human translocation produces a dominant negative effect by yielding a protein having an intact DNA-binding domain but lacking the regulatory PAS domain. Finally, we cannot exclude the possibility that the human mutation produces a gain-of-function effect that somehow disrupts NPAS3 regulation.
Regardless of mechanism, the current findings suggest that schizophrenia in the affected family members described by Kamnasaran et al. (6) results from an abnormality in the widely distributed interneurons that normally express the NPAS3 protein. Further study of the behavioral deficits in the NPAS1-, NPAS3-, and doubly deficient mice should help elucidate the regulatory defects that underlie this mouse model of schizophrenia, thereby providing further clues into this important human disease.
Aside from behavioral deficits tangentially pointing toward schizophrenia, we report two additional observations supportive of the prediction that the NPAS1 and NPAS3 transcription factors may regulate pathways relevant to psychotic illness. First, both transcription factors are expressed in inhibitory interneurons of the adult mouse brain. The processing of complex informational input into the brain is undoubtedly guided and gated by inhibitory interneurons. As such, changes in perception and inferential thinking diagnostic of schizophrenia can logically be predicted to be affected in patients suffering deficits in the function of inhibitory interneurons. Indeed, it is almost certainly the case that prepulse inhibition depends on proper function of inhibitory circuitry in the CNS (12). Because both NPAS1 and NPAS3 are expressed in inhibitory interneurons, we speculate that they serve to control regulatory pathways important to the function of this class of neurons.
Second, among numerous markers of inhibitory interneurons sampled in this study, the sole gene product thus far observed to be affected in NPAS3 homozygotes and NPAS1:NPAS3 double-null animals was reelin. Reelin expression was attenuated in the cortex, dentate gyrus, and amygdala of NPAS3 homozygotes and NPAS1:NPAS3 double-null mice. During embryonic patterning of the brain, reelin plays a critical role in the guidance of newborn neurons to proper cortical layers (13). In the adult, reelin is found in a subset of GABAergic interneurons (14), where it may act to modulate synaptic plasticity by serving as an intercellular signaling molecule (15), as an extracellular protease (16), or both. More relevant to the present results is the report that functional and neurochemical deficits in the reeler heterozygous mice are similar to those observed in schizophrenia (17).
The fact that reelin expression was reduced in widespread regions of the adult brain in NPAS3 homozygotes and NPAS1:NPAS3 double-null animals is consistent with numerous reports indicating an attenuation in reelin protein and mRNA in postmortem brain tissue of patients with schizophrenia (18). In a multivariate analysis of data from the Stanley Foundation Neuropathology Consortium involving 14 different laboratories that evaluated postmortem, prefrontal brain tissue of patients with schizophrenia, a reduction in reelin mRNA emerged as the single most statistically significant variable identified in the entire study (19). This same study identified abnormalities in cortical interneurons as one of the principal deficits in postmortem brain tissue of individuals with schizophrenia. These correlative observations articulate the challenge of understanding the functional role of reelin in the adult and the relationship of its expression to the function of the NPAS1 and NPAS3 transcription factors.
In conclusion, we tentatively identify the regulatory pathways controlled by the NPAS1 and NPAS3 transcription factors as being selectively relevant to psychotic disease. If correct, it is predictable that studies focused on the signals that activate NPAS1 and NPAS3, and the regulatory pathways downstream of these transcription factors, may provide valuable footing for the study of human psychosis.
Supplementary Material
Acknowledgments
All experimental observations reported herein were performed by members of the McKnight laboratory at the University of Texas Southwestern Medical Center (UTSWMC). We thank Dr. Kebreten Manaye at Howard University (Washington, DC) for the use of a Nikon microscope equipped with the neurolucida program (MicroBright-Field, Williston, VT), Dr. Joachim Herz and Uwe Beffert (UTSWMC) for advice on reelin antibody staining, and Drs. Herz, Michael Brown, Carol Tamminga (UTSWMC), and Huda Zoghbi (Baylor College of Medicine) for critical comments on the manuscript. This work was supported by National Institute of Mental Health Grant 4R37MH59388 (to S.L.M.), by a 2-year grant from the McKnight Foundation for Neurosciences, and by unrestricted funds from the Morton H. Meyerson Family Tzedakah Fund and an anonymous donor.
Abbreviations: GAD, glutamic acid decarboxylase; GABA, γ-aminobutyric acid; wt, wild-type.
References
- 1.Zhou, Y.-D., Barnard, M., Tian, H., Li, X., Ring, H.Z., Francke, U., Shelton, J., Richardson, J., Russell, D. W. & McKnight, S. L. (1997) Proc. Natl. Acad. Sci. USA 94, 713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brunskill, E. W., Witte, D. P., Shreiner, A. B. & Potter, S. S. (1999) Mech. Dev. 88, 237–241. [DOI] [PubMed] [Google Scholar]
- 3.Dudley, C. A., Erbel-Sieler, C., Estill, S. J., Reick, M., Franken, P., Pitts, S. & McKnight, S. L. (2003) Science 301, 379–383. [DOI] [PubMed] [Google Scholar]
- 4.Rutter, J., Reick, M., Wu, L. C. & McKnight, S. L. (2001) Science 293, 510–514. [DOI] [PubMed] [Google Scholar]
- 5.Taylor, B. L. & Zhulin, I. B. (1999) Microbiol. Mol. Biol. Rev. 63, 479–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamnasaran, D., Muir, W. J., Ferguson-Smith, M. A. & Cox, D. W. (2003) J. Med. Genet. 40, 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gulyás, A. I., Hájos, N. & Freund, T. F. (1992) J. Neurosci. 16, 3997–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miettinen, R., Gulyás, A. I., Baimbridge, K. G., Jacobowitz, D. M. & Freund, T. F. (1992) Neuroscience 48, 29–43. [DOI] [PubMed] [Google Scholar]
- 9.Geyer, M. A., McIlwain, K. L. & Paylor, R. (2002) Mol. Psychiatry 7, 1039–1053. [DOI] [PubMed] [Google Scholar]
- 10.Torres, G., Hallas, B. H., Bernace, V. A., Jones, C., Gross, K. W. & Horowitz, J. M. (2004) Brain Res. Bull. 62, 315–326. [DOI] [PubMed] [Google Scholar]
- 11.Young, L. J. (2001) Am. J. Med. Genet. 105, 53–54. [PubMed] [Google Scholar]
- 12.Frost, W. N., Tian, L.-M., Hoppe, T. A., Mongeluzi, D. L. & Wang, J. (2003) Neuron 40, 991–1001. [DOI] [PubMed] [Google Scholar]
- 13.Tissir, F. & Goffinet, A. M. (2003) Nat. Rev. Neurosci. 4, 496–505. [DOI] [PubMed] [Google Scholar]
- 14.Alcántara, S., Ruiz, M., D'Arcangelo, G., Ezan, F., deLecea, L., Curran, T., Sotelo, C. & Soriano, E. (1998) J. Neurosci. 18, 7779–7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pesold, C., Liu, W. S., Guidotti, A., Costa, E. & Caruncho, H. J. (1999) Proc. Natl. Acad. Sci. USA 96, 3217–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quattrocchi, C. C., Wannenes, F., Perscio, A. M., Ciafre, S. A., D'Arcangelo, G., Farace, M. G. & Keller, F. (2002) J. Biol. Chem. 277, 303–309. [DOI] [PubMed] [Google Scholar]
- 17.Costa, E., Davis, J., Pesold, C., Tueting, P. & Guidotti, A. (2002) Curr. Opin. Pharmacol. 2, 56–62. [DOI] [PubMed] [Google Scholar]
- 18.Impagnatiello, F., Guidotti, A. R., Pesold, C., Dwivedi, Y., Caruncho, H., Pisu, M. G., Uzunov, D. P., Smalheiser, N. R., Davis, J. M., Pandey, G. N., et al. (1998) Proc. Natl. Acad. Sci. USA 95, 15718–15723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knable, M. B., Torrey, E. F., Webster, M. J. & Bartko, J. J. (2001) Brain Res. Bull. 55, 651–659. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}