

Abstract
NMR experiments of 129Xe adsorbed on an iridium single crystal surface are reported. Very high nuclear polarization (Pz ≈ 0.7) makes the experiment possible. A coverage of less then one monolayer is investigated on the Ir(111) surface with an area of 0.8 cm2. The observed resonance line shifts are very large and highly anisotropic. We find σiso = 1,032 ± 11 ppm and σan = 291 ± 33 ppm, which are far above the typical range of physisorption. The highly ordered substrate leads to homogeneous conditions for the xenon atoms, as seen in the narrow linewidth of 20 ppm. Chemical shifts under physisorption conditions are not large enough to totally explain the results. Knight shift can clearly be identified as the cause of the findings. This shift shows the presence of conduction electrons of the metallic substrate at the xenon nucleus and thus the mixing of metallic and atomic states at the Fermi level. Such mixing is in accordance with recent Hartree–Fock and density functional calculations of similar van der Waals adsorption systems. Quantitative comparisons, however, fail completely. The size and ratio of σan and σiso are pure ground-state properties in a structurally simple system. They are accessible to theory and provide detailed local information that can serve as a benchmark for theory.
The world contains far more objects then hard solids (1), namely objects that tend to be bound together by van der Waals (vdW) interaction. Physical adsorption is an extremely important and very general phenomenon; examples include the way geckos run up a wall or how new sticky tape might work (2, 3). Physical adsorption phenomena are also found in the omnipresent friction forces (4) or in noble gas adsorption on surfaces (5). The latter often serves as a prime example for vdW interaction and is studied experimentally and theoretically. Detailed spectroscopic information is difficult to obtain because vdW interaction is hardly visible in IR or photo electron spectroscopy. NMR provides better information as the chemical shift is highly sensitive to the local environment, regardless of whether it is chemically bound or not. NMR therefore yields detailed spectroscopic information even for systems held together by vdW interaction. 129Xe is especially suited as an NMR probe because of the large shift range it experiences when physisorbed. Therefore, it has found many applications in the study of supported catalysts or porous media among others (6–8). Here, we present previously unreported NMR data of 129Xe adsorbed on a single crystal metal surface, namely Ir(111). Conceptually, this adsorption system constitutes a simple enough situation to be treated by theory. Experimentally, it is a long-sought-after goal (9) that is not so easily reached (10, 11).
The theoretical treatment, especially of the xenon/metal adsorption, has been the subject of much discussion and development (1, 5, 12–15). Density functional theory (DFT) treatment would be possible, if one only had the correct functional (1, 13). Hartree–Fock approaches cannot cope with the infinite surface geometry (15). Many adsorption properties are correctly calculated by DFT, but large deficiencies, some of a fundamental nature, remain (13). New experimental input derived from structurally simple systems may be very helpful. Therefore, we think that the ground-state properties of the 129Xe/Ir(111) NMR shifts can be a good testing ground for theory and serve as benchmark data in the field.
The distinction between chemisorption and physisorption is not easy. The ambivalence of this situation can be seen in xenon very well. On the one hand, the xenon–xenon or xenon–solid interaction are used as prime examples of vdW interaction. On the other hand, xenon does undergo true chemical bonding in many fluoro and oxofluoro compounds. This situation is clearly reflected in NMR experiments. NMR chemical shifts are in the range of 0–300 ppm for physisorbed 129Xe atoms and between 2,400 and 7,500 ppm for true chemical compounds (8, 16).
Here we present entirely new experimental results of xenon adsorbed on an iridium single crystal surface, Ir(111), using 129Xe NMR. Chemical shift measurements are used in many liquid, solid, or biological systems to obtain detailed information on structural, electronic, or geometric properties. For adsorbates on a metallic single crystal surface these experiments were not feasible until now.
Recently, a breakthrough was achieved when it was shown that NMR experiments of xenon adsorbed in monolayer quantities on a 1-cm2 nonmetallic surface are possible (11). This method has now been further developed and adapted to metallic surfaces. Iridium has a fcc crystal structure and the (111) face is hexagonally close-packed. Iridium was chosen as a substrate material because of its relatively low magnetic susceptibility (χ = 38·10–6) when compared with Pt, Ru, or Pd. Nevertheless, the Ir(111) surface is reasonably similar to the more commonly studied Pt(111) or Ru(001) surfaces, as seen in many aspects like the adsorption energy of xenon and CO or the reactivity toward ethylene. For Xe/Pt (111) and Xe/Ru (001), many experimental and theoretical investigations exist to which we will try to compare our results.
The experimental apparatus has been described in more detail (refs. 10 and 17–19; http://archiv.ub.uni-marburg.de/diss/z2004/0085). An ultrahigh vacuum chamber is connected to an NMR setup and to an optical pumping system. The base pressure of the ultrahigh vacuum system is 5·10–10 mbar (1 bar = 100 kPa). The chamber contains the crystal, its support structure for heating, cooling, and positioning, a sputter gun, a quadrupole mass spectrometer, and a gas-handling manifold. Cleaning of the crystal is achieved by argon ion sputtering and/or oxygen treatment before annealing at 1,150 K. The temperature is measured by a chromel/constantan (type E) thermocouple. The 75-μm thin wires are spot welded to the backside of the crystal. Thin wires are mandatory to keep magnetic distortions due to the low-temperature ferromagnetic material below our detection limit. The NMR spectrometer is essentially home-built and consists of a single channel irradiation and quadrature detection system. We use an electromagnet with a 1.98 T field that gives a 129Xe resonance frequency of 23.275 MHz. The xenon used is isotopically enriched to 71% 129Xe. The excitation pulse length for 90° is 16 μs. After a dead time of 24 μs, data acquisition starts for 10 ms. The spectrometer bandwidth is 100 kHz, and the data are taken in oversampling at 500 kHz. For a single measurement a few hundred induction scans are added up. The rather strong ringing of our probe head is suppressed by using a simplified version of the RIDE sequence (20). We alternately sum the induction signals of a 90x° excitation and a 180x° – τ – 90–°x excitation with an appropriately long τ of 400 μs. This adds the signal coherently and eliminates the ringing substantially. The overall repetition rate for scans is 10 per s or less. The time between the excitation pulses is necessary for the exchange of the used xenon atoms (spent polarization) by fresh ones and to interlace heating intervals into the time sequence to stabilize the sample temperature.
The optical pumping setup produces highly polarized, so-called hyperpolarized, 129Xe by spin-exchange optical pumping with rubidium vapor by using N2 as a buffer/quench gas. The latter is removed by a freeze-pump thaw cycle (18). The achieved polarization is 0.4–0.8, i.e., an increase of 5 orders of magnitude compared with the thermal equilibrium value. This high polarization is the prime reason for achieving the necessary sensitivity gain that allows the present experiments. The polarization can be monitored in situ in the ultrahigh vacuum chamber by observing the bulk xenon NMR line shift because of the nuclear magnetization (19, 21).
The hyperpolarized xenon is stored in a reservoir in frozen form and dosed onto the crystal by using its own vapor pressure. At 87 K reservoir temperature, the boiling point of liquid argon, the xenon flux is ≈40 monolayers (MLs) per s. For a reservoir temperature of 77 K, 4 MLs per s is achieved. All experiments are performed in a batch mode, which means that the polarized gas is produced and stored and then continuously released so that data can be taken for 1–2 min at high flux or 10 times longer at low flux. The crystal temperature is such that the xenon on the surface is continuously replenished and many NMR scans can be taken with one batch.
Fig. 1 shows temperature-programmed desorption spectra of xenon adsorbed on Ir(111). The surface is exposed to various amounts of xenon at a low temperature and then heated at a constant rate (4 K/s). The xenon desorption rate is recorded as a function of temperature. The least bound atoms leave at the lowest temperature. In the figure, multilayer and second-layer xenon atoms desorb at ≈60 K. The desorption up to 100 K comes from the first atomic layer. The results are typical for xenon on transition metals and very similar to those from Ru(001), from which the peak assignment is taken (22). The thermal desorption parameters of Xe/Ru(001) are well known (22). Using those parameters and shifting the temperature scale so that the high temperature peaks coincide, we estimate the mean residence time for xenon on Ir(111) at 92 K to be 4 s and at 104 K to be 0.1 s. The mean residence time of second-layer atoms (xenon on xenon) would be below 10–4 s at 100 K. Hence, one achieves roughly full monolayer coverage at a xenon flux of 40 MLs per s but virtually no atoms are present in the second layer.
Fig. 1.

Temperature-programmed desorption (TPD) spectra. The Ir(111) surface is dosed with xenon at low temperature. Upon crystal heating, the xenon desorbs, which is recorded in a quadrupole mass spectrometer. The various traces show different initial coverages. At 60 K the physisorbed multilayer xenon desorbs; up to 100 K, the atoms directly bound to the metal surface desorb.
NMR signals were obtained between 91 and 104 K at 40 MLs per s. Fig. 2 shows a resonance line obtained at 91 K. The signal-to-noise ratio is ≈7 and the linewidth is 400 Hz (17 ppm). The crystal normal, i.e., [111] direction, was oriented at 90° with respect to the magnetic field which maximizes the exposure to polarized xenon gas in our experimental setup. Data were also taken at smaller angles down to 50°. Fig. 3 shows the line position as a function of angle at 104 K. The functional dependence of the shift is known to be cos2 θ if cylindrical symmetry is present (23). This is certainly the case here, because the substrate temperature is even above the temperature where xenon desorption occurs in temperature-programmed desorption (Fig. 1). The atoms are therefore highly mobile. The hopping rate can be estimated to 108 per s (24), clearly in the motional narrowing regime. Therefore, the xenon experiences a cylindrically symmetric local environment. The data are fitted with σ(θ) = σiso + σan·(3 cos2 θ – 1)/2. From the data in Fig. 3 one obtains for the isotropic part σiso = 1,032 ± 11 ppm and for the anisotropic part σan = 291 ± 33 ppm. With these values the shift at 0° is σ∥ = 1,323 ± 35 ppm and at the perpendicular orientation σ⊥ = 886 ± 35 ppm; the difference of both is often called Δσ = σ∥ – σ⊥ = 437 ppm. All shifts are given with respect to the xenon gas line at low pressure being at 0 ppm. The estimated error bars are quite small and may be fortuitous, but the experimental data at 90° and at 50° show little variance and dominate the fit.
Fig. 2.

NMR line of 129Xe adsorbed on Ir(111) at 91 K. The shift is given in ppm in the upper axis relative to low-pressure xenon gas at 0 ppm. The position of bulk xenon is also given. The crystal normal was at 90° with respect to the magnetic field. The xenon flux used was 40 MLs per s.
Fig. 3.

Angular dependence of the NMR line of 129Xe adsorbed on Ir(111) at 104 K. The shift scale is the same as in Fig. 2. For technical reasons the angular range is very restricted; 90° is the normal position in the experiment because the xenon flux is admitted into the experimental chamber at that angle. The fit shown as a solid line is σ = σiso + σan·[3 cos2(θ – 5.2°) – 1]/2 with σiso = 1,032 ± 11 ppm and σan = 291 ± 33 ppm. The angular correction was not fitted but determined separately. All data points represent several measurements; the numerical values of the shifts and standard deviations (in ppm) are given in Inset.
We attempted but failed to observe the NMR resonances at a xenon flux of 4 MLs per s and surface temperatures of 83–89 K. This failure will be discussed further below in an attempt to estimate the T1 time.
Clearly, the observed shifts are far above the physisorption regime of 0–300 ppm. Next, we briefly discuss some minor contributions to the shift and then focus on the major effects, namely the Knight and chemical shifts.
As mentioned above, iridium has a small but nonzero susceptibility. On the outside of the crystal the bulk demagnetizing field leads to a distortion of the magnetic field, resulting in an angle-dependent line shift and broadening. The latter is part of the observed experimental linewidth. The angle-dependent part of the shift has been obtained from measurements on a xenon film condensed on the iridium single crystal. The analysis showed an anisotropy of Δσ = 15 ppm due to the substrate susceptibility. This effect is well understood quantitatively (19, 21). It is not discussed further in this article because 15 ppm is very small compared with the monolayer Δσ of 437 ppm.
In a monolayer the xenon–xenon interaction is lateral only and, therefore, highly anisotropic. A good estimate of this contribution can be taken from our experiments on xenon adsorbed on CO-covered Ir(111) (11). Here, the metallic interaction is strongly shielded by the molecular CO spacer. The measured shift difference between 0 and 90° is 43 ppm (19). Also, the anisotropic Xe–CO interaction is much smaller as can be seen from results of a Hartree–Fock calculation (25) of the Xe–CO system, which finds a rather small chemical shift anisotropy (<10 ppm) for the equilibrium Xe–CO distance. Thus, the Xe–Xe interaction cannot explain the observed shift anisotropy on the metal.
The moment density of the highly polarized xenon nuclei generates another anisotropic contribution to the total field. For a thin film the shift has an angular dependence of (3 cos2(θ) – 1)/2 as has been shown for highly polarized 3He films (26, 27) or 129Xe films (28). Here, the shift and anisotropy are in the order of 10–20 ppm even for highest nuclear polarization (21). Therefore, this also does not serve as an explanation of the experimental anisotropy.
The largest contribution and most likely explanation for our observations is the simultaneous presence of a Knight shift and a possibly strong chemical shift. Experimentally, the Knight shift and chemical shift contributions cannot be disentangled with our setup. Relaxation time measurements might be helpful but are not feasible for us at this point.
To judge the relative weight of the chemical shift and the Knight shift contributions in the current experiment it is useful to look at results from studies on iridium clusters of various sizes. Xenon interacting with small isolated clusters (Ir4 and Ir6) shows shifts between 100 and 140 ppm, indicative of physisorption (29). The linewidth observed is <20 ppm. When nanometer size particles are prepared, the shift goes up to 800 ppm (30). The difference between small and large clusters is essentially the occurrence of free electrons and an established Fermi level, so that paramagnetic metallic behavior occurs. The Pauli susceptibility and the accompanying Fermi contact interaction with adsorbed xenon atoms leads to a large Knight shift.
Theoretical studies of xenon interacting with isolated metal ions (31) (Li, Na, K, Cu, and Ag) show binding energies from 0.2 to 0.6 eV, which are not too different from xenon adsorption energies on metal single crystals (32). The experimental and calculated chemical shifts lie between –100 and +100 ppm. This shows that relatively high binding energies do not necessarily mean large chemical shifts for xenon. The interaction of xenon with single halogen ions can however lead to enormous shifts like 47,00 ppm for Cl+ (33).
From the discussion above we conclude that the physical adsorption of xenon on a metal causes proper chemical shifts in the range of 140 ppm. Thus, the observed shift of σiso = 1,032 ppm here must then originate to a large extent from the Knight shift alone. This compares reasonably well with the 800 ppm reported from supported catalyst work of xenon on iridium clusters (30). The latter results are room-temperature measurements of the chemical shift at very low pressure. The higher binding energy on the metal than the zeolite support allows for the extraction of the pure metal contribution. However, this is an averaged value over all sites occurring on the cluster surface. As argued by Bifone et al. (9) the shifts from the terraces are likely to be the largest ones, because they show the largest local density of states (34). It is therefore not too astonishing that the single crystal results give an ≈230-ppm larger shift than the averaged cluster work.
The interaction of xenon with a metal substrate is highly anisotropic. In supported catalyst work (large particles) at elevated temperatures this is masked by motional narrowing. At lower temperatures substantial broadening occurs [>1,000 ppm (9)] that cannot be disentangled from the inhomogeneous broadening through differences in the local binding site. Here, we have measured the anisotropy of the xenon on metal free of those broadenings and on a defined crystal face. The obtained shift anisotropy is σan = 291 ppm or Δσ = 437 ppm. The xenon shift measurements on small iridium and rhodium clusters already mentioned (29) show little line broadening and thus small anisotropies even at 100 K. The anisotropy resulting from Xe–Xe interaction is by far not large enough (see above). In restricted geometries (cages and channels) rather large anisotropies have been seen (35); the largest being in clathrates of almost 200 ppm (36). Nevertheless, this is still far below 437 ppm and it is questionable if those examples can help us understand the flat and open geometry on the single crystal surface. Altogether we think that pure chemical interaction cannot produce shift anisotropies of the observed magnitude here. Therefore, the anisotropic Knight shift is a much more likely physical mechanism. The contact interaction of the s electrons give rise to an isotropic shift. Assuming that the orbital moments of the p electrons are frozen (37), the anisotropy results from the dipolar interaction of the p electron spins with the nucleus. Abragam gives an estimate for the ratio of the anisotropic part of the chemical shift divided by the isotropic one (23):
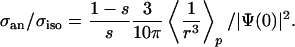 |
[1] |
Here, 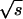 and
and 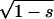 are the amplitudes of s and p waves of the wave function relevant at the Fermi level. For hydrogen wave functions the ratio can be calculated (38) with ns and np being the principle quantum numbers of the states:
are the amplitudes of s and p waves of the wave function relevant at the Fermi level. For hydrogen wave functions the ratio can be calculated (38) with ns and np being the principle quantum numbers of the states:
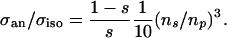 |
[2] |
In a free atom the ratio of the hyperfine interaction of a p state versus that of an s state is given by Eq. 2 when (1 – s)/s is omitted. For the same “n” this gives 1/10. Experimentally, this is well fulfilled even for heavy atoms as one can see in Na, Rb, and Cs. There, the ratio of P1/2 vs. S1/2 hyperfine constants are 0.11, 0.12, and 0.13, respectively (39), quite close to the estimate. This finding shows that hydrogen atomic wave functions can be used to estimate the anisotropy of the Knight shift, even for cesium, because the dominant contribution to 〈1/r3〉p occurs for r < 0.03 Å, where the effective electrostatic potential is almost spherically symmetric.
For the xenon adsorption system the character of the wave function at the Fermi level can only be taken from a calculation. Recently, an all-electron DFT calculation was performed for xenon adsorbed on Pt(111) (5). The local density of states was calculated. From this the relative occupancy at the Fermi level of the 6s vs. 5p vs. 5d states was determined. The result was ≈1:16:1.6, respectively, at the geometrically optimized binding position. The calculation well explains the experimentally observed on-top binding site and work function changes. Ignoring the d state for the moment, the relative occupancy of only 0.06 in the s state as compared with the p state gives σan/σiso = 0.94/0.06·1/10·1.7 = 2.8. The factor 1.7 arises from the ns = 6 and np = 5 in the calculation (5). Experimentally (Fig. 3) this ratio is (estimating the proper chemical shift at 150 ppm) 291/(1,032–150) = 0.33, i.e., an order of magnitude away from the simple estimate above. The core polarization effect due to the 5d electrons is typically opposite of the s-contribution (40), so that the isotropic contribution would be smaller and the theoretical σan/σiso estimate even larger. The discrepancy between experiment and theory is quite large and cannot be removed by a small adjustment in the relative occupancy of the states. Possible reasons can only be speculated about. Most certainly, the analysis along the Abragam formula may be too simple. The nonhydrogenic nature of the wave functions could be substantial even though it seemed not too bad for Cs. Most likely, however, a (possibly fundamental) problem exists in DFT treating weakly interacting systems as xenon on metals (13) especially when finer (23), i.e., point-like properties of matter, are probed. The currently used energy functionals in DFT tend to overestimate the binding energy somewhat, but find the equilibrium bond length much too short by ≈0.5 Å (13). In addition, the binding potential toward the vacuum is steeper than the best empirical ones (13). At finite temperature this leads to a further underestimation of the bond length.
An independent measurement of additional properties of the wave functions would certainly be most welcomed. The measurement of the relaxation rate (1/T1) of the adsorbed xenon on Ir(111) would be useful information, when the metal electrons dominate the relaxation process (41). With the current experiment this is not possible, but a reasonable estimate can be made. We have observed the resonance line at a xenon flux of 40 MLs per s and failed to do so at 4 MLs per s (at a reduced temperature to have full surface coverage). This observation makes a T1 time between 25 ms and 250 ms plausible. For a further estimate, literature data can be used. In xenon adsorbed on Pt clusters, T1 was measured to be 14 ms at 100 K (9) at 1,300 ppm shift. Assuming a chemical shift of 150 ppm, this leaves the Knight shift contribution to be 1,150 ppm. The Korringa ratio, B, can therefore be determined from the Korringa relation (41) T1TK2 = SB, with the Knight shift, K, and the constant 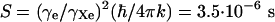 K for 129Xe. With T1 and K from above this gives B = 0.9, very close to the ideal value of 1. B can range from substantially <1 up to 30, with no apparent trend on nuclear charge (40). With this value of B = 0.9 for the Xe/Ir(111) system and also allowing 150 ppm for the proper chemical shift, i.e., K = (1,032–150) ppm, one estimates T1 = 41 ms, well within the range given above. Forty-one milliseconds is about a factor of 10 larger than results obtained from density functional calculations (10), which in that article led to the wrong conclusion that the current experiment would not be feasible. These calculations might have been flawed by the mentioned tendency of DFT calculations to overbind and to give bond lengths that are too short (13). Redoing the calculations with manually increased bond lengths (≈0.5 Å) one easily obtains T1 > 30 ms (W. Mannstadt, private communication, 2003).
K for 129Xe. With T1 and K from above this gives B = 0.9, very close to the ideal value of 1. B can range from substantially <1 up to 30, with no apparent trend on nuclear charge (40). With this value of B = 0.9 for the Xe/Ir(111) system and also allowing 150 ppm for the proper chemical shift, i.e., K = (1,032–150) ppm, one estimates T1 = 41 ms, well within the range given above. Forty-one milliseconds is about a factor of 10 larger than results obtained from density functional calculations (10), which in that article led to the wrong conclusion that the current experiment would not be feasible. These calculations might have been flawed by the mentioned tendency of DFT calculations to overbind and to give bond lengths that are too short (13). Redoing the calculations with manually increased bond lengths (≈0.5 Å) one easily obtains T1 > 30 ms (W. Mannstadt, private communication, 2003).
The equilibrium DFT calculations of the T1 time are an order of magnitude away from the admittedly crude experimental estimate. The analysis of the Knight shift anisotropy in terms of the s vs. p state occupancy also finds a discrepancy of an order of magnitude. This finding certainly shows that the NMR observables are far from being quantitatively understood and can very well serve as sensitive test cases for the theory on its way to fully understanding vdW systems.
In summary, we have measured the NMR signal of a submonolayer amount of 129Xe adsorbed on an Ir(111) single crystal surface. Large isotropic shifts (1,032 ppm) and shift anisotropies (291 ppm) have been found in narrow resonances. Isotropic and anisotropic Knight shifts have been identified as the probable mechanisms to produce these shifts. The T1 time of 129Xe adsorbed on Ir(111) is estimated to be ≈40 ms. Comparison with theoretical results shows large discrepancies in σan/σiso and T1, which might derive from the difficulties to treat the vdW adsorption system with DFT in view of the fine details accessible to NMR.
Acknowledgments
We thank D. Fick for many fruitful discussions and continuous support. This work was supported by the Deutsche Forschungsgemeinschaft (Bonn).
Abbreviations: DFT, density functional theory; vdW, van der Waals; ML, monolayer.
Data deposition: The NMR chemical shifts have been deposited in the Physics and Astronomy Classification Scheme database, www.aip.org/pacs (accession nos. 33.25.+k, 68.43.–h, and 76.60.Cq).
References
- 1.Hult, E., Rydberg, H., Lundquist, B. I. & Langreth, D. C. (1999) Phys. Rev. B Solid State 59, 4708–4713. [Google Scholar]
- 2.Autumn, K., Sitti, M., Liang, Y. A., Peattie, A. M., Hansen, W. R., Sponberg, S., Kenny, T. W., Fearing, R., Israelachvili, J. N. & Full, R. J. (2002) Proc. Natl. Acad. Sci. USA 99, 12252–12256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geim, A. K., Dubonos, S. V., Grigorieva, I. V., Novoselov, K. S., Zhukov, A. A. & Shapoval, S. Y. (2003) Nat. Mater. 2, 461–463. [DOI] [PubMed] [Google Scholar]
- 4.Volokitin, A. I. & Persson, B. N. J. (2003) Phys. Rev. Lett. 91, 106101 (lett.). [DOI] [PubMed] [Google Scholar]
- 5.Da Silva, J. L. F., Stampfl, C. & Scheffler, M. (2003) Phys. Rev. Lett. 90, 066104 (lett.). [DOI] [PubMed] [Google Scholar]
- 6.Ito, T. & Fraissard, J. (1982) J. Chem. Phys. 76, 5225–5229. [Google Scholar]
- 7.Pietrass, T., Bifone, A. & Pines, A. (1995) Surf. Sci. Lett. 334, L730–L734. [Google Scholar]
- 8.Goodson, B. M. (2002) J. Magn. Reson. 155, 157–216. [DOI] [PubMed] [Google Scholar]
- 9.Bifone, A., Pietrass, T., Kritzenberger, J., Pines, A. & Chmelka, B. F. (1995) Phys. Rev. Lett. 74, 3277–3280. [DOI] [PubMed] [Google Scholar]
- 10.Stahl, D., Mannstadt, W., Gerhard, P., Koch, M. & Jänsch, H. J. (2002) J. Magn. Reson. 159, 1–12. [DOI] [PubMed] [Google Scholar]
- 11.Jänsch, H. J., Gerhard, P., Koch, M. & Stahl, D. (2003) Chem. Phys. Lett. 372, 325–330. [Google Scholar]
- 12.Müller, J. E. (1990) Phys. Rev. Lett. 65, 3021–3024. [DOI] [PubMed] [Google Scholar]
- 13.Betancourt, A. E. & Bird, D. M. (2000) J. Phys. Condens. Matter 12, 7077–7088. [Google Scholar]
- 14.Clarke, S., Bihlmayer, G. & Blügel, S. (2001) Phys. Rev. B Solid State 63, 085416. [Google Scholar]
- 15.Bagus, P. S., Staemmler, V. & Wöll, C. (2002) Phys. Rev. Lett. 89, 096104 (lett.). [DOI] [PubMed] [Google Scholar]
- 16.Pietrass, T. & Gaede, H. C. (1995) Adv. Mater. 7, 826–838. [Google Scholar]
- 17.Jänsch, H. J., Hof, T., Ruth, U., Schmidt, J., Stahl, D. & Fick, D. (1998) Chem. Phys. Lett. 296, 146–150. [Google Scholar]
- 18.Ruth, U., Hof, T., Schmidt, J., Fick, D. & Jänsch, H. J. (1999) Appl. Phys. B Photophys. Laser Chem. 68, 93–97. [Google Scholar]
- 19.Gerhard, P. (2003) Ph.D. thesis (Philipps-Universität, Marburg, Germany).
- 20.Braun, S., Kalinowski, H.-O. & Berger, S. (1996) 100 and More Basic NMR Experiments (VCH, Weinheim, Germany).
- 21.Gerhard, P., Koch, M. & Jänsch, H. J. (2004) C. R. Physique 5, 297–304. [Google Scholar]
- 22.Schlichting, H., Menzel, D., Brunner, T. & Brenig, W. (1992) J. Chem. Phys. 97, 4453–4467. [Google Scholar]
- 23.Abragam, A. (1961) The Principles of Nuclear Magnetism (Oxford Univ. Press, Oxford).
- 24.Meixner, D. L. & George, S. M. (1993) J. Chem. Phys. 98, 9115–9125. [Google Scholar]
- 25.deDios, A. C. & Jameson, C. J. (1997) J. Chem. Phys. 107, 4253–4269. [Google Scholar]
- 26.Tastevin, G., Nacher, P. J., Wiesenfeld, L., Leduc, M. & Laloë, F. (1988) J. Phys. [French] 49, 1–6. [Google Scholar]
- 27.Candela, D., Hayden, M. E. & Nacher, P. J. (1994) Phys. Rev. Lett. 73, 2587–2590. [DOI] [PubMed] [Google Scholar]
- 28.Raftery, D., Long, H., Reven, L., Tang, P. & Pines, A. (1992) Chem. Phys. Lett. 191, 385–390. [Google Scholar]
- 29.Labouriau, A., Panjabi, G., Enderle, B., Gates, T. Pietrass B. C., Earl, W. L. & Ott, K. C. (1999) J. Am. Chem. Soc. 121, 7674–7681. [Google Scholar]
- 30.Ryoo, R., Cho, S. J., Pak, C., Kim, J.-G., Ihm, S.-K. & Lee, J. Y. (1992) J. Am. Chem. Soc. 114, 76–82. [Google Scholar]
- 31.Freitag, A., van Wüllen, C. & Staemmler, V. (1995) Chem. Phys. 192, 267–280. [Google Scholar]
- 32.Bruch, L. W., Cole, M. W. & Zaremba, E. (1997) Physical Adsorption: Forces and Phenomena, The International Series of Monographs on Chemistry, eds. Rowlinson, J. S., Green, M. L. H., Halpern, J., Ley, S. V., Mukaiyama, T. & Simons, J. P. (Clarendon Press, Oxford),
- 33.Bagno, A. & Saielli, G. (2003) Chem. Eur. J. 9, 1486–1495. [DOI] [PubMed] [Google Scholar]
- 34.Bachelet, G. B., Bassani, F., Bourg, M. & Julg, A. (1983) J. Phys. C. Solid State Phys. 16, 4305–4320. [Google Scholar]
- 35.Jameson, C. J. (2002) J. Chem. Phys. 116, 8912–8929. [Google Scholar]
- 36.Ripmeester, J. A. (1982) J. Am. Chem. Soc. 104, 289. [Google Scholar]
- 37.Bloembergen, N. & Rowland, T. J. (1953) Acta Metallurgica 1, 731–746. [Google Scholar]
- 38.Bransden, B. H. & Joachain, C. J. (1983) Physics of Atoms and Molecules (Longman, London).
- 39.Arimondo, E., Inguscio, M. & Violino, P. (1977) Rev. Mod. Phys. 49, 31–75. [Google Scholar]
- 40.Carter, G. C., Bennett, L. H. & Kahan, D. J. (1977) Metallic Shifts in NMR, Progress in Materials Science, eds. Chalmers, B., Christian, J. W. & Massalski, T. B. (Pergamon Press, Oxford).
- 41.Korringa, J. (1950) Physica 16, 601. [Google Scholar]