

Abstract
N-substituted formamide was produced through the hydration of an isonitrile by isonitrile hydratase in the isonitrile metabolism. The former compound was further degraded by a microorganism, strain F164, which was isolated from soil through an acclimatization culture. The N-substituted formamide-degrading microorganism was identified as Arthrobacter pascens. The microbial degradation was found to proceed through an enzymatic reaction, the N-substituted formamide being hydrolyzed to yield the corresponding amine and formate. The enzyme, designated as N-substituted formamide deformylase (NfdA), was purified and characterized. The native enzyme had a molecular mass of ≈61 kDa and consisted of two identical subunits. It stoichiometrically catalyzed the hydrolysis of N-benzylformamide (an N-substituted formamide) to benzylamine and formate. Of all of the N-substituted formamides tested, N-benzylformamide was the most suitable substrate for the enzyme. However, no amides were accepted as substrates. The gene (nfdA) encoding this enzyme was also cloned. The deduced amino acid sequence of nfdA exhibited the highest overall sequence identity (28%) with those of regulatory proteins among known proteins. Only the N-terminal region (residues 58–72) of NfdA also showed significant sequence identity (27–73%) to that of each member of the amidohydrolase superfamily, although there was no similarity in the overall sequence except in the above limited region.
Nitriles are very toxic and generally unbiodegradable organic compounds containing a C N moiety. We have studied nitrile metabolism (1–3) and clarified the structures and functions of enzymes [i.e., nitrilase (4–7), nitrile hydratase (8–11), and amidase (12–14)] involved in metabolism and their genes and regulation mechanisms.
N moiety. We have studied nitrile metabolism (1–3) and clarified the structures and functions of enzymes [i.e., nitrilase (4–7), nitrile hydratase (8–11), and amidase (12–14)] involved in metabolism and their genes and regulation mechanisms.
On the other hand, information is quite limited on the metabolism of an isonitrile (more generally called an isocyanide) containing an isocyano group (–NC), which is an isomer of a nitrile. Isonitriles as well as nitriles are generally highly toxic and produced in nature by various organisms, including bacteria, fungi, marine sponges, etc. (15–17). An isocyanide metabolite, xanthocillin, was first isolated from Penicillium notatum (18). This isonitrile exhibits a wide antibiotic activity spectrum (19). Although parts of the metabolic intermediates of some isonitriles have been elucidated through incorporation experiments (20–24), their synthetic and degradative pathways remained entirely undetermined. Furthermore, to our knowledge none of the enzymes involved in isonitrile metabolism, except for the enzyme we describe below, has yet been identified.
Recently, we isolated a microorganism, Pseudomonas putida strain N19–2, that is able to degrade isonitriles from soil and discovered an isonitrile-metabolizing enzyme, designated as isonitrile hydratase (EC 4.2.1.103), in this strain (25, 26). This enzyme catalyzes the hydration of an isonitrile [R-NC] to the corresponding N-substituted formamide [R-NH-CH( O)], and confers the ability of isonitrile-degradation on the microorganism. However, there have been no reports on the metabolic pathway for N-substituted formamide produced from the isonitrile or the enzymes acting on the former in the metabolism.
O)], and confers the ability of isonitrile-degradation on the microorganism. However, there have been no reports on the metabolic pathway for N-substituted formamide produced from the isonitrile or the enzymes acting on the former in the metabolism.
We are interested in how C-N hydrolases evolved. Because the structure of N-substituted formamide contains a N—C bond, an N-substituted formamide-degrading enzyme would also belong to the category of C-N hydrolases. A search for such an enzyme and its functional analysis would contribute to the clarification of the metabolism of an isonitrile in nature and provide us with new knowledge about C-N hydrolases, which might facilitate elucidation of their functional and structural evolution. Here, we report the isolation of a microorganism, Arthrobacter pascens strain F164, that catabolizes each of N-substituted formamide and isonitrile from soil, and the purification and characterization of enzyme N-substituted formamide deformylase (NfdA) that catalyzes the hydrolysis of an N-substituted formamide to the corresponding amine and formate. Moreover, we describe the cloning of the gene of this enzyme (nfdA) and interesting evolutionary relationships between this enzyme, regulatory proteins, and amidohydrolase superfamily members.
Materials and Methods
Isolation of Bacteria Catabolizing N-Substituted Formamide and Isonitrile. N-Substituted formamide- and isonitrile-catabolizing microorganisms were isolated from soil by the enrichment culture technique described in ref. 25 with the following modifications. N-benzylformamide (NBFA) instead of benzyl isocyanide was used as the sole nitrogen source at a final concentration of 0.02% (wt/vol). After one month of cultivation, the microorganisms were spread on agar plates and isolated. Each of the isolated strains was inoculated into a test tube containing 10 ml of a synthetic medium (25) containing NBFA at a final concentration of 0.05% (wt/vol) as the sole nitrogen source and then incubated at 28°C for 72 h with reciprocal shaking. The cells were then harvested by centrifugation, washed twice with 10 mM potassium phosphate buffer (pH 7.5), and suspended in 0.1 M potassium phosphate buffer (pH 7.5).
The NBFA-catabolizing abilities of the isolated strains were assayed by means of the resting-cell reaction. The reaction mixture was composed of 0.1 M potassium phosphate buffer (pH 7.5), 10 mM NBFA, and an appropriate amount of cell suspension in a total volume of 1 ml. The reaction was carried out at 25°C for 2 h and stopped by placing the reaction mixture on ice water and then rapidly removing the cells by centrifugation at 0–4°C. The residual amount of NBFA in the reaction mixture was determined by HPLC with a Shimadzu LC-6A system (Kyoto) equipped with a Cosmosil 5C18-AR-II column (reversed-phase; 4.6 × 150 mm; Nacalai Tesque, Kyoto). The following solvent system was used at a flow rate of 1.0 ml/min and 40°C: 10 mM KH2PO4-H3PO4 buffer (pH 2.7)/acetonitrile, 1:1 (vol/vol). The absorbance was measured at 198 nm. The isonitrile-catabolizing abilities of the isolated strains were assayed by the method described in ref. 25.
Purification of NfdA. All purification procedures were performed at 0–4°C. Potassium phosphate buffer (pH 7.5) containing 10% (wt/vol) glycerol was used throughout the purification, unless noted otherwise. Centrifugation was carried out for 15 min at 13,000 × g.
Step 1: Preparation of a cell-free extract. Washed cells from 12 liters of culture broth were resuspended in 480 ml of 0.1 M buffer and then disrupted by sonication at 200 W for 60 min with an Insonator model 201M (Kubota, Tokyo). The cell debris was removed by centrifugation.
Step 2: Ammonium sulfate fractionation. The resulting supernatant solution was fractionated with ammonium sulfate (40–50% saturation), followed by dialysis against 10 mM buffer.
Step 3: DEAE-Sephacel column chromatography. The dialyzed solution was applied to a DEAE-Sephacel column (5 × 40 cm) equilibrated with 10 mM buffer. Protein was eluted from the column with 1 liter of the same buffer, the concentration of KCl being increased linearly from 0.2 to 0.5 M. The active fractions were collected and then ammonium sulfate was added to give 70% saturation. After centrifugation of the suspension, the precipitate was dissolved in 10 mM buffer, followed by dialysis against 10 mM buffer.
Step 4: Resource ISO column chromatography. The enzyme solution from step 3 was mixed with an equal amount of 10 mM buffer containing 50% saturated ammonium sulfate and then placed on a 1.6- × 3-cm Resource ISO column (Amersham Biosciences) equilibrated with 10 mM buffer containing 25% saturated ammonium sulfate. The enzyme was eluted by lowering the concentration of ammonium sulfate (25–15% saturation) in 180 ml of the same buffer. The active fractions were combined and precipitated with ammonium sulfate at 70% saturation. The precipitate was collected by centrifugation, dissolved in 10 mM buffer, and then dialyzed against 10 mM buffer.
Step 5: Resource Q column chromatography. The enzyme solution from step 4 was applied to a 0.64- × 3-cm Resource Q column (Amersham Biosciences) equilibrated with 10 mM buffer. Protein was eluted from the column with 60 ml of the same buffer, the concentration of KCl being increased linearly from 0.2 to 0.4 M. The active fractions were pooled and then precipitated with ammonium sulfate at 70% saturation. The precipitate was collected by centrifugation and then dissolved in 10 mM buffer. The resultant solution was dialyzed against 10 mM buffer containing 10% (vol/vol) glycerol and then centrifuged. The homogeneity of the purified protein was confirmed by SDS/PAGE.
Supporting Materials and Methods. The materials used in this study, the culture conditions for A. pascens strain F164, and the two enzyme assay systems used to measure NfdA activity are described in Supporting Materials and Methods, which is published as supporting information on the PNAS web site. Additionally, electrophoresis; molecular mass determination; metal analysis; amino acid sequencing of NfdA; tests of the substrate specificity of N-substituted formamides, amides, and other compounds; and cloning and nucleotide sequencing of the NfdA gene were performed as described in Supporting Materials and Methods.
Results
Isolation and Identification of A. pascens Strain F164. At ≈1 month from the start of the study, by using the acclimatization culture method described in Materials and Methods, we isolated 13 microorganisms that are able to use NBFA as the sole nitrogen source. We next examined whether these microorganisms can grow on a culture medium containing benzyl isocyanide (an isonitrile) as the sole nitrogen source or not and finally selected one positive strain, F164. As shown by the resting-cell reaction, strain F164 degraded NBFA and produced an unknown compound concomitantly with the N-substituted formamide-degradation; it also degraded benzyl isocyanide to the corresponding N-substituted formamide.
Morphologically, strain F164 is a Gram-positive rod that is non-endospore-forming and nonmotile. Strain F164 is negative for oxidase, catalase, β-glucuronidase, urease, oxidative/fermentative dissimilation of glucose, and reduction of nitrate and positive for pyrazinamidase, pyrrolidonyl arylamidase, alkaline phosphatase, β-galactosidase, α-glucosidase, and N-acetyl-β-glucosaminidase. Additionally, strain F164 is negative for growth on sole carbon sources with glucose, ribose, xylose, mannitol, maltose, milk sugar, white sugar, and glycogen, and it is negative for growth at 42°C. Strain F164 is positive for decomposition of aesculin and gelatin and rod-coccus cycle. The GC content of the genomic DNA of this strain was determined as 63.0% with a DNA-GC kit (Yamasa Shoyu, Chosin, Japan) according to the same method as described in ref. 25. Based on these characteristics, together with its 16S rRNA sequence, which was determined (data not shown) with a Microseq 500 16S rRNA-encoding DNA Bacterial Sequencing kit (Applied Biosystems) according to the manufacturer's specifications, strain F164 was identified as A. pascens.
Identification of the Reaction Products and Stoichiometry. The unknown compound produced from NBFA through the reaction of the enzyme purified from A. pascens strain F164 was examined by GC-MS, as described in Supporting Materials and Methods.As a result, it was found that both the retention time (1.8 min under the experimental conditions used) on the GC and the MS spectrum of the reaction product agreed with those of authentic benzylamine (data not shown). Furthermore, the reaction product and authentic benzylamine retention times on HPLC were the same as each other (data not shown). On the other hand, the other product that was not detected on GC-MS was found to be formate on HPLC analysis; the retention time of the reaction product agreed with that of authentic formate (6.1 min under the standard assay B conditions). No other compounds (that may be produced from NBFA), such as benzoic acid, benzylalcohol, formamide, and ammonia, exhibited any similarity to the reaction product on HPLC or GC-MS analysis. Thus, the reaction products were identified as benzylamine and formate.
The stoichiometry of N-substituted formamide consumption, and benzylamine and formate formation during the hydrolysis of N-substituted formamides was examined in a reaction mixture consisting of 100 mM potassium phosphate buffer (pH 7.5), 4 mM NBFA, and 1.67 μg/ml enzyme in a final volume of 800 μl. After a 30-min incubation, the amounts of NBFA, benzylamine, and formate were determined. The amounts of benzylamine and formate formed and the NBFA remaining were 1.12 mM, 1.18 mM, and 2.83 mM, respectively. The formation of other compounds was not noted. The results demonstrated that benzylamine and formate were formed stoichiometrically with the consumption of NBFA and that the enzyme catalyzes the hydrolysis of an N-substituted formamide to the corresponding amine and formate: the deformylation of an N-substituted formamide. Therefore, we named this enzyme N-substituted formamide deformylase:
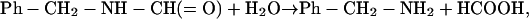 |
where Ph is the phenyl group (C6H5–).
Purification of NfdA. We found that the NfdA activity of A. pascens strain F164 was only induced when NBFA was added to the culture medium (unpublished results). Among the formamides tested, NBFA was the most effective inducer. Therefore, purification of the enzyme was carried out from an extract of NBFA-induced cells.
Through the purification steps described in Materials and Methods, the enzyme was purified 99.7-fold with a yield of 7.1% (Table 1). The purified enzyme gave only one band on SDS/PAGE (Fig. 1), corresponding to a molecular mass of 61 kDa. The molecular mass of the native enzyme was 121 kDa according to the results of gel filtration chromatography, indicating that the enzyme consists of two identical subunits. The purified enzyme showed specific activity of 28.9 units per mg (Table 1).
Table 1. Purification of NfdA.
| Step | Total protein, mg | Total activity, units | Specific activity, units per mg | Yield, % |
|---|---|---|---|---|
| Cell-free extract | 2,430 | 705 | 0.29 | 100 |
| (NH4)2SO4 | 752 | 564 | 0.75 | 80.0 |
| DEAE-Sephacel | 53.2 | 277 | 5.2 | 39.3 |
| Resource ISO | 3.81 | 76.2 | 20.0 | 10.8 |
| Resource Q | 1.72 | 49.7 | 28.9 | 7.1 |
Enzyme assay was performed by means of standard assay A.
Fig. 1.

SDS/PAGE of the purified NfdA. Row A, the purified enzyme (4 μg); row B, marker proteins.
Qualitative and quantitative analyses of the following metals in the purified enzyme solution were performed with an inductively coupled radiofrequency plasma spectrophotometer: Be, B, Mg, Al, Si, P, S, Ca, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Se, Sr, Zr, Mo, Pd, Ag, Cd, Sn, Sb, Ba, Ta, W, Pt, Au, Hg, Pb, La, and Ce. Because the purified enzyme could not be obtained in a large amount from Arthrobacter and metal analysis required a large amount of the enzyme, we established an enzyme overproduction system in a Streptomyces transformant containing the nfdA gene that succeeded in the preparation of a lot of the purified enzyme and used this enzyme [the properties of which are the same as those of the Arthrobacter enzyme (unpublished data)] only for the metal analyses. As a result, it was found that the enzyme contained 2.94 mol of zinc per mol of subunit. On the other hand, none of the other 34 metals was detected within the limits of the assay (10 ng/ml).
The absorption spectrum of the purified enzyme in 10 mM potassium phosphate buffer (pH 7.5) showed maximum absorbance at 282 nm. No other absorption peak or shoulder was observed, suggesting that no cofactor other than zinc would be bound to the enzyme.
Effects of Temperature and pH on the Activity and Stability of the Enzyme. The effects of pH and temperature on the enzyme activity were examined. The enzyme exhibited maximum activity at pH 7.0. The optimal temperature was 35°C (Fig. 4, which is published as supporting information on the PNAS web site). The stability of the enzyme was examined at various temperatures. After the enzyme had been preincubated for 30 min in 10 mM potassium phosphate buffer (pH 7.5) containing 10% (wt/vol) glycerol, an aliquot of the enzyme solution was taken, and then the enzyme activity was assayed under the standard assay A conditions. The enzyme solution exhibited the following activities: 60°C, 0%; 55°C, 0%; 50°C, 51%; 45°C, 75%; 40°C, 84%; 35°C, 94%; 30°C, 98%; 25°C, 100%; 20°C, 100%; 10°C, 100%. The stability of the enzyme was examined at various pH values. After the enzyme had been incubated at 25°C for 30 min in four buffers at a concentration of 0.1 M (citrate-Na2HPO4 buffer, pH 4.0–8.0; potassium phosphate buffer, pH 6.0–8.0; Tris·HCl buffer, pH 7.0–9.0; and NH4OH-NH4Cl buffer, pH 8.5–10.5), an aliquot of the enzyme solution was taken, and then the enzyme activity was assayed under the standard assay A conditions. NfdA was most stable in the pH range of 7.5–8.5.
Substrate Specificity. The ability of the enzyme to catalyze the hydrolysis of various N-substituted formamides, amides, and other compounds was examined. As described below, NfdA from A. pascens strain F164 has a narrow substrate spectrum. Among the tested N-substituted formamides, NBFA was the most suitable substrate for the enzyme. N-butylformamide (3.4%) was hydrolyzed at significantly lower rates compared with the activity toward NBFA (100%). The hydrolysis of NBFA followed Michaelis–Menten-type kinetics, the Km and Vmax values being 0.075 mM and 52.7 μmol·min–1·mg–1, respectively. On the other hand, The Km and Vmax values for N-butylformamide were 7.5 mM and 6.3 μmol·min–1·mg–1, respectively; N-butylformamide was turned over more slowly and bound with much lower affinity by the enzyme. Allylformamide, N-(2-cyclohex-1-enylethyl)formamide, and N-(α-methylbenzyl)formamide were rather poor substrates (Table 2, which is published as supporting information on the PNAS web site). Neither N-substituted formamides nor all of the tested compounds (except the mentioned ones above), which are listed in Supporting Materials and Methods, acted as substrates for our enzyme, despite the addition of a large amount of the enzyme and a long incubation period.
Inhibitors. Various compounds were investigated as to their inhibitory effect on the enzyme activity. Each compound was added to the standard reaction mixture without the substrate, and then assaying of the enzyme was performed by adding the substrate. The final concentration of each of the tested compounds was 1 mM, unless otherwise stated. The enzyme was very sensitive to HgCl2, CuCl (at 0.25 mM), CuCl2, and AgNO3, the inhibition being 100%. ZnCl2 and SnCl2 also showed inhibitory effects on the enzymatic activity (25% and 34%, respectively). The enzyme was completely inhibited by thiol-specific reagents, such as N-ethylmaleimide and p-chloromercuribenzoate, whereas iodoacetate and 5,5′-dithio-bis-2-nitrobenzoate did not inhibit the activity at all. Carbonyl-specific reagents, e.g., aminoguanidine and semicarbazide, hardly inhibited the enzyme, but phenylhydrazine caused partial inhibition (25%). Chelating agents, such as α,α′-dipyridyl, KCN, diethyldithiocarbamate, and EDTA, did not influence the activity at all, but o-phenanthroline and 8-hydroxyquinoline caused appreciable inhibition (38% and 54%, respectively). The enzyme was unaffected by oxidizing reagents and serine-modifying reagents, such as H2O2, ammonium persulfate phenylmethanesulfonyl fluoride, and diisopropyl fluorophosphates. However, reducing reagents such as DTT caused remarkable inhibition (90% inhibition).
Cloning and Nucleotide Sequencing of the nfdA Gene. NfdA was purified to homogeneity from A. pascens strain F164, and the amino acid sequences of peptides were determined by digesting the enzyme with lysyl endopeptidase or endoproteinase Glu-C. Two oligonucleotide primers were synthesized based on the NH2-terminal and internal sequences (corresponding to the amino acid residues 3–11 and 347–354 deduced from the nfdA gene, respectively) and used for PCR amplification with genomic DNA of A. pascens strain F164 as a template, resulting in the generation of a 1,057-bp fragment. The deduced amino acid sequence of the amplified fragment was consistent with the internal sequences of the enzyme determined by Edman degradation, indicating that the fragment was a portion of the enzyme gene.
To obtain the entire nfdA gene, after digestion of the genomic DNA with several restriction enzymes, Southern hybridization was performed by using the 1,057-bp fragment as a probe. A single 9-kb BamHI fragment was positively detected, and this fragment was recovered and ligated with BamHI-digested pUC19 to transform Escherichia coli DH10B. After screening the recombinant plasmids by colony hybridization, a positive clone, designated as pNFD10, was obtained.
The nucleotide sequencing of pNFD10 revealed a 1,629-bp ORF encoding 542 amino acids (DNA Data Bank of Japan accession no. AB164325), which precisely corresponded to those determined with the purified NfdA. Although the molecular mass of the protein encoded by this gene (nfdA) was calculated to be 58,694 Da, which was consistent with that of the enzyme subunit (Mr = 58,556) determined by matrix-assisted laser desorption–time-of-flight MS, it was slightly different from that of the enzyme subunit (Mr = 61 kDa) determined by SDS/PAGE. This discrepancy may be explained by the unusual mobility of the enzyme protein by SDS/PAGE, which was caused by its small SDS-binding capacity, because some proteins exhibit greater resistance to binding than others (27).
A search of protein sequence databases with the blast server revealed that NfdA exhibits <30% overall amino acid sequence identity with known proteins. Interestingly, the highest identities were observed with regulatory proteins, e.g., LAF3 isoform 1 (LAF3–1) from Arabidopsis thaliana (GenBank accession no. AAP55749, 26%) (28) and AepA from Brucella melitensis 16M (GenBank accession no. NP_541100.1, 28%) (29) (Fig. 5, which is published as supporting information on the PNAS web site). Only the N-terminal region (residues 58–72) of NfdA showed significant sequence identity (27–73%) to those of members of the amidohydrolase superfamily (30), including imidazolonepropionase (31), atrazine chlorohydrolase (32), cytosine deaminase (33), dihydroorotase (34), and urease (35) (Fig. 2), although there is no similarity in the overall sequence except in the above limited region.
Fig. 2.

Sequence alignment of NfdA with distantly related proteins. Each region of the amino acid sequences of cytosine deaminase (CodA), dihydroorotase (PyrC), and urease α-subunit (UreC) contains a β-strand secondary structure in which the functional residues (histidine and aspartic acid) map to the C terminus of strands 1, 5, 6, and 8 in each enzyme (30). For NfdA, LAF3–1, HutI, and AtzA, the regions containing the predicted secondary structures that correspond to β1, 5, 6, or 8 of CodA, PyrC, and UreC are shown. LAF3 isoform 1 (LAF3–1) is from A. thaliana (GenBank accession no. AAP55749), AepA precursor (AepA) is from B. melitensis 16M (GenBank accession no. NP_541100.1), imidazolonepropionase (HutI) is from Caulobacter crescentus (SwissProt accession no. P58079), atrazine chlorohydrolase (AtzA) is from Pseudomonas sp. ADP (SwissProt accession no. P72156), cytosine deaminase (CodA) is from E. coli (SwissProt accession no. P25524), dihydroorotase (PyrC) is from E. coli (SwissProt accession no. P05020), and urease α-subunit (UreC) is from Klebsiella aerogenes (SwissProt accession no. P18314). The residues with amino acid numbers and vertical arrows are metal ligands established by x-ray crystallography of CodA (PDB ID code 1K6W), PyrC (PDB ID code 1J79), UreC (PDB ID code 2KAU), and the corresponding residues in the other proteins. The residues highlighted in reverse type are conserved in NfdA and all of the other proteins. Except for the residues highlighted in reverse type, identical amino acid residues in either of the regulatory proteins and the members of the amidohydrolase superfamily are boxed.
Discussion
Isonitriles are elaborated by various organisms, including bacteria, fungi, and marine sponges. Most isonitriles show a wide antibiotic activity spectrum and have potential as possible agents of practical use, e.g., a series of isocyanoterpenes isolated from marine sponges exhibit antimalarial activity (36, 37) and an antifouling effect similar to those of copper sulfate (38). Interest in the biosynthesis of these metabolites, particularly in the origin of the isocyano group, has led to some experimental studies on them (20–24). However, the metabolism of isonitriles had been completely unknown at the protein and gene levels before we discovered an isonitrile-degrading enzyme, which was designated isonitrile hydratase (25). The enzyme catalyzes the hydration of an isonitrile to the corresponding N-substituted formamide, indicating that isonitrile hydratase is responsible for the initial committed step in isonitrile catabolism. However, no enzyme has been reported to be involved in the further metabolism of an N-substituted formamide produced from the corresponding isonitrile. In this work, we initially isolated an N-substituted formamide-degrading microorganism that was also able to degrade the corresponding isonitrile, and we characterized the NfdA in the cells at the protein and gene levels.
In living organisms, many enzymes [e.g., amine oxidase (39, 40)] are responsible for amine metabolism (41). Among them, amine-synthesizing enzymes have received increasing attention in various fields. There are several types of amine-forming deformylases involved in the metabolism of N-substituted formamides that are present in nature. (i) Kynurenine formamidase (EC 3.5.1.9) involved in the tryptophan degradation pathway (42) converts N-formyl kynurenine to kynurenine. This enzyme is known to be responsible for the synthesis of the ommochrome pigment in insects (43, 44); it also plays a role in maintaining an appropriate level of NAD in chicken embryos and is an essential enzyme in their normal development (45, 46). (ii) Formylmethionine deformylase (EC 3.5.1.31) deformylates N-formylmethionine (which is the free formyl amino acid produced on degradation of proinflammatory N-formylpeptide) but does not act on formyl dipeptides or tripeptides (47, 48). On the other hand, N-formylmethionine, which is generated through enzymatic transformylation of methionyl-tRNA and is an important amino acid for the initiation of all protein synthesis in prokaryotes, is deformylated by (iii) peptide deformylase (EC 3.5.1.88) (49, 50). (iv) 10-Formyltetrahydrofolate deformylase (EC 3.5.1.10) hydrolyzes 10-formyltetrahydrofolate to tetrahydrofolate and formate, resulting in regulation of the ratio of tetrahydrofolate and one-carbon tetrahydrofolate pools in prokaryotic cells (51). (v) Formamidase (EC 3.5.1.49) hydrolyzes formamide (which is the shortest N-substituted formamide and the shortest amide) to formate and ammonium (52). (vi) N,N-dimethylformamidase (EC 3.5.1.56) deformylates N,N-dimethylformamide, which is a useful chemical compound in the chemical industry, but there is concern about it being a possible environmental pollutant because of its toxicity to human beings and other organisms (53). (vii) Formylaspartate deformylase (EC 3.5.1.8) involved in the histidine degradation pathway (54) acts on N-formyl-l-aspartic acid. (viii) Formylglutamate deformylase (EC 3.5.1.68) involved in the terminal reaction of the five-step pathway for histidine utilization in P. putida (55) degrades N-formylglutamate to glutamate and formate. However, the substrate specificity of our NfdA enzyme is completely different from those of the above deformylases. The enzyme utilizes no amides as substrates. Moreover, the deduced amino acid sequences of NfdA showed no significant sequence similarity to ones of any other deformylases mentioned above. These findings demonstrate that our enzyme is a deformylase for N-substituted formamides.
We found that A. pascens strain F164 grew on a medium containing benzyl isocyanide, which was the isonitrile corresponding to NBFA, as the sole nitrogen source. In the cell-free extract reaction, the isonitrile was confirmed to be hydrated to NBFA, which was further converted into benzylamine and formate by NfdA (unpublished data). This observation suggests that isonitrile hydratase and NfdA are closely related in sequential isonitrile metabolism. Although the isonitrile hydratase gene has not been cloned from A. pascens strain F164, yet there could be a genetic relationship between NfdA and isonitrile hydratase. It would be interesting to elucidate the potential genetic link between them for a greater understanding of the biological metabolism of isonitriles in nature.
On the other hand, only the N-terminal region (residues 58–72) of NfdA exhibited significant conservation with those of some enzymes, including imidazolonepropionase (HutI) (31), atrazine chlorohydrolase (AtzA) (32), cytosine deaminase (CodA) (33), dihydroorotase (PyrC) (34), and urease α-subunit (UreC) (35) (Fig. 2), although the entire translated NfdA showed no sequence similarity with other enzymes, except in the above limited region. All these enzymes showing the weak similarity with NfdA belong to the amidohydrolase superfamily, which is characterized by members with a structural context of alternating α-helix and β-strand secondary structure elements, designated as (α/β)8 barrel structure, where the highly conserved residues (histidine and aspartic acid) locate in the C terminus of β-strands 1, 5, 6, and 8 of CodA, PyrC, and UreC (30) (Fig. 2). These signature residues consisted of four histidine and one aspartic acid (where two histidine in the N-terminal region of each enzyme are designated as the HXH motif) are involved in metal-binding, catalytic function, and stability of the enzyme structure (30). The blast search revealed that NfdA has the HXH motif and the signature aspartic acid, which correspond to the metal-binding residues present in β-strands 1 and 8 of each member of the amidohydrolase superfamily (e.g., urease α-subunit), respectively. We then attempted to compare the secondary structure of NfdA with those of CodA, PyrC, and UreC to identify possible residual signature residues of NfdA. The secondary structural prediction of NfdA with the psi-pred (56) and profsec (57, 58) programs revealed that the distribution pattern of the predicted secondary structure for NfdA was similar to those of CodA, PyrC, and UreC. The residual signature histidine residues were also found to exist in the predicted β-strands of NfdA, which correspond to β5 and β6 of the amidohydrolase superfamily members, respectively (Fig. 6, which is published as supporting information on the PNAS web site).
Moreover, our enzyme contains three zinc ions per subunit, although all other members of the amidohydrolase superfamily contain one or two metal ions per subunit; there have been no reports on another member containing three metal ions (30). From these findings, together with the presence of three zinc ions in the NfdA subunit, we propose that NfdA is a member of the amidohydrolase superfamily and that the zinc ions coordinated by the above five conserved residues would be concerned with the catalytic activity and stability of the enzyme structure. Mutant analyses will elucidate whether these amino acid residues actually serve as the zinc-binding ligands, and determination of the enzyme structure will provide the information to clarify the reaction mechanism.
Furthermore, to our surprise, NfdA also exhibits the highest overall sequence identity with known regulatory proteins, such as LAF3 isoform 1 (26%) and AepA (28%). LAF3 is one participant in the light-signaling pathway that is essential for photoperiod sensing that regulates plant growth and development. This protein is involved in activation of the transcription of XTR7, which encodes a xyloglucan endotransglycosylase-related protein that breaks down the cell wall to allow seedling stems to elongate, and is an important for termination of hypocotyl elongation (28). AepA is produced by a plant-pathogenic bacterium, Erwinia carotovora subspecies carotovora strain Ecc71 and is an activator protein that regulates the production of extracellular enzymes, which are required for maceration of plant tissues, and triggers the transcription of pel-1, which specifies a pectate lyase isozyme (59). As shown in Fig. 5, sequence alignment of NfdA and these proteins for the first time revealed that all of the possible five conserved metal-binding residues are present in these proteins. On the other hand, each distribution pattern of their predicted secondary structure was found to be an (α/β)8 barrel structure, which was consistent with those of the amidohydrolase superfamily members (Fig. 6). These findings, together with the sequence alignment, support the assignment of the β-secondary structure of NfdA as described above and suggest that these regulatory proteins belong to the amidohydrolase superfamily as well as NfdA. The precise roles of these regulatory proteins in the regulation of gene expression are not yet known. Based on the analogy to NfdA, these regulatory proteins may be involved in the binding and degradation of a molecule chemically related to NBFA.
Unexpectedly, we have found that the histidine-aspartic acid signature in the amidohydrolase superfamily are conserved in NfdA and the regulatory proteins (Fig. 3), suggesting that these enzymes and proteins might have evolved from a common ancestral gene. It would be of interest to determine how these proteins diverged and became differentiated as to biological function. Further studies on each enzyme and protein from the standpoints of its reaction mechanism and its three-dimensional structure could provide information on their evolutionary relationships.
Fig. 3.

Schematic representation of NfdA and distantly related proteins. The five highly conserved residues are indicated, and the number for each histidine and aspartic acid residue indicates the deduced amino acid number in NfdA. Numbers on the right indicate the total numbers of amino acid residues in each molecule.
Supplementary Material
Acknowledgments
We thank Drs. Y. Akutsu-Shigeno and T. Nakajima-Kambe (University of Tsukuba) for help with the GC-MS analysis. This work was supported in part by the 21st Century Center of Excellence Program of the Ministry of Education, Culture, Sports, Science, and Technology of Japan; by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, and Culture of Japan; by the Industrial Technology Research Grant Program in 2002 of the New Energy and Industrial Technology Development Organization of Japan; by the National Project on Protein Structural and Functional Analyses; and by a Research Grant (A) of the University Research Projects.
Abbreviations: NBFA, N-benzylformamide; NfdA, N-substituted formamide deformylase.
Data deposition: The sequence reported in this paper has been deposited in DNA Data Bank of Japan (accession no. AB164325).
References
- 1.Kobayashi, M. & Shimizu, S. (1998) Nat. Biotechnol. 16, 733–736. [DOI] [PubMed] [Google Scholar]
- 2.Yamada, H. & Kobayashi, M. (1996) Biosci. Biotech. Biochem. 60, 1391–1400. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi, M., Nagasawa, T. & Yamada, H. (1992) Trends Biotechnol. 10, 402–408. [DOI] [PubMed] [Google Scholar]
- 4.Komeda, H., Hori, Y., Kobayashi, M. & Shimizu, S. (1996) Proc. Natl. Acad. Sci. USA 93, 10572–10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kobayashi, M., Komeda, H., Yanaka, N., Nagasawa, T. & Yamada, H. (1992) J. Biol. Chem. 267, 20746–20751. [PubMed] [Google Scholar]
- 6.Kobayashi, M., Izui, H., Nagasawa, T. & Yamada, H. (1993) Proc. Natl. Acad. Sci. USA 90, 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi, M. & Shimizu, S. (1994) FEMS Microbiol. Lett. 120, 217–224. [Google Scholar]
- 8.Komeda, H., Kobayashi, M. & Shimizu, S. (1996) Proc. Natl. Acad. Sci. USA 93, 4267–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komeda, H., Kobayashi, M. & Shimizu, S. (1996) J. Biol. Chem. 271, 15796–15802. [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi, M., Suzuki, T., Fujita, T., Masuda, M. & Shimizu, S. (1995) Proc. Natl. Acad. Sci. USA 92, 714–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi, M. & Shimizu, S. (1999) Eur. J. Biochem. 261, 1–9. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi, M., Fujiwara, Y., Goda, M., Komeda, H. & Shimizu, S. (1997) Proc. Natl. Acad. Sci. USA 94, 11986–11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi, M., Goda, M. & Shimizu, S. (1998) FEBS Lett. 439, 325–328. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi, M., Komeda, H., Nagasawa, T., Nishiyama, M., Horinouchi, S., Beppu, T., Yamada, H. & Shimizu, S. (1993) Eur. J. Biochem. 217, 327–336. [DOI] [PubMed] [Google Scholar]
- 15.Edenborough, M. S. & Herbert, R. B. (1988) Nat. Prod. Rep. 5, 229–245. [DOI] [PubMed] [Google Scholar]
- 16.Scheuer, P. J. (1992) Acc. Chem. Res. 25, 433–439. [Google Scholar]
- 17.Garson, M. J. & Simpson, J. S. (2004) Nat. Prod. Rep. 21, 164–179. [DOI] [PubMed] [Google Scholar]
- 18.Rothe, W. (1950) Pharmazie 5, 190. [Google Scholar]
- 19.Hagedorn, I. & Tonjes, H. (1957) Pharmazie 12, 567–580. [PubMed] [Google Scholar]
- 20.Hagadone, M. R., Scheuer, P. J. & Holm, A. (1984) J. Am. Chem. Soc. 106, 2447–2448. [Google Scholar]
- 21.Achenbach, H. & Grisebach, H. (1965) Z. Naturforsch. 20B, 137–140. [PubMed] [Google Scholar]
- 22.Achenbach, H. & König, F. (1972) Chem. Ber. 105, 784–793. [Google Scholar]
- 23.Pfeifer, S., Bär, H. & Zarnack, J. (1972) Pharmazie 27, 536–542. [PubMed] [Google Scholar]
- 24.Simpson, J. S., Brust, A. & Garson, M. J. (2004) Org. Biomol. Chem. 2, 949–956. [DOI] [PubMed] [Google Scholar]
- 25.Goda, M., Hashimoto, Y., Shimizu, S. & Kobayashi, M. (2001) J. Biol. Chem. 276, 23480–23485. [DOI] [PubMed] [Google Scholar]
- 26.Goda, M., Hashimoto, Y., Takase, M., Herai, S., Iwahara, Y., Higashibata, H. & Kobayashi, M. (2002) J. Biol. Chem. 277, 45860–45865. [DOI] [PubMed] [Google Scholar]
- 27.Nelson, C.A. (1971) J. Biol. Chem. 246, 3895–3901. [PubMed] [Google Scholar]
- 28.Hare, P. D., Møller, S. G., Huang, L.-F. & Chua, N.-H. (2003) Plant Physiol. 133, 1592–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paulsen, I. T., Seshadri, S., Nelson, K. E., Eisen, J. A., Heidelberg, J. F., Read, T. D., Dodson, R. J., Umayam, L., Brinkac, L. M., Beanan, M. J., et al. (2002) Proc. Nat. Acad. Sci. USA 99, 13148–13153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holm, L. & Sander, C. (1997) Proteins 28, 72–82. [PubMed] [Google Scholar]
- 31.Nierman, W. C., Feldblyum, T. V., Laub, M. T., Paulsen, I. T., Nelson, K. E., Eisen, J., Heidelberg, J. F., Alley, M. R. K., Ohta, N., Maddock, J. R., et al. (2001) Pro. Natl. Acad. Sci. USA 98, 4136–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Souza, M. L., Sadowsky, M. J. & Wackett, L. P. (1996) J. Bacteriol. 178, 4894–4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ireton, G. C., McDermott, G., Black, M. E. & Stoddard, B. L. (2002) J. Mol. Biol. 315, 687–697. [DOI] [PubMed] [Google Scholar]
- 34.Thoden, J. B., Phillips, G. N., Neal, T. M., Raushel, F. M. & Holden, H. M. (2001) Biochemistry 40, 6989–6997. [DOI] [PubMed] [Google Scholar]
- 35.Jabri, E., Carr, M. B., Hausinger, R. P. & Karplus, P. A. (1995) Science 268, 998–1004. [PubMed] [Google Scholar]
- 36.Angerhofer, C. K., Pezzuto, J. M., König, G. M., Wright, A. D. & Sticher, O. (1992) J. Nat. Prod. 55, 1787–1789. [DOI] [PubMed] [Google Scholar]
- 37.Wright, A. D., Wang, H., Gurrath, M., König, G. M., Kocak, G., Neumann, G., Loria, P., Foley, M. & Tilley, L. (2001) J. Med. Chem. 44, 873–885. [DOI] [PubMed] [Google Scholar]
- 38.Fusetani, N. (1997) Curr. Org. Chem. 1, 127–152. [Google Scholar]
- 39.Yamashita, M., Azakami, H., Yokoro, N., Roh, J. H., Suzuki, H., Kumagai, H. & Murooka, Y. (1996) J. Bacteriol. 178, 2941–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frebort, I., Sebela, M., Hirota, S., Yamada, M., Tamaki, H., Kumagai, H., Adachi, O. & Pec, P. (2003) Biol. Chem. 384, 1451–1461. [DOI] [PubMed] [Google Scholar]
- 41.Chattopadhyay, M. K., Tabor, C. W. & Tabor, H. (2003) Proc. Natl. Acad. Sci. USA. (2003) 100, 13869–13874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bailey, C. B. & Wagner, C. (1974) J. Biol. Chem. 249, 4439–4444. [PubMed] [Google Scholar]
- 43.Cochran, D. G. (1976) Insect Biochem. 6, 267–272. [Google Scholar]
- 44.Glassman, E. (1956) Genetics 41, 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seifert, J. & Casida, J. E. (1978) Biochem. Pharmacol. 27, 2611–2615. [DOI] [PubMed] [Google Scholar]
- 46.Eto, M., Seifert, J., Engel, J. L. & Casida, J. E. (1980) Toxicol. Appl. Pharmacol. 54, 20–30. [DOI] [PubMed] [Google Scholar]
- 47.Woodhouse, A. F., Anderson, R. P., Myers, D. B., Broom, M. F., Hobson, C. H. & Chadwick, V. S. (1987) J. Gastroenterol. Hepatol. 2, 35–43. [Google Scholar]
- 48.Broom, M. F., Sherriff, R. M., Tate, W. P., Collings, J. & Chadwick, V. S. (1989) Biochem. J. 257, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ragusa, S., Blanquet, S. & Meinnel, T. (1998) J. Mol. Biol. 280, 515–523. [DOI] [PubMed] [Google Scholar]
- 50.Sereo, A., Giglione, C., Sardini, A., Maritinezsanz, J. & Meinnel, T. (2003) J. Biol. Chem. 278, 52953–52963. [DOI] [PubMed] [Google Scholar]
- 51.Nagy, P. L., Marolewski, A., Benkovic, S. J. & Zalkin, H. (1995) J. Bacteriol. 177, 1292–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fraser, J. A., Davis, M. A. & Hynes, M. J. (2001) Genetics, 157, 119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schär, H.-P., Holzmann, W., Rasmostombo, G. M. & Ghisalba, O. (1986) Eur. J. Biochem. 158, 469–475. [DOI] [PubMed] [Google Scholar]
- 54.Ohmura, E. & Hayaishi, O. (1957) J. Biol. Chem. 227, 181–190. [PubMed] [Google Scholar]
- 55.Hu, L., Mulfinger, L. M. & Phillips, A. T. (1987) J. Bacteriol. 69, 4696–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cuff, J. A., Clamp, M. E., Siddigni, A. S., Finlay, M. & Barton, G. J. (1998) Bioinformatics 14, 892–893. [DOI] [PubMed] [Google Scholar]
- 57.Rost, B. (1996) Methods Enzymol. 266, 525–539. [DOI] [PubMed] [Google Scholar]
- 58.Rost, B. & Liu, J. (2003) Nucleic Acids Res. 31, 3300–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu, Y., Murata, H., Chatterjee, A. & Chatterjee, A. K. (1993) Mol. Plant–Microb. Interact. 6, 299–308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}