

Abstract
The C terminus of transcription factor NusA from Escherichia coli comprises two repeat units, which bind during antitermination to protein N from phage λ. To delineate the structural basis of the NusA–λN interaction, we attempted to crystallize the NusA C-terminal repeats in complex with a λN peptide (residues 34–47). The two NusA domains became proteolytically separated during crystallization, and crystals contained two copies of the first repeat unit in contact with a single λN fragment. The NusA modules employ identical regions to contact the peptide but approach the ligand from opposite sides. In contrast to the α-helical conformation of the λN N terminus in complex with boxB RNA, residues 34–40 of λN remain extended upon interaction with NusA. Mutational analyses indicated that only one of the observed NusA–λN interaction modes is biologically significant, supporting an equimolar ratio of NusA and λN in antitermination complexes. Solution studies indicated that additional interactions are fostered by the second NusA repeat unit, consistent with known compensatory mutations in NusA and λN. Contrary to the RNA polymerase α subunit, λN binding does not stimulate RNA interaction of NusA. The results demonstrate that λN serves as a scaffold to closely oppose NusA and the mRNA in antitermination complexes.
Phage λ regulates transcriptional termination events to switch between its life cycles (1, 2). Early gene expression is regulated by a 107-residue unstructured phage-encoded polypeptide, λN, which recognizes a signal sequence, boxB, in the untranslated region of the mRNA and thus initiates the buildup of a multifactor ribonucleoprotein complex on the surface of the RNA polymerase (RNAP). This protein–RNA assembly bestows on RNAP the capability to read through ρ-dependent and intrinsic termination sites. Apart from λN and the consecutive boxA and boxB mRNA sequences (the nut site), the host-derived N-utilization substances (NusA, B, E, and G) are components of the antitermination complex.
The λN-dependent antitermination complex is characterized by a multitude of mutual interactions between its protein and RNA components (3, 4). Within this network, different regions of λN interact with boxB RNA (λN residues 1–22), NusA (λN residues 34–47), and RNAP (λN residues 73–107) (5, 6). The interaction of a λN fragment with boxB RNA is accompanied by the local folding of the peptide into a kinked α-helix (7–9). The atomic structures of other regions of λN in complex with their binding partners are presently unknown.
Apart from participating in the λN-dependent antitermination process, NusA is a general modifier of RNAP during the elongation phase of transcription. Bacterial NusA harbors an N-terminal domain (NTD) that constitutes a major interaction site with RNAP (10). Furthermore, it features in tandem one S1 and two K-homology (KH) RNA binding domains. NusA proteins from Escherichia coli and other γ-proteobacteria carry an additional C-terminal extension, which is made up of a dually repeated acidic domain of ≈70 residues (10, 11). These modules are referred to as acidic repeats (AR) 1 and 2. Although isolated E. coli NusA does not bind well to nut site RNA (3, 12), the factor is known to contact the nascent transcript in transcription complexes (13, 14). Recently, it was shown that the NusA C-terminal extension hinders RNA interaction in isolation but is sequestered in transcription complexes by the C-terminal domain (CTD) of RNAP subunit α (α-CTD) (10, 12, 15). λN binds to the same region of NusA as α-CTD, but it has not been ascertained whether this interaction enhances NusA–RNA binding as well. In any case, the C-terminal NusA repeats constitute a versatile protein–protein interaction region because they can form a complex with the α-CTD, λN, and, presumably, the remainder of NusA during inhibition of RNA binding. Whether the interaction sites for these ligands on AR1–AR2 are the same or different is not known.
We have attempted to crystallize the dual AR of E. coli NusA in complex with the cognate region of λN. The two NusA domains became proteolytically separated during crystallization, and the structure revealed two binding modes of AR1 to λN. Mutation analyses suggested that only one AR1–λN contact is physiologically relevant and that an additional λN interaction site exists on AR2. Our studies suggest that λN serves as a scaffold that brings NusA into proximity of the mRNA nut site.
Materials and Methods
Cloning, Overexpression, and Purification of NusA Constructs. Cloning of the E. coli nusa gene and gene fragments was performed by using standard molecular biological techniques. The NusA proteins were overexpressed in E. coli M15 and purified by various column chromatographic steps. Details of these and the following protocols are given in Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Crystallographic Procedures. A mixture of NusA AR1–AR2 and λN(34–47) crystallized under ammonium sulfate conditions in space group P43212. Crystals were derivatized with potassium bis(cyano)aurate, and anomalous diffraction data were collected at beamline BW6 of the Deutsches Elektronen Synchrotron (Table 2, which is published as supporting information on the PNAS web site). Indexing, integration, and reduction of the diffraction data were carried out with denzo/scalepack (16).
Heavy atom sites were determined with shelxd (17). Programs from the Collaborative Computational Project Number 4 (ccp4) suite (18) were used for phase calculations (mlphare) and density modification (dm). arp/warp (19) was used to generate a first atomic model, which was manually modified with main (www-bmb.ijs.si/doc/index.html) and refined by standard procedures. The structure coordinates have been deposited in the Protein Data Bank (PDB ID code 1U9L).
Mass Spectrometric Analyses. Mass spectra were recorded with a matrix-assisted laser desorption ionization/time-of-flight mass spectrometer (Bruker Daltronic, Bremen, Germany) with α-cyano-cinnamic acid as the matrix. The spectra were recorded in reflection mode (fingerprinting) or linear mode (full-length sample) with external spectral calibration (20).
Molecular Modeling Studies. Protein sequences were aligned by using blast (21), and homology modeling was performed by using the homology package of insight ii (Accelrys, San Diego) with AR1 residues 353–419 as the template. Relaxation of all protein atoms except atoms of the secondary structure elements was allowed during minimization. Energy minimization was performed by using the discover module of insight ii for 9,990 cycles, employing a steepest-descent gradient until the average derivative fell <0.01 kcal/mol per Å.
Isothermal Titration Calorimetry (ITC). ITC measurements were carried out at 20°C by using an MCS-ITC Instrument (Microcal Software, Northampton, MA) to obtain enthalpy and heat capacity changes (22). The data were analyzed with origin (Version 5.0, Microcal). For interpretation of the data, exact protein and peptide concentrations were determined by UV spectroscopy and quantitative amino acid analyses, respectively.
CD Measurements. CD spectra for NusA AR1–AR2 were recorded with a J-715 spectropolarimeter (Jasco, Tokyo) and the protein at a concentration of 0.2 mg/ml in 50 mM Tris·HCl (pH 7.0)/150 mM NaCl/2 mM DTT. The spectra were interpreted as a mixture of helical, sheet, and random-coil structure. For interpretation of the spectra, the exact protein concentration was determined by UV spectroscopy.
Results and Discussion
Crystal Contents and Quality of the Structure. For crystallization, we mixed a C-terminal NusA fragment, residues A350–A495, which covered the entire dual AR region (AR1–AR2), with a synthetic peptide spanning residues 34–47 of λN. Crystals were obtained only after prolonged incubation times. Their structure could be solved by a two-wavelength multiple anomalous diffraction strategy, using a gold derivative (Table 2). The experimental electron-density map could be interpreted in terms of two copies of AR1 (AR1A and AR1B) in complex with a single λN fragment (Fig. 1 A and B). The AR2 domain was not seen in the structure. Refinement converged at an R/Rfree factor of 21.5%/24.0% for all data up to a 1.9-Å resolution, maintaining the model in excellent stereochemistry (Table 2). The mean residual coordinate error estimated from Cruickshank's diffraction data precision indicator amounted to 0.17 Å.
Fig. 1.

Structure of the (NusA AR1)2–λN complex. (A) Ribbon plot of the (NusA AR1)2–λN complex (blue, AR1A; cyan, AR1B). The λN(34–40) peptide (red sticks) is sandwiched between the two AR1 α1–α2 loops. N and C termini are labeled. Structure figures were prepared with pymol (www.pymol.org). (B) Portion of the final 2Fo – Fc electron-density map, which covers residues 34–40 of the λN peptide, contoured at the 1σ level. (C Left) Electrostatic surface potentials calculated separately for the binding partners in the physiological complex. Red, negative potential; blue, positive potential. A charge complementarity at the peptide interface can be clearly discerned. (C Right) Surface representation of the λN(34–40) peptide with NusA AR1B displayed as a gray ribbon (prepared with swisspdbviewer, http://us.expasy.org/spdbv/mainpage.html). (D) Sequence alignment of AR1 and AR2. Numbering above and below the alignment corresponds to AR1 and AR2, respectively. The background of identical residues is red, and the background of conserved amino acids is yellow. Secondary structure elements as found for AR1 are indicated. Blue arrows indicate residues of AR1 (D364–D366), which align in a β-like arrangement with I37–S39 of the peptide. Prepared with alscript (36).
Extensively washed crystals and mother liquor were analyzed by MS (data not shown). Three major peaks were seen for the crystalline sample, which corresponded to the entire synthetic λN peptide (residues N34–R47) and to regions A350–Q421 and A350–S424 of NusA AR1. In drops that had not yet developed crystals or in mother liquor surrounding the crystals, a NusA fragment corresponding to residues A418–A495, matching the region of AR2, was seen in addition. All NusA-derived peaks were verified by peptide mass fingerprinting. Therefore, the λN peptide survived the crystallization period intact so that part of the peptide (residues N41–R47) must be disordered in the crystals. In contrast, the link between the two NusA domains was proteolytically severed, and only AR1 crystallized in complex with the λN fragment, whereas AR2 remained in the supernatant fraction. Because their connection can be cleaved, the two C-terminal NusA domains probably are linked by a flexible element.
Structure of the NusA Acidic C-Terminal Repeats. Although the crystal structure of the NTD and the S1/KH region of NusA has been determined (23), no experimental structure is available so far for the C-terminal repeats. The two crystallographically independent AR1 molecules of the current structure display an almost identical, all-helical fold (rms deviation of 0.64 Å for all common Cα atoms). Five α-helices (α1–α5) are arranged as two perpendicularly packed helix–hairpin–helix motifs (Fig. 1 A). The axes of the helices within the two pairs, α1–α2 and α4–α5, are almost parallel, different from the approximately perpendicular arrangement of helices in helix–turn–helix motifs (24). Helix α3 serves as a connector (Fig. 1 A). Duplication of a helix–hairpin–helix motif to a (helix–hairpin–helix)2 domain is also known from other proteins (24). The structure of AR1 is consistent with previous motif predictions (12) and recent NMR assignments for the NusA C-terminal extension (25).
Structural similarity searches (26, 27) assigned AR1 to the sterile α motif fold, an important protein–protein interaction module found in diverse protein families (28). In addition, various nucleic acid-binding proteins were identified that carry closely related motifs to NusA AR1. In these latter proteins, the helix–hairpin–helix portions are known or expected to interact with nucleic acids. The hairpin loops of these proteins, which are contacting the nucleic acids, are rich in positively charged residues and usually contain a consensus GhG (h, hydrophobic amino acid) fingerprint (24, 29). In contrast, the surface of AR1 is decisively negatively charged (Fig. 1C). Therefore, it is unlikely that AR1 constitutes a nucleic acid-binding module besides its known function as a protein-binding domain (see also below).
Based on the crystal structure of AR1, we have devised a 3D homology model of AR2. Consistent with the high sequence similarity (Fig. 1D), the AR2 model closely resembled the AR1 fold with an rms deviation of 1.33 Å between all common Cα atoms. It maintained a proper hydrophobic core during energy minimization and displayed conserved residues at similar positions to AR1. The surface electrostatic features of the AR2 model matched those of AR1. The secondary structure content determined from a CD spectrum of the AR1–AR2 repeat (47.1% helix) closely corresponded to the helical content predicted from the crystal structure of AR1 and the model of AR2 (51% helix; data not shown). Therefore, AR2 presumably harbors a very similar fold to AR1.
Structure of the (NusA AR1)2–λN Peptide Complex. In an asymmetric unit of the present crystal structure, the α1–α2 loops of the two AR1 domains run alongside each other in opposite directions. The λN peptide is sandwiched between these loops (Fig. 1 A). Peptide residues I37–S39 build up a short parallel β-like arrangement with residues D364–D366 of one AR1 domain and engage in a short antiparallel β-like association with the same residues of the other AR1 molecule (Fig. 1 A). Thus, the same regions of the two NusA fragments are engaged in the contacts to the peptide, but these interactions aim at opposite surfaces of λN. Upon complex formation, ≈700 Å2 of combined surface area are covered by the AR1A–λN contact, whereas ≈850 Å2 are buried in the AR1B–λN complex.
The peptide is seen in an extended conformation and thus exposes a large number of chemical functionalities to the environment, explaining how a rather short stretch of λN suffices to build up a specific complex with NusA AR1. This conformation is quite different from that of the first 22 residues of λN in complex with a boxB RNA hairpin. Although protein λN is unfolded in isolation, its N-terminal region assumes an α-helical structure upon interaction with boxB RNA (9). Van Gilst, von Hippel, and coworkers (8) have shown that NusA interacts with an unfolded region of λN and that the conformation of this region is independent of boxB RNA binding, consistent with the present findings.
The present structure indicates that AR1 comprises a major λN binding site of NusA, in agreement with other biochemical studies. Mah et al. (10) demonstrated that a NusA fragment containing AR1 is necessary for binding to λN, whereas the C-terminal 80 amino acids, encompassing AR2, are dispensable. However, it could not be excluded that AR2 does engage in additional contacts to λN that were not reflected in the assays.
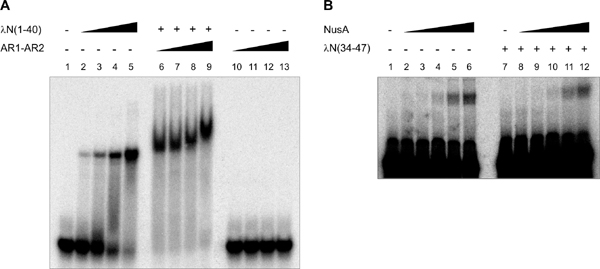
To test whether the observed AR1–λN(34–40) interaction is stable in solution, we conducted electrophoretic gel-mobility shift assays. A peptide corresponding to λN (residues 1–40) efficiently bound to synthetic boxB RNA (Fig. 4, which is published as supporting information on the PNAS web site). NusA AR1–AR2 alone did not interact with the RNA, even at very high concentrations, consistent with the acidic nature of the protein (see above and Fig. 4). However, NusA AR1–AR2 was capable of supershifting the λN(1–40)–boxB complex (Fig. 4). These results indicate that region 41–47 of λN is dispensable for rudimentary binding to NusA AR1–AR2.
λN Peptides Interact Exclusively and Stoichiometrically with AR1–AR2 of NusA. Quantitative Western blot analysis of antitermination complexes has suggested that two NusA molecules may be in contact with λN during antitermination (30). These results could explain the binding of two AR1 modules to a single λN peptide seen in the present crystal structure. Alternatively, only one of the observed AR1–λN interaction modes may be physiologically relevant. To distinguish between these possibilities, we studied the interactions between various NusA and λN constructs in solution by ITC. The amount of heat released after injections of peptides into solutions of NusA was used to derive the dissociation constants, enthalpies and entropies of the interactions, and stoichiometries between the binding partners (Table 1 and Fig. 2).
Table 1. Interactions of NusA AR1–AR2 with λN peptides.
| λN peptide | Sequence | Kd, μM | ΔHa, cal/mol | ΔSa, cal/mol per K | Stoichiometry |
|---|---|---|---|---|---|
| λN(34-47) | NRPILSLNRKPKSR | 3.55 ± 0.11 | -9,452 ± 24.01 | -27.77 | 0.965 ± 0.003 |
| λN(31-43) | KPVNRPILSLNRK | 9.65 ± 0.23 | -13,350 ± 50.92 | -22.58 | 1.263 ± 0.003 |
| N34A | KPVARPILSLNRK | 13.48 ± 0.23 | 12,700 ± 53.09 | -21.21 | 0.862 ± 0.002 |
| R35A | KPVNAPILSLNRK | Below DL | Below DL | Below DL | Below DL |
| P36A | KPVNRAILSLNRK | 12.86 ± 0.24 | 15,340 ± 56.77 | -29.96 | 1.087 ± 0.003 |
| 137A | KPVNRPALSLNRK | Below DL | Below DL | Below DL | Below DL |
| L38A | KPVNRPIASLNRK | Below DL | Below DL | Below DL | Below DL |
| S39A | KPVNRPILALNRK | 41.24 ± 0.46 | -10,650 ± 41.32 | -16.25 | 1.229 ± 0.003 |
| L40A | KPVNRPILSANRK | Below DL | Below DL | Below DL | Below DL |
Below DL, below detection limit; A, mutated residue.
Fig. 2.

Probing of the NusA–λN interactions by ITC. (A) Reaction of NusA AR1–AR2 with λN(31–43). (B) Reaction of NusA AR1–AR2 with λN(34–47). The negative peaks indicate an exothermic reaction. The area under each peak represents the heat released after an injection of the peptide into the solution of NusA. (Lower) Binding isotherms obtained by plotting peak areas against the molar ratio of λN peptide to NusA. The lines represent the best-fit curves obtained from least-squares regression analyses assuming a one-site binding model.
For these binding studies, we chose a fragment of λN spanning residues K31–K43. It expands the region that is in contact with AR1 in the crystal structure by three residues on either side to reduce end effects. First, we verified in solution that the λN peptides interacted within the C-terminal region of NusA. The binding affinity of λN(31–43) to the NusA AR1–AR2 region (Kd = 9.6 μM) was comparable to its affinity for full-length NusA (Kd = 7.9 μM). In contrast, a NusA fragment covering only the NTD and the S1/KH region, but lacking AR1–AR2, completely lost its ability to bind the λN peptide (data not shown). Therefore, the AR1–AR2 region is exclusively responsible for the binding of λN.
Because full-length NusA binds the peptide equally well or slightly better than AR1–AR2, the λN binding site on the C-terminal repeats must be directly accessible in the framework of the full-length NusA protein. ITC results reveal that the enthalpic contribution to peptide binding is less favorable for full-length NusA than for the AR1–AR2 fragment (–8,544 cal/mol vs. –13,350 cal/mol, respectively), but the difference is made up by a more favorable entropic term (–5.8 cal/mol per K vs. –22.58 cal/mol per K, respectively). Thus, in full-length NusA, the AR1–AR2 module may be fixed by intramolecular contacts so that a loss in entropy upon interaction with the peptide is reduced as compared with free AR1–AR2. These results support the notion that the AR1–AR2 region interacts with the remainder of NusA, as suggested for the autoinhibition of RNA binding (12).
Approximately equimolar interaction stoichiometries were observed for various λN peptides and NusA constructs (Table 1 and Fig. 2), indicating that in solution one NusA monomer binds one peptide substrate. This finding contradicts the idea that the present crystal structure reflects an in vivo situation, in which two NusA molecules bind by means of AR1 to the same region of λN. The NusA/λN binding stoichiometry in antitermination complexes is an unresolved issue. In agreement with our results, other studies have shown that NusA binds λN in a 1:1 complex in vitro (3, 5, 31). Furthermore, NusA binds stoichiometrically to RNAP in the absence of λN both in vitro (32, 33) and in vivo (30). Therefore, the 2:1:1 interaction of NusA/λN/RNAP may result from the purification of the complexes under low salt (30).
Identification of the Biologically Relevant Complex by Alanine Scanning. In the next series of experiments, mutated λN(31–43) peptides were tested for binding to NusA AR1–AR2 (Table 1 and Fig. 2). Significantly, λN residues, which upon mutation to alanine displayed reduced affinity, were found to preferentially or exclusively engage with domain AR1B of the crystal structure (cyan in Fig. 1 A).
In detail, N34 is not in direct contact with either of the AR1 modules (Fig. 3), and, consequently, binding of N34A is similar to that of the wild-type peptide (Table 1). The R35 side chain, alternatively, is poking deeply into a crevice of AR1B. It forms a salt bridge to Oδ2 of D364B (superscripts identify the AR1 molecule) and hydrogen bonds to the carbonyl oxygen of R409B (Fig. 3). In addition, a water molecule mediates an interaction to the carbonyl atom of Y362B. A second water molecule connects Nω2 and the carboxyl function of E408B. The intimate contacts to AR1B are responsible for the complete loss of binding upon deletion of the R35 side chain (Table 1). Mutation of the peptide residue P36 to alanine shows only a marginal reduction in binding affinity (Table 1). P36 stacks on the aromatic ring of F369 from AR1A (Fig. 3). If this interaction were important, a more severe loss of affinity would have been expected, discrediting the significance of the λN–AR1A interaction. A proline at position 36 may be preferred slightly over other residues because of a more favorable entropy of interaction, as supported by the data (Table 1). I37 engages in very similar hydrophobic contracts to both AR1 molecules, so the loss of affinity of the I37A mutant cannot discriminate the relevant interaction. Replacement of L38 with an alanine results in complete loss of binding to NusA AR1–AR2 (Table 1). One side of residue L38 favorably interacts with I365 and F369 of AR1B (Fig. 3). At the same time, L38 points to the same side of the peptide as its P36 and L40 side chains. It thus seems to stabilize the extended peptide conformation through hydrophobic interactions with the latter two residues. S39 displays only a single specific contact, a hydrogen bond to D366 Oδ1 of AR1B (Fig. 3). The dissociation constant of the S39A mutant is ≈4-fold higher than that of the wild-type peptide (Table 1), again identifying AR1B as the relevant interaction partner. The side chain of L40 exclusively interacts with AR1B. It is in hydrophobic contact with L398B and V372B and stacks on the aromatic ring of F369B (Fig. 3). A smaller residue would not be able to fill this hydrophobic pocket. In agreement, the corresponding L40A mutant has lost its affinity for NusA AR1–AR2 entirely (Table 1).
Fig. 3.

Stereoview detailing the AR1–λN(34–40) interaction. The backbone ribbons of the AR1A and AR1B molecules are in blue and cyan, respectively. Residues interacting specifically with the peptide are shown as sticks and are color-coded by atom type (gray, carbon; red, oxygen; blue, nitrogen). The λN peptide is drawn as a stick figure (pink, carbon). Three water molecules, which are mediating interactions, are shown as green spheres. Hydrogen bonds or salt bridges discussed in the text are indicated as dashed lines.
Taken together, the AR1B–λN complex of the current crystal structure constitutes a specific and biologically important interaction, because four of the seven peptide residues tested are absolutely required for NusA binding (Table 1). This finding is consistent with the larger surface area buried by the AR1B–λN complex (see above). Furthermore, the electrostatic surface potentials of AR1B and the peptide are compatible with an intimate interaction (Fig. 1C). In particular, the electropositive patch around R35 of the peptide fits nicely into an electronegative pocket of AR1B. Single-residue mutations in the peptides, which completely abolished binding, support the biological relevance of the observed interactions despite the rather high dissociation constants. The NusA–λN contacts occur within the framework of a large number of protein–protein and protein–RNA interactions (3, 4), and the antitermination complexes are assembled on the surface of RNAP. Therefore, an enhancement of the individual binary interactions can be expected because of networking, proximity, and local concentration effects in vivo. Furthermore, additional NusA–λN interactions may lead to an enhanced affinity in vivo (see below).
AR2 Contributes to λN Binding. Full-length λN binds to full-length NusA with a Kd of 70 nM (5), demonstrating contacts beyond those characterized herein. λN(34–47), which was used for crystallization, corresponds to a mapped NusA binding site on λN (6). The seven amino acids, 41–47, which are contained in the present crystals (see above) but do not engage in contacts to AR1, may harbor an additional binding site for AR2. We tested the affinity of λN(34–47) toward AR1–AR2 by ITC. Indeed, the dissociation constant of λN(34–47) was 3-fold lower than that of λN(31–43) (3.5 μM vs. 9.6 μM) (Table 1 and Fig. 2). A concomitant and independent binding of the two halves of λN(34–47) to AR1 and AR2, respectively, would explain that loss of one of the two ARs in NusA has only a minor effect on λN binding (10). In support of this notion, the homology model of AR2 maintains an electronegative surface similar to that of AR1 and can be seen easily to engage in a complex with the positively charged region 41–47 of λN. In addition, the sequence of λN(34–40) (NRPILSL) resembles that of λN(41–47) (NRK-PKSR), suggesting that residues 41–47 of λN may bind in a similarly extended conformation to AR2 as seen in the AR1–λN(34–40) complex.
Previous results have indicated that the entire NusA interaction region of λN is contained in residues 34–107 (5, 6) and that residues 241–495 of NusA hold its entire λN interaction region (10). Our results demonstrate that region 34–47 of λN and full-length NusA interact with a Kd of 3.5 μM. Therefore, additional interactions that confer the 70 nM dissociation constant must encompass NusA 241–495 and λN 47–107. The pun mutations of λN, which restore λN function in a nusA1 mutant background (34), map within and adjacent to the suspected AR2-interaction region of λN (35) (K45R, punA134; S50R, punA150; and I55M, punA165). NusA1 is a point mutation in the core of the NusA S1 domain and may lead to a reduction in RNA affinity (23). Assuming that tight NusA–λN binding is the result of an interaction between AR1 and λN(34–40) (this crystal structure) of a similar AR2–λN(41–47) interaction and of additional, uncharacterized interactions between λN(48–55) and AR2 yields a simple interpretation of the genetic data: The pun mutations could compensate for weakened S1 domain–RNA interactions in NusA1 with increased AR2–λN(pun) binding affinity.
λN Binding to NusA Does Not Enhance Its RNA Affinity. Full-length NusA is capable of efficient RNA binding only in the presence of α-CTD of RNAP, whereas NusA lacking the last 80 amino acids (i.e., AR2) can bind RNA without the intervention of the RNAP α subunit (12). Thus, AR2 in the full-length protein in some fashion occludes the RNA binding domains, e.g., by directly folding back onto the S1/KH region. The activation of RNA binding may occur by alternative interaction of AR2 with α-CTD. The present studies infer that AR2, in addition, interacts with λN. The dual role of AR2 as a main interaction site for the α-CTD and a binding site for λN could be the main switch through which NusA is either directed to stimulate pausing and termination (α-CTD) or antitermination (λN). We therefore tested whether λN binding also stimulates NusA–RNA interaction similarly to the α-CTD (12) through gel-mobility shift experiments, using radiolabeled nut site RNA, full-length E. coli NusA, and various λN peptides. We restricted the λN peptides to all or part of the NusA binding region because the strong boxB RNA affinity of the N-terminal part of λN (Fig. 4) would mask any RNA binding by NusA. Contrary to the findings with α-CTD, none of the λN peptides tested stimulated nut site RNA binding by NusA (Fig. 4). Similar results were obtained with boxA RNA or boxB RNA oligomers instead of the entire nut site (data not shown). Complementary evidence comes from the work of others, in which the interaction of NusA and λN was found to be independent of RNA binding (5, 31). These results demonstrate that the interactions of λN or α-CTD with AR2 have different physiological consequences, possibly because the two proteins interact with different surfaces of AR2. The aforementioned observation that full-length NusA binds equally well or better than the AR1–AR2 fragment to λN is consistent with this notion. λN can interact with the AR1–AR2 region without liberating AR2 from its interaction with the S1/KH portion; in contrast, α-CTD may target the surface of AR2, which is used to block RNA binding pockets on NusA.
The NusA binding site on λN (residues 34–47) directly neighbors the λN interaction site for boxB RNA (residues 1–22). Although residues 34–47 alone do not stimulate RNA binding by NusA, it is likely that the close apposition of the nut site and NusA through the λN scaffold will facilitate NusA–mRNA interactions.
Supplementary Material
Acknowledgments
We thank Peter H. von Hippel and Clarke R. Conant (University of Oregon, Eugene) for valuable comments on the manuscript; Michael Krause (Institut für Molekularbiologie und Tumorforschung, Marburg, Germany) for the synthesis of the λN-derived peptides; Henning Urlaub (Max-Planck-Institut für Biophysikalische Chemie) for mass spectrometric analyses; Karl-Heinz Mann (Max-Planck-Institut für Biochemie) for quantitative amino acid analyses; and R. Lührmann (Max-Planck-Insitut für Biophysikalische Chemie) for continued interest and support. This work was supported by the Max Planck Society, the Stiftung Volkswagenwerk, and the Deutsche Forschungsgemeinschaft.
Abbreviations: AR, acidic repeat; CTD, C-terminal domain; α-CTD, CTD of RNAP subunit α; ITC, isothermal titration calorimetry; KH, K-homology; NTD, N-terminal domain; RNAP, RNA polymerase.
Data deposition: The structure coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 1U9L).
References
- 1.Greenblatt, J., Mah, T. F., Legault, P., Mogridge, J., Li, J. & Kay, L. E. (1998) Cold Spring Harbor Symp. Quant. Biol. 63, 327–336. [DOI] [PubMed] [Google Scholar]
- 2.Nudler, E. & Gottesman, M. E. (2002) Genes Cells 7, 755–768. [DOI] [PubMed] [Google Scholar]
- 3.Mogridge, J., Mah, T. F. & Greenblatt, J. (1995) Genes Dev. 9, 2831–2845. [DOI] [PubMed] [Google Scholar]
- 4.Mogridge, J., Mah, T. F. & Greenblatt, J. (1998) J. Biol. Chem. 273, 4143–4148. [DOI] [PubMed] [Google Scholar]
- 5.Van Gilst, M. R. & von Hippel, P. H. (1997) J. Mol. Biol. 274, 160–173. [DOI] [PubMed] [Google Scholar]
- 6.Mogridge, J., Legault, P., Li, J., Van Oene, M. D., Kay, L. E. & Greenblatt, J. (1998) Mol. Cell 1, 265–275. [DOI] [PubMed] [Google Scholar]
- 7.Van Gilst, M., Rees, W. A. & von Hippel, P. H. (1995) Nucleic Acids Symp. Ser. 33, 145–147. [PubMed] [Google Scholar]
- 8.Van Gilst, M. R., Rees, W. A., Das, A. & von Hippel, P. H. (1997) Biochemistry 36, 1514–1524. [DOI] [PubMed] [Google Scholar]
- 9.Legault, P., Li, J., Mogridge, J., Kay, L. E. & Greenblatt, J. (1998) Cell 93, 289–299. [DOI] [PubMed] [Google Scholar]
- 10.Mah, T. F., Li, J., Davidson, A. R. & Greenblatt, J. (1999) Mol. Microbiol. 34, 523–537. [DOI] [PubMed] [Google Scholar]
- 11.Craven, M. G., Granston, A. E., Schauer, A. T., Zheng, C., Gray, T. A. & Friedman, D. I. (1994) J. Bacteriol. 176, 1394–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mah, T. F., Kuznedelov, K., Mushegian, A., Severinov, K. & Greenblatt, J. (2000) Genes Dev. 14, 2664–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Landick, R. & Yanofsky, C. (1987) J. Mol. Biol. 196, 363–377. [DOI] [PubMed] [Google Scholar]
- 14.Liu, K. & Hanna, M. M. (1995) J. Mol. Biol. 247, 547–558. [DOI] [PubMed] [Google Scholar]
- 15.Liu, K., Zhang, Y., Severinov, K., Das, A. & Hanna, M. M. (1996) EMBO J. 15, 150–161. [PMC free article] [PubMed] [Google Scholar]
- 16.Otwinowski, Z. & Minor, W. (1996) Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 17.Schneider, T. R. & Sheldrick, G. M. (2002) Acta Crystallogr. D 58, 1772–1779. [DOI] [PubMed] [Google Scholar]
- 18.Collaborative Computational Project Number 4 (1994) Acta Crystallogr. D 50, 760–763.15299374 [Google Scholar]
- 19.Morris, R. J., Perrakis, A. & Lamzin, V. S. (2003) Methods Enzymol. 374, 229–244. [DOI] [PubMed] [Google Scholar]
- 20.Hartmuth, K., Urlaub, H., Vornlocher, H. P., Will, C. L., Gentzel, M., Wilm, M. & Lührmann, R. (2002) Proc. Natl. Acad. Sci. USA 99, 16719–16724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997) Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wiseman, T., Williston, S., Brandts, J. F. & Lin, L. N. (1989) Anal. Biochem. 179, 131–137. [DOI] [PubMed] [Google Scholar]
- 23.Worbs, M., Bourenkov, G. P., Bartunik, H. D., Huber, R. & Wahl, M. C. (2001) Mol. Cell 7, 1177–1189. [DOI] [PubMed] [Google Scholar]
- 24.Shao, X. & Grishin, N. V. (2000) Nucleic Acids Res. 28, 2643–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eisenmann, A., Schwarz, S., Rösch, P. & Schweimer, K. (2004) J. Biomol. NMR 28, 193–194. [DOI] [PubMed] [Google Scholar]
- 26.Holm, L. & Sander, C. (1993) J. Mol. Biol. 233, 123–138. [DOI] [PubMed] [Google Scholar]
- 27.Krissinel, E. & Henrick, K. (2003) in Proceedings of the 5th International Conference on Molecular Structural Biology, eds. Kungl, A. J. & Kungl, P. J. (Austrian Chem. Soc., Vienna), p. 88.
- 28.Thanos, C. D., Goodwill, K. E. & Bowie, J. U. (1999) Science 283, 833–836. [DOI] [PubMed] [Google Scholar]
- 29.Drohat, A. C., Kwon, K., Krosky, D. J. & Stivers, J. T. (2002) Nat. Struct. Biol. 9, 659–664. [DOI] [PubMed] [Google Scholar]
- 30.Horwitz, R. J., Li, J. & Greenblatt, J. (1987) Cell 51, 631–641. [DOI] [PubMed] [Google Scholar]
- 31.Xia, T., Frankel, A., Takahashi, T. T., Ren, J. & Roberts, R. W. (2003) Nat. Struct. Biol. 10, 812–819. [DOI] [PubMed] [Google Scholar]
- 32.Greenblatt, J. & Li, J. (1981) Cell 24, 421–428. [DOI] [PubMed] [Google Scholar]
- 33.Gill, S. C., Weitzel, S. E. & von Hippel, P. H. (1991) J. Mol. Biol. 220, 307–324. [DOI] [PubMed] [Google Scholar]
- 34.Sparkowski, J. & Das, A. (1992) Genetics 130, 411–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Friedman, D. I., Schauer, A. T., Baumann, M. R., Baron, L. S. & Adhya, S. L. (1981) Proc. Natl. Acad. Sci. USA 78, 1115–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barton, G. J. (1993) Protein Eng. 6, 37–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}