

Abstract
Many methods have been developed to search for homologous members of a protein family in databases, and the reliability of results and conclusions may be compromised if only one method is used, neglecting the others. Here we introduce a general scheme for combining such methods. Based on this scheme, we implemented a tool called comparative homology agreement search (chase) that integrates different search strategies to obtain a combined “E value.” Our results show that a consensus method integrating distinct strategies easily outperforms any of its component algorithms. More specifically, an evaluation based on the Structural Classification of Proteins database reveals that, on average, a coverage of 47% can be obtained in searches for distantly related homologues (i.e., members of the same superfamily but not the same family, which is a very difficult task), accepting only 10 false positives, whereas the individual methods obtain a coverage of 28–38%.
Sequence-homology search algorithms are important computational tools in molecular biology. There exist at least three general classes of techniques used in searches for protein homologues, namely pairwise sequence comparisons such as basic local alignment search tool (blast), profile-based searches such as hmmsearch, and motifor pattern-based analyses such as pattern-hit-initiated blast (phi-blast) (1–5).
In a pairwise search, a query sequence is compared to any database sequence, yielding a confidence estimate that is supposed to indicate the probability of finding a comparably similar sequence (of the same size) in a database of random sequences. The comparison is done for every sequence in the database, and the sequences with highest confidence (“hits”) are reported. The most popular pairwise-search tool is blast (1).
Simple profile searches make use of position-specific scoring statistics and are usually more sensitive than pairwise comparisons. The introduction of hidden Markov models (HMMs) seems to provide a firmer statistical basis for profile search. The majority of currently available profile tools use HMMs (for example, the hmmer package) (6).
Kinship between protein sequences can also lead to (and thus be recognized by) the occurrence of particular amino acid motifs (also known as patterns, signatures, or fingerprints) that were conserved throughout the evolution of the protein family in question and are believed to correlate with specific structural features and function. Motif analysis, therefore, can also be used for identifying new members of a protein family (7–10). Motifs are the backbone of homology-search methods such as phi-blast (5). In contrast to profiles, motifs are usually short, they include a short stretch of very specific amino acids deemed relevant for function, and they are denoted by specific regular expressions.
In this study, we will show that the overall performance of homology searches can be improved if these methods are combined appropriately. The combination of methods is an advanced form of a metastudy. Important medical questions are typically studied more than once, and a metastudy compiles and analyzes the results of all relevant studies. interpro (11) and metafam (12) present such compilations in protein-family research. Combining methods directly to generate a consensus result is also common practice in some areas of bioinformatics. Two algorithms that combine different methods are pcons (13) (for fold recognition) and jpred (14) (for secondary structure prediction). They improve the accuracy of results considerably.
Here we restricted ourselves to combining the following five homology-search methods: hmmsearch (6), treesearch (15), position-specific iterated blast (psi-blast) (16), phi-blast (5), and motif alignment and search tool (mast) (17). All of them can use a collection of sequences as search input and report a confidence estimate for each hit. The first two methods perform profile-based searches by transforming the sequences into an HMM, and treesearch also uses a phylogenetic tree of the input sequences. Although psi-blast can be used iteratively, each new run being based on the output of the previous one, we use one step of this algorithm only, using a profile read off from a multiple clustalw alignment of the input sequences as input. For phi-blast, we use a motif in the form of a “regular expression” (18) designed to represent a family-specific pattern. Such an expression can be derived from the input sequences by using pratt (10). Then, following a suggestion from the blast software “read-me” file for improving the competitiveness of phi-blast, we apply psi-blast just once, using a profile derived from the phi-blast result. Finally, mast uses profiles derived from motif analysis.
We combine these five methods as follows: First, given a collection of query sequences, method-specific input queries structured according to the specific requirements of the individual search algorithms are automatically derived for each of the five component algorithms. Then, after these algorithms have been applied by using their respective input queries, we compute and report a “consensus hit list.”
In addition to detailing the resulting consensus tool dubbed comparative homology agreement search (chase), we present a comparative evaluation of its performance. Needless to say, the evaluation is performed by testing chase on a database that is disjoint from the database used to calibrate this tool. A standalone version of the chase software is available upon request.
Materials and Methods
Homology-Search Methods. All of the five homology-search methods that we use provide confidence estimates (E values, as described below) for their results. To perform their task, they require a query and a target database such as swissprot (19) or structural classification of proteins (scop) (20). The exact query format requirements, however, vary from method to method. We developed scripts called “input processors” (IPs) that take a collection of sequences and process them as follows to obtain the specific type of input for each of these homology-search methods.
hmmsearch IP. We use clustalw (21) to generate a multiple alignment that in turn is used by hmmbuild, available with the hmmer package, to build an HMM. We calibrate the required HMM by using hmmcalibrate, also available as part of hmmer.
treesearch IP. We use build_compound, available with treesearch, to generate, as required, a sequence alignment (using clustalw), a phylogenetic tree [using fitch (22)], and an HMM (using hmmbuild).
psi-blast IP. We use clustalw to align the input sequences and format the alignment such that it can be used to “jump-start” a “single-run” psi-blast search.
phi-blast IP. We use pratt to generate a prosite-like pattern and a clustalw alignment to generate a consensus sequence by relative majority rule for starting a phi-blast search, followed by a single run of psi-blast.
mast IP. We use multiple expectation maximization for motif elicitation (meme) (23) to generate motifs and convert them into the required profiles.
Automatic Evaluation of Database-Search Methods and Calculation of Performance Weights. phase4 (24) is a system for the automatic evaluation of database-search methods. In phase4, the performance of a method is evaluated by its ability to find a test set of sequences in a target database by using a training set of sequences for “learning” (e.g., for calculating an HMM). To construct test and training sets, phase4 relies on target databases such as scop 1.53 (20) that classify proteins [in a strictly Linnean, i.e., a binary or (according to proper Linnean terminology) “binomial” hierarchical, fashion] according to membership in families (of closely related sequences) and in superfamilies (of not-so-closely related sequences). An “evaluation scenario” is provided by specifying a training and a test set in the target database. For example, the scenario “distant family one model” is used to evaluate a homology search method for its ability to report distant relationships in protein families by splitting off one family from a given superfamily to provide the test sequences and keeping the rest of the superfamily as training sequences. Such a test is executed, for each family in turn, for every superfamily (see Table 1 for commonly used scenarios and ref. 24 for more details).
Table 1. Evaluation scenarios defined by phase4, given a protein sequence database that is organized into families and superfamilies.
| Scenario | Description |
|---|---|
| Distant relationship (distant family one model) | From a superfamily, each family in turn is chosen to provide the test sequences. |
| The remaining families within that superfamily provide the training sequences. | |
| Close relationship (family halves one model) | For each superfamily, half of the sequences of each of its families are chosen as training sequences and the remaining ones are chosen as test sequences. |
| Very close relationship (family half one model) | For each superfamily: For each family, half of its sequences are chosen as test sequences, and the remaining ones are chosen as training sequences. The sequences of the surrounding superfamily are ignored in the evaluation. |
Note that training sequences are always ignored in the evaluation and that the division into test and training sequences as described above is performed for each superfamily in turn. For the last model, average performance is calculated over an additional inner loop that considers each family in turn.
To evaluate the performance of any method numerically, phase4 offers “evaluators,” which make use of the list of sequences found that are ranked according to a confidence estimate, called an E value, or according to a score. In the simplest pairwise case of standard blast searches, given a normalized pairwise-comparison score S, the E value estimates the expected number of distinct local matches with normalized score at least S in an equally large database (16). This concept can be generalized to other search methods with different degrees of mathematical rigor. E values are reported by each of the search methods we want to combine, and our combination scheme will report a combined E value. For a given test, the “coverage vs. false-positive counts” evaluator compares the “good” and the “bad” guys as follows: It calculates the percentage P(k) of true positives (relative to the set of all true positives in the database) with an E value smaller than or equal to that threshold value for which exactly k false positives are found, thus rendering the percentage coverage P as a function P = P(k) of the absolute number k of misclassifications considered acceptable. Finally, results are averaged over all tests executed.
We use the phase4 system first for evaluating the individual homology-search methods to be combined in chase to derive a “weighting scheme” for the methods that is based on their performance. Among several available scenarios offered by phase4 that define training and test sequences using the scop database as described before, we use one for detecting distant relationships, one for detecting close relationships, and one for detecting very close relationships (see Table 1 for details). An E value EC = 1,000 was set as a cutoff for all individual homology-search methods; sequences with a larger E value are not listed. For each method i, consider the average percentage Pi(k) of coverage of true positives while considering k misclassifications (false positives) acceptable. In this case, the average is taken over the coverage for all three scenarios mentioned above, and the coverage for a scenario is in turn the average taken over all tests. Using some fixed number k (in our case, we used k = 50), this gives rise to the weighting scheme W = W1,..., Wn (as listed in Table 2), where n is the number of methods, and the weight Wi of method i is set to Pi/(P1 +... + Pn), with the average coverage Pi divided by the ntotal sum of the average coverages of all n methods so that 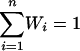 holds.
holds.
Table 2. Estimated weights for different homology-search methods based on the performance of the methods using the odd half of the scop database.
| Method | Weight |
|---|---|
| hmmsearch | 0.22048 |
| treesearch | 0.20404 |
| psi-blast | 0.20328 |
| mast | 0.17713 |
| phi-blast | 0.19507 |
A Scheme for Combining Homology-Search Methods. The results of each homology-search method are parsed to extract specific information such as the unique sequence identifiers of the hits and the corresponding E values. Tallying data for all methods, we obtain a preliminary list of hits, each row containing one sequence identifier and the corresponding E values reported by the different methods. This list is similar to the one presented in Table 3 except for the rescaling and reordering described below.
Table 3. Sample chase result.

A major problem in combining confidence estimates is the variability in the size of the E values estimated by different homology-search methods. We rescale E values to homogenize the confidence estimates to combine them. More precisely, to construct a consensus hit list from these data, we first rescale the E values Ei(s) obtained by the individual methods i = 1,..., n for each sequence s to produce E values of comparable size. We then use the weights as described in Automatic Evaluation of Database-Search Methods and Calculation of Performance Weights to obtain a weighted average E value. These two steps are now described in detail.
Placing methods on a common scale. For each method i and each sequence s in the database, we report the sequence, provided its E value Ei(s) is below or equal to a cutoff value EC of 1,000. Then, one method is chosen to be used as a reference method, on the basis of which the E values of the other methods are rescaled (25). In chase, we use hmmsearch as our reference method. Next, before doing any E value manipulation, we take the logarithm to base 10 to transform the E values for all methods. This transformation is necessary, because E values may be very close to zero for good database hits, and we must avoid rounding problems. This way, we obtain for each sequence s taken into consideration and each method i = 1,..., n a number ei(s):= log10Ei(s) that we call the “e value” of the sequence (with a small e) for conciseness. Next, we use a regression procedure yielding the slopes and the intercepts for hmmsearch versus treesearch, psi-blast, phi-blast, and mast to rescale the e values. For example, ordinary leastsquares regression (26) applied to hmmsearch e values eHMM(s) and corresponding psi-blast e values ePSI(s) provides the slope a and the intercept b for which the error term Σ[eHMM(s) – a·ePSI(s) – b]2 is minimized. Here, the sum is taken over all sequences s with both e values eHMM(s) and ePSI(s) below or equal to a certain threshold e0. This procedure is repeated each time chase is applied to search for a protein family. Slope and intercept depend on the specific data under consideration: there is no universal data-independent regression line for the various methods. For each sequence s, we then put
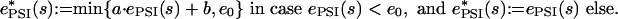 |
[1] |
For a small scaling threshold e0, the formula rescales small e values according to the regression line and keeps large e values as they are. Keeping large e values as they are may be useful, because they may be “downscaled” otherwise, suggesting a significance that is not there. In the rare case that rescaled e values exceed the threshold, they are set to precisely this threshold to keep the ranking as is. For larger e0, fewer e values are kept as they are. In Results and Discussion, we set e0 = log10(EC) = 3. Because no hits are considered for which the E value exceeds the E value cutoff EC = 1,000, all values are rescaled in this case. Nevertheless, results improve slightly for smaller e0, as discussed later.
The same scaling procedure is applied to the e values reported by the other three methods. For notational consistency, we set 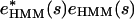 for our reference method hmmsearch. Calculating the C value. Once we have the rescaled e values
for our reference method hmmsearch. Calculating the C value. Once we have the rescaled e values 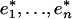 for all n methods, we calculate the c value for each sequence s as the W-weighted sum:
for all n methods, we calculate the c value for each sequence s as the W-weighted sum:
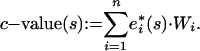 |
The final C value (on the original E value scale) is then obtained as C value(s):= 10c-value(s), which yields a consensus over individual homology-search methods. “Missing E values” arise if a homology search method finds a sequence not found by another, given the E value cutoff EC. In the C value formula, these missing E values are set to the cutoff E value EC.
Evaluation of chase. As noted above, our tool chase implements the above scheme by using five homology-search methods. Using the weights W1,..., Wn of the component search algorithms calculated once and for all, we compute the regression lines and the resulting C values of the sequences in each database search. Treating the C values as E values, we can use phase4 again to evaluate the performance of chase and compare its performance with that of the component algorithms. Clearly, the weights that are incorporated in (and thus the performance of) chase depend on the database that was used for determining these weights. In particular, if a component algorithm does very well on that database, it will get a high weight, implying that it will strongly influence the outcome of the consensus method, making it look good on that particular database, too.
To avoid this kind of circularity, we split the scop 1.53 database into two separate databases: the odd database, containing every second scop superfamily starting with the first one, and the even database, containing the rest. We used the odd database to compute the weights, W1,..., Wn, as listed in Table 2, and the even database to evaluate the performance of the resulting consensus method and compare this performance with that of its component algorithms, using again the three scenarios offered in phase4 as described in Table 1. As before, we used coverage vs. false-positive counts in phase4 as a performance evaluator, and sorting of chase hits was based on the C value. Sequences with a C value exceeding EC = 1,000 are not listed. By default, chase sets the E value cutoff EC to 1,000, and the e value threshold used for rescaling e0 to 3 (= log101,000) so that all values are rescaled. However, other cutoff values can be specified also.
Results and Discussion
We conducted a comparative evaluation of five homology-search methods and our consensus method, chase. We used three different scenarios offered by phase4, as listed in Table 1, to define distant, close, and very close relationship between scop database entries. If one considers the averaged coverage of true positives at the cost of zero false positives, as shown in Fig. 1, and ranks the methods according to their ability to find distant homologous proteins, chase obtains a coverage of 34%, and hmmsearch comes next with a coverage of 28%. Then comes Mast, psi-blast, treesearch, and phi-blast, with coverages between 27% and 21%. It is important to note that we do not claim to conduct a valid comparison of these individual methods. Such a comparison would need to do more justice to their different input requirements. The comparative analysis of the individual methods, starting with the same training data of sequences for each, suffers from the application of the IPs (described above), by which some of the input information may be lost. It is also worth noting that methods that do not perform well on average can still give excellent results in specific instances, a remarkable fact that clearly needs to be investigated further.
Fig. 1.

Average coverage of chase and its component algorithms. Shown is the averaged coverage of true positives permitting 0 and 50 false positives using scop (even half) as the target database and evaluation scenarios provided by phase4 (as described in Table 1).
If we plot coverages of true positives at the cost of 10 false positives, performance of chase goes up, covering 47% on average in case of distant relationship, compared to 38% coverage by hmmsearch (see Fig. 2). Permitting 50 false positives, as presented in Fig. 1, these numbers go up to 59% and 49%, respectively.
Fig. 2.

Coverage versus false-positive counts. Shown is the phase4 evaluation in the form of coverage vs. false-positive counts for chase as well as for the five component algorithms using three different scenarios (as described in Table 1) offered in phase4. Averaging is done over all scop families included in the even half of the database. (The odd half was used to determine the weights used by the chase combination scheme.)
The advantage of chase is smaller in the case of close and very close relationship, but it still outperforms all component methods by a good margin. The coverage vs. false-positive count plots in Fig. 2 for the various phase4 scenarios give a more detailed picture of the coverage of true positives for up to 200 false positives. If the e value threshold used for rescaling is set to –1 instead of 3, not all values are rescaled anymore in the C value formula (Eq. 1). Remarkably, chase seems to perform even slightly better in this case. For example, chase obtains 36% coverage of distant relatives at a cost of 0 false positives (+2%), 50% coverage permitting 10 false positives (+3%), and 60% coverage permitting 50 false positives (+1%).
The results of running chase for the scop superfamily featuring the FAD/NAD(P)-binding domain are shown in Table 3. C values along with rescaled E values from different methods are listed. The names of the sequences from the given family are shown in black (in the “description” column), and the others (the names of the false positives) are shown in red. We consider a family member to be classified correctly by method i if its rescaled E value is smaller than the rescaled E value of the first false positive. For the false positives listed by method i, the minimum rescaled E value is shown in red. Rescaled E values of family members f that would not be classified correctly using method i alone are marked in orange. They are larger than the smallest rescaled E value of the false positives for method i (printed in red) so that the false positive with the smallest rescaled E value would precede the family members f in the ranking based on method i. In the twilight zone of rows 15–24, chase performs well, triggered by the rescaled E values marked in green, which indicate success for at least one method. Inspecting the consensus hit lists for all protein families under consideration in the “distant relationship” scenario, we noted that each method detects specific true positives that would not be detected if we had restricted ourselves to combining the other four.
The evaluation that we report is one scenario for chase. In another scenario, chase can be used to search for a maximum number of members of a protein family by providing expert rather then automatic input information to the component methods. Such information could, for example, be patterns described in the prosite database (18). These patterns may be found by ps_scan (18). We provide an advanced user interface by which one can submit protein sequence(s), a sequence alignment, a profile, or a pattern as input for the underlying search methods.
In the future, we would like to include more search methods. In some evaluation scenarios and for some data, methods based on support vector machines seem to be superior to some of the methods we combine (e.g., ref. 27), but they lack E values.
Conclusion
Our results show that combining homology-search methods provides improved performance over an entire set of scenarios ranging from the detection of distant to very close relationships between protein sequences. This observation corroborates, in the context of protein family research, the frequent claim that appropriately designed consensus methods can be more reliable than any of their component algorithms.
Acknowledgments
We are grateful to Mohammed Shahid for developing the interface for chase and Inge Jonassen for valuable discussions and advice on pratt. The ordinary least-squares formula presented in ref. 26 was implemented by S. Morton (Globewide Network Academy, Austin, TX) in the statistics::ols perl module available from www.cpan.org. The perl script “consensus.pl” used as part of the phi-blast input processor was written by N. Brown (EMBL, Heidelberg, Germany) and obtained from www.bork.embl-heidelberg.de/Alignment/consensus.html. This work was supported by the International NRW Graduate School in Bioinformatics and Genome Research and the Deutsche Forschungsgemeinschaft.
Abbreviations: HMM, hidden Markov model; chase, comparative homology agreement search; IP, input processor.
References
- 1.Altschul, S., Gish, W., Miller, W., Myers, E. W. & Lipman, D. (1990) J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 2.Bork, P. & Gibson, T. J. (1996) Methods Enzymol. 266, 162–183. [DOI] [PubMed] [Google Scholar]
- 3.Eddy, S. R. (1998) Bioinformatics 14, 755–763. [DOI] [PubMed] [Google Scholar]
- 4.Grundy, W. N. (1998) J. Comput. Biol. 5, 479–492. [DOI] [PubMed] [Google Scholar]
- 5.Zhang, Z., Schaffer, A. A., Miller, W., Madden, T. L., Lipman, D. J., Koonin, E. V. & Altschul, S. F. (1998) Nucleic Acids Res. 26, 3986–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eddy, S. R. (1996) Curr. Opin. Struct. Biol. 6, 361–365. [DOI] [PubMed] [Google Scholar]
- 7.Bairoch, A., Bucher, P. & Hofmann, K. (1997) Nucleic Acids Res. 25, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hudak, J. & McClure, M. A. (1999) Pac. Symp. Biocomput. 4, 138–149. [DOI] [PubMed] [Google Scholar]
- 9.Jonassen, I., Collins, J. F. & Higgins, D. G. (1995) Protein Sci. 4, 1587–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jonassen, I. (1997) Comput. Appl. Biosci. 13, 509–522. [DOI] [PubMed] [Google Scholar]
- 11.Apweiler, R., Attwood, T. K., Bairoch, A., Bateman, A., Birney, E., Biswas, M., Bucher, P., Cerutti, L., Corpet, F., Croning, M. D. R., et al. (2001) Nucleic Acids Res. 29, 37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silverstein, K. A., Shoop, E., Johnson, J. E., Kilian, A., Freeman, J. L., Kunau, T. M., Awad, I. A., Mayer, M. & Retzel, E. F. (2001) Nucleic Acids Res. 29, 49–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lundström, J., Rychlewski, L., Bujnicki, J. & Elofsson, A. (2001) Protein Sci. 10, 2354–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cuff, J. A., Clamp, M. E. & Barton, G. J. (1998) Bioinformatics 14, 892–893. [DOI] [PubMed] [Google Scholar]
- 15.Rehmsmeier, M. & Vingron, M. (2001) Proteins Struct. Funct. Genet. 45, 360–371. [DOI] [PubMed] [Google Scholar]
- 16.Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997) Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailey, T. L. & Gribskov, M. (1998) Bioinformatics 14, 48–54. [DOI] [PubMed] [Google Scholar]
- 18.Falquet, L., Pagni, M., Bucher, P., Hulo, N., Sigrist, C. J., Hofmann, K. & Bairoch, A. (2002) Nucleic Acids Res. 30, 235–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bairoch, A. & Apweiler, R. (2000) Nucleic Acids Res. 28, 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murzin, A., Brenner, S. E., Hubbard, T. & Chothia, C. (1995) J. Mol. Biol. 247, 536–540. [DOI] [PubMed] [Google Scholar]
- 21.Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994) Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Felsenstein, J. (1989) Cladistics 5, 164–166. [Google Scholar]
- 23.Bailey, T. L. & Elkan, C. (1994) Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology (Am. Assoc. Artificial Intelligence Press, Menlo Park, CA), pp. 28–36.
- 24.Rehmsmeier, M. (2002) Brief. Bioinform. 3, 342–352. [DOI] [PubMed] [Google Scholar]
- 25.Yona, G., Linial, N. & Linial, M. (2000) Nucleic Acids Res. 28, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gujarati, D. (1988) Basic Econometrics (McGraw–Hill, New York).
- 27.Liao, L. & Noble, W. S. (2002) Proceedings of the Sixth International Conference on Computational Molecular Biology (ACM Press, New York), pp. 225–232.