

Abstract
The Wilms' tumor gene WT1 is overexpressed in leukemias and various types of solid tumors, and the WT1 protein was demonstrated to be an attractive target antigen for immunotherapy against these malignancies. Here, we report the outcome of a phase I clinical study of WT1 peptide-based immunotherapy for patients with breast or lung cancer, myelodysplastic syndrome, or acute myeloid leukemia. Patients were intradermally injected with an HLA-A*2402-restricted, natural, or modified 9-mer WT1 peptide emulsified with Montanide ISA51 adjuvant at 0.3, 1.0, or 3.0 mg per body at 2-week intervals, with toxicity and clinical and immunological responses as the principal endpoints. Twenty-six patients received one or more WT1 vaccinations, and 18 of the 26 patients completed WT1 vaccination protocol with three or more injections of WT1 peptides. Toxicity consisted only of local erythema at the WT1 vaccine injection sites in patients with breast or lung cancer or acute myeloid leukemia with adequate normal hematopoiesis, whereas severe leukocytopenia occurred in patients with myelodysplastic syndrome with abnormal hematopoiesis derived from WT1-expressing, transformed hematopoietic stem cells. Twelve of the 20 patients for whom the efficacy of WT1 vaccination could be assessed showed clinical responses such as reduction in leukemic blast cells or tumor sizes and/or tumor markers. A clear correlation was observed between an increase in the frequencies of WT1-specific cytotoxic T lymphocytes after WT1 vaccination and clinical responses. It was therefore demonstrated that WT1 vaccination could induce WT1-specific cytotoxic T lymphocytes and result in cancer regression without damage to normal tissues.
Recent advances in the field of molecular biology and tumor immunology have resulted in the identification of a large number of tumor-associated antigens (TAAs) and their epitopes recognized by HLA class I-restricted cytotoxic T lymphocytes (CTLs) from various kinds of malignant neoplasms. One of the TAAs thus identified is Wilms' tumor gene WT1 product (1, 2). These advances have led to the possibility of the development of a new peptide-based cancer immunotherapy.
The WT1 gene was isolated as a gene responsible for Wilms' tumor, a pediatric renal cancer, and encodes a zinc finger transcription factor, which is involved in cell proliferation and differentiation, apoptosis, and organ development (3–6). Although the WT1 gene was first categorized as a tumor suppressor gene, we have proposed that the wild-type WT1 gene functions as an oncogene rather than a tumor-suppressor gene on the basis of the following findings. The first is high expression of the wild-type WT1 gene in both leukemias and solid tumors (7–18), the second is growth inhibition of leukemic and solid tumor cells by treatment with WT1 antisense oligomers (14, 19), and the third is block of differentiation, but induction of proliferation, of wild-type WT1 gene-transfected myeloid progenitor cells in response to granulocyte colony-stimulating factor (20, 21). The last two are block of thymocyte differentiation but induction of thymocyte proliferation in the transgenic mice with the lck promoter-driven WT1 gene (22), and WT1 gene expression in the majority of dimethylbenzanthracene-induced erythroblastic leukemia and a stronger tendency of the cells with high levels of WT1 to develop into leukemias (23).
Expression of the wild-type WT1 gene has been found in most cases of acute myelocytic leukemia (AML), acute lymphocytic leukemia, chronic myelocytic leukemia, and myelodysplastic syndrome (MDS) at higher levels than those in normal bone marrow (BM) or peripheral blood (7–13). Furthermore, various types of solid tumors, including lung, breast, thyroid, and colorectal cancers, expressed the wild-type WT1 gene at higher levels compared to those in corresponding normal tissues (15–18). These results indicated that the wild-type WT1 gene product may be a promising target for cancer immunotherapy (24, 25).
We tested the potential of the WT1 gene product to serve as a target antigen for tumor-specific immunotherapy. Human WT1-specific CTLs have been found to induce lysis of endogenously WT1-expressing tumor cells in vitro, but not to cause damage to physiologically WT1-expressing normal cells (24, 26–28). We used a mouse in vivo system to demonstrate that immunization of mice with either MHC class I-restricted WT1 peptide or WT1 cDNA induced WT1-specific CTLs. We also showed that the immunized mice rejected challenges of WT1-expressing tumor cells, whereas the induced CTLs did not affect normal healthy tissues that physiologically expressed WT1 nor damaged the normal tissues (25, 29). These results indicated that the WT1 protein could be a novel tumor rejection antigen for cancer immunotherapy (24–32).
In view of these various findings, we performed a phase I clinical study of cancer immunotherapy targeting the WT1 protein in patients with leukemia, MDS, lung cancer, or breast cancer (33, 34). The study presented here demonstrates that WT1 vaccination can induce WT1-specific CTLs and result in cancer regression without damage to normal tissues in the clinical setting.
Methods
Patients. The WT1 peptide-based phase I clinical study was approved by the Ethical Review Boards of the Faculty of Medicine, Osaka University and Hiroshima Red Cross and Atomic Bomb Survivor Hospital. Patients aged 20–80 with leukemia, MDS, and lung or breast cancer were eligible if their diseases proved to be resistant to conventional chemotherapy, radiotherapy, or hormonal therapy (breast cancer), including cases of relapse after hematopoietic stem cell transplantation or operation. Patients who had refused such treatments but wanted to receive WT1 vaccine therapy under the auspices of this clinical study were also eligible. Other inclusion criteria were: (i) overexpression of the WT1 gene in leukemic cells or cancer tissues determined by RT-PCR and/or immunohistochemistry, (ii) HLA-A*2402-positivity, (iii) estimated survival of >2 months, (iv) performance status from 0 to 2 (Eastern Cooperative Oncology Group), (v) no severe impairment of organ function, and (vi) no administration of chemotherapy, immunotherapy, immunosuppressive therapy, or radiotherapy within 4 weeks before WT1 vaccination.
WT1 Peptides. A natural 9-mer WT1 peptide (amino acids 235–243 CMTWNQMNL) and the modified 9-mer WT1 peptide (amino acids 235–243 CYTWNQMNL), in which Y was substituted for M at amino acid position 2 (anchor position) of the natural WT1 peptide were used for immunization (26, 28, 33, 34). The modified 9-mer WT1 peptide was shown to induce much stronger CTL activity than the natural peptide against WT1-expressing tumor cells (28, 33). The WT1 peptides (GMP grade) were purchased from Multiple Peptide Systems (San Diego) as lyophilized peptides.
Vaccine Preparation and Vaccination. After written informed consent had been obtained, skin test-negative patients were intradermally injected with increasing doses of 0.3, 1.0, or 3.0 mg of HLA-A*2402-restricted, natural, or modified 9-mer WT1 peptide emulsified with Montanide ISA51 adjuvant (33–35). WT1 vaccination was scheduled to be performed three times at 2-week intervals (33, 34). Four weeks after the third injection, the toxicity was evaluated without the administration of any other treatments. If some effect was observed after fewer than three injections, further WT1 vaccinations at 2-week intervals were administered only with patients' informed consent.
RT-PCR for Quantitation of WT1 Expression Levels. RNA from cancer tissues, BM cells, and peripheral blood mononuclear cells (PBMCs) was isolated and converted into cDNA. PCR with optimized cycles was performed with a DNA thermal cycler as described (13). WT1 expression levels in the samples were shown relative to those in K562 leukemia cells, which was defined as 1.0, as described (7, 12, 13).
Immunohistochemistry. The procedure was performed as described (15–17, 34). Formalin-fixed tissue sections were cut from a paraffin block and stained with anti-WT1 rabbit polyclonal antibody C-19 (Santa Cruz Biotechnology). Immunoreactive WT1 protein was visualized with the Vectastain ABC kit (Vector Laboratories). The sections were then counterstained with hematoxylin.
WT1 Peptide/HLA-A*2402 Tetramer Assay of WT1-Specific CTLs. The WT1 (a natural, HLA-A*2402-restricted, 9-mer WT1 peptide)/HLA-A*2402 tetramer was kindly provided by M. Gotoh (Sumitomo Pharmaceuticals, Osaka) (33, 34). This tetramer stained >90% of TAK-1 cells, which were WT1-specific CTLs that could recognize the complex of the natural 9-mer WT1 peptide and HLA-A*2402 molecules (26, 33, 34). PBMCs from HLA-A*2402-positive patients were double-stained with PerCP-CD8 antibody (BD Pharmingen) and phycoerythrin-tetramer and analyzed by fluorescence-activated cell sorting. A double-positive fraction was considered to represent WT1-specific CD8+ CTLs (33, 34, 36).
Cytoplasmic IFN-γ Staining Assay of HLA-A*2402-Restricted, WT1-Specific CTLs. Cytoplasmic IFN-γ staining assay was performed as described (37). PBMCs, stocked in aliquots before WT1 vaccination, were thawed, pulsed with natural 9-mer WT1 peptide, irradiated, and used as stimulator cells. Responder cells were PBMCs obtained before and after WT1 vaccination.
Statistical Analysis. The statistical significance of the increases in the amount of WT1-specific tetramer-positive or IFN-γ-producing CTLs after the WT1 vaccination was determined with the t test. Correlations between immunological and clinical responses were examined with the χ2 test.
Results
Patient Accrual. Twenty-six patients (2 breast and 10 lung cancers in advanced stages, 1 MDS with myelofibrosis, 1 AML from MDS, and 12 de novo AML in hematological complete remission) were accrued for this study (Table 1). Severe leukocytopenia occurred in two patients, one with MDS with myelofibrosis (patient 13) and the other with MDS-derived AML (patient 14), as a result of the injection of a single dose of 0.3 mg of the modified WT1 peptide (33). Thereafter, MDS patients, whose hematopoiesis was largely sustained by blood cells differentiated from WT1-expressing, transformed hematopoietic stem cells (33, 38, 39), were excluded from this study and only patients with de novo AML in hematological CR (blast cells in BM ≤ 5%) and normal hematopoiesis were accrued for this study for hematopoietic malignancies.
Table 1. Summary of clinical responses of WT1 peptide-based immunotherapy.
| Patient no. | WT1 peptide (mg per body) | Clinical responses |
|---|---|---|
| Breast cancer | ||
| 1 | M(0.3) | + (Tumor size ↓) |
| 2 | N(1.0) | + (Tumor size ↓) |
| Lung cancer | ||
| 3 | N(0.3) | + (CEA ↓) |
| 4 | N(0.3) | SD |
| 5 | N(0.3) | Unevaluable† |
| 6 | N(0.3) | + (SLX ↓) |
| 7 | M(0.3) | Unevaluable† |
| 8 | M(0.3) | PD |
| 9 | M(0.3) | PD |
| 10 | M(0.3) | PD |
| 11 | M(0.3) | PD |
| 12 | M(1.0) | + (SCC ↓) |
| Leukemia* | ||
| 13 | M(0.3) | + (WT1 ↓) |
| 14 | M(0.3) | + (Leukemic blast cells ↓) |
| 15 | N(1.0) | Unevaluable‡ |
| 16 | N(1.0) | PD |
| 17 | N(1.0) | Unevaluable‡ |
| 18 | M(1.0) | Unevaluable‡ |
| 19 | M(1.0) | Unevaluable‡ |
| 20 | N(3.0) | + (Leukemic blast cells ↓) |
| 21 | N(3.0) | + (WT1 ↓) |
| 22 | N(3.0) | + (WT1 ↓) |
| 23 | M(3.0) | + (WT1 ↓) |
| 24 | M(3.0) | + (WT1 ↓) |
| 25 | M(3.0) | PD |
| 26 | M(3.0) | SD |
M, modified WT1 peptide; N, natural WT1 peptide; SD, stable disease; PD, progressive disease; CEA, chorio-embryonic antigen.
Patients 13 and 14 are MDSs, and the others are AMLs.
Clinical responses could not be evaluated because WT1 vaccination was simultaneously stopped when severe leukocytopenia occurred in patients 13 and 14.
Clinical responses could not be evaluated because of the absence of evaluable markers for efficacy.
When severe leukocytopenia occurred in the two MDS and MDS-derived AML patients, WT1 vaccination was terminated not only for the two MDS patients but also for two of the patients with lung cancer (patients 5 and 7), although they were in this study. Two patients with lung cancer (patients 8 and 9) and two patients with AML (patients 16 and 25) were dropped from this study because of their progressive disease. A final total of 18 patients (nos. 1–4, 6, 10–12, 15, 17–24, and 26) completed this study with three or more injections of WT1 peptides (Table 1).
Toxicity and Adverse Effect. All 26 enrolled patients showed local inflammatory response with erythema at the WT1 vaccine injection sites (33, 34). Leukocytopenia occurred in the two patients of MDS with myelofibrosis and MDS-derived AML (patients 13 and 14) as described (33). Such patients have very few normal hematopoietic stem cells, and most of the peripheral blood cells with a normal appearance are derived from leukemic, transformed stem cells with a high level of WT1 expression (33, 38, 39). Therefore, the killing of the WT1-expressing leukemic, transformed stem cells by the WT1-specific CTLs elicited by WT1 vaccination was expected to result in the eradication of the leukemic, transformed stem cells, followed by a resultant reduction in peripheral blood cells, including leukocytes. From this point of view, the leukocytopenia occurring in the MDS and MDS-derived AML patients can be considered evidence of efficacy rather than an adverse effect (33). No leukocytopenia occurred in the remaining 24 patients with adequate and normal hematopoiesis. One patient with AML (patient 17) experienced subfever (37.6°C) that persisted only for several hours after the first WT1 vaccination, but that did not occur after the second and the third ones, indicating that WT1 vaccination was not necessarily the cause of subfever. Routine examinations such as renal and liver functional tests, BM aspiration, chest x-ray, and ECG revealed that WT1 vaccination did not damage normal organs, including kidney, pleura, and BM, that physiologically expressed WT1 (data not shown). Thus, the remaining 24 patients with normal hematopoiesis did not suffer any severe toxicity or other adverse effects.
Clinical Responses. Both patients with breast cancer (patients 1 and 2) showed regression of metastatic tumors (Fig. 1). Because patient 1, who had undergone radical mastectomy for right breast cancer, followed by hormonal therapy, chemotherapy, and radiotherapy, had metastatic tumors in both lungs, the left supraclavicular lymph nodes, and the cerebellum, WT1 vaccination was performed. After the second WT1 vaccination, the size of lung and supraclavicular lymph node tumors began to decrease, accompanied by a decrease in serum chorio-embryonic antigen (CEA) levels (data not shown). After the fifth WT1 vaccination, computed tomography (CT) scan showed a striking regression of the metastatic tumors in S2 and S3 of the right lung and in S6 of the left lung (Fig. 1a). Despite the reduction in the size of the metastatic tumors, serum CEA levels began to increase again after the third WT1 vaccination due to the progression of the metastatic tumor in the cerebellum. This case was considered to be “mixed response” with a marked clinical response to the metastatic tumors in lung but weak or no responses to that in the cerebellum. Patient 2 with breast cancer had undergone a partial bilateral mastectomy, followed by hormonal and chemotherapy. However, her disease was progressive, and ileus occurred due to the metastatic tumors in the colon, so that WT1 vaccination was initiated. The increase in NCC-ST-439, a tumor marker, was suppressed, after which the levels started to decease with the repeated WT1 vaccinations (data not shown). After the second WT1 vaccination, CT scan showed that thickening of walls of the colon tract was significantly improved (Fig. 1b), accompanied by improvement of appetite and relief from meteorism. She was not able to eat any foods because of ileus caused by the metastatic tumors in the colon before WT1 vaccination. However, she now spends her daily life without limitation of daily activities, including oral ingestion, and without regrowth of breast cancer at the present time, 16 months after the start of WT1 vaccination.
Fig. 1.

Tumor regression in patients with breast cancer. (a) Computed tomography of chest before (Left) and after (Right) WT1 vaccination in patient 1. Arrows indicate metastastic tumor masses. (b) Computed tomography of abdomen before (Left) and after (Right) WT1 vaccination in patient 2. Arrows indicate walls of bowel tract. Before WT1 vaccination, the walls were thickened by metastatic tumor cells.
In three patients with lung cancer (patients 3, 6, and 12), a decrease in tumor markers caused by WT1 vaccination was observed. In patient 3, whose lung cancer was completely resistant to chemotherapy and radiotherapy, chorio-embryonic antigen levels began to decrease from 2,048 ng/ml immediately after the WT1 vaccination and reached 805 ng/ml after the seventh WT1 vaccination. The tumor masses in the lung fields regressed after the second WT1 vaccination (34). In patient 6, SLX, a tumor marker that had returned to normal range after the operation, began to rise again, indicating the regrowth of residual lung cancer. For this reason, WT1 vaccination was performed. SLX began to decrease after the first WT1 vaccination and returned to and stayed at normal levels after the second WT1 vaccination (34). WT1 vaccination has been repeated for ≈2 years without significant adverse effect. In patient 12, the tumor markers, SCC and CYFRA, gradually decreased after the WT1 vaccination (Fig. 2). These three patients (patients 3, 6, and 12) received repeated WT1 injections after the completion of the protocol's three initial ones because WT1 vaccination was considered to be effective.
Fig. 2.
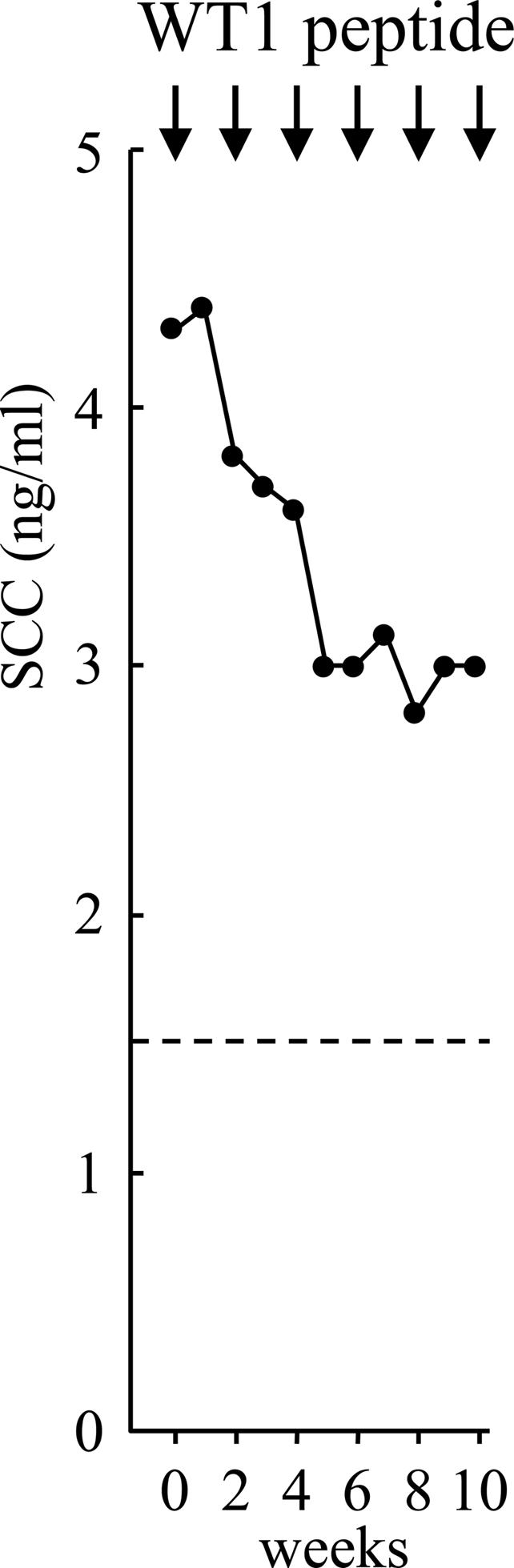
Reduction in a tumor marker (SCC) in patient 12 with lung cancer. The dotted line indicates an upper limit of normal range.
In patient 13, who had MDS with myelofibrosis, WT1 expression levels reflecting the amount of leukemic cells (7, 12, 13) started to decrease in peripheral blood and leukocytopenia occurred after the first WT1 vaccination (33). In patient 14, who had MDS-derived AML, leukocytes began to decrease from the day after the first WT1 vaccination (day 0) and reached minimal levels of 700 per μl on day 3. Leukemic blast cells accounted for 50% in BM before WT1 vaccination, but they were reduced to 11% along with a decrease in WT1 expression levels after only one WT1 vaccination (33).
In the five de novo AML patients with hematological complete remission (patients 20–24), a decrease in residual leukemic cells and/or WT1 expression levels was observed. In patient 20, residual leukemic cells in BM were gradually reduced from 2.7 to 1.6 and further to 0% in association with a decrease in WT1 expression levels during the repeated WT1 vaccination (Fig. 3a). The remaining four AML patients (patients 21–24) did not have microscopically detectable residual leukemic cells, but did have abnormal levels of WT1 expression that indicated persistent residual leukemic cells in BM (7, 12, 13). Their WT1 expression levels decreased in response to the repeated WT1 vaccinations, indicating a reduction in residual leukemic cells (Fig. 3 b and c). In patient 23, WT1 expression levels decreased to normal levels (<1 × 10–3) after the three WT1 vaccinations (Fig. 3b). In patient 21, WT1 expression levels returned to normal after three injections of WT1 vaccine but increased again to abnormal levels that were higher than those at prevaccination, because of delay in the fourth WT1 vaccination (Fig. 3c). After the fifth WT1 vaccination, however, WT1 expression levels returned to normal again. These findings strongly indicated that WT1 vaccination resulted in a reduction in WT1 expression levels, i.e., eradication of residual leukemic cells. In contrast to the MDS patients, none of the five de novo AML patients showed evidence of leukocytopenia, and normal hematopoiesis was maintained regardless of the repeated WT1 vaccinations.
Fig. 3.

Clinical course of patients with de novo AML. Clinical course of patients 20 (a), 23 (b), and 21(c) are shown. WT1 expression levels (solid lines) and percentages of leukemic blast cells in BM (filled columns) are shown. The dotted line indicates the upper limit of normal range in WT1 expression levels. U.D., Undetectable.
Altogether, the efficiency of WT1 vaccination could be assessed for 20 of the 26 patients who received one or more WT1 vaccinations. Of those 20 patients, 12 (60%) showed clinical responses. The metastastic tumor masses of both patients with breast cancer (patients 1 and 2) were reduced, tumor markers decreased in three (patients 3, 6, and 12) of the eight patients with lung cancer, and leukemic blast cells and/or WT1 expression levels decreased in seven (patients 13, 14, 20, 21, 22, 23, and 24) of the 10 patients with MDS, MDS-derived AML, or de novo AML. Of the remaining eight patients, two (patients 4 and 26) had stable disease and six (patients 8, 9, 10, 11, 16, and 25) showed progressive disease.
Immunological Responses. Immunological responses to WT1 vaccine were evaluated with HLA tetramer and cytoplasmic IFN-γ assays with the PBMCs. The assays were performed at prevaccination and at one or several postvaccination time points (33, 34). Immunological responses were defined to be positive when the frequencies of WT1-specific CTLs determined by tetramer assay increased by >1.5 times at least at one time point after the WT1 vaccination compared to those of WT1-specific CTLs before the WT1 vaccination. The immunological responses were positive in 1 of the 2 patients with breast cancer, 3 of the 8 patients with lung cancer, and 9 of the 13 patients with leukemia (Table 2). Possible correlations between clinical and immunological responses were examined for 19 patients with both clinical and immunological responses available for assessment (among the 20 patients whose clinical responses could be assessed, the immunological responses of patient 13 could not be assessed because not enough PBMC samples could be obtained for immunological analysis because of the patient's pancytopenia). A clear correlation (P = 0.0397) between clinical and immunological responses was observed in the evaluable 19 patients (10 with solid tumors and 9 with leukemia) (Table 3).
Table 2. Immunological responses in patients who received WT1 vaccine.
| WT1 tetramer, %
|
|||
|---|---|---|---|
| Patient no. | Before | After* | Immunological responses |
| Breast cancer | |||
| 1 | 0.78 | 0.34 | - |
| 2 | 0.12 | 0.25 | + |
| Lung cancer | |||
| 3 | 0.25 | 0.76 | + |
| 4 | 0.28 | 0.52 | + |
| 5 | 0.86 | ND | UE |
| 6 | 0.17 | 0.33 | + |
| 7 | 1.31 | ND | UE |
| 8 | 0.13 | 0.17 | - |
| 9 | 0.19 | 0.25 | - |
| 10 | 0.47 | 0.65 | - |
| 11 | 0.20 | 0.29 | - |
| 12 | 0.08 | 0.11 | - |
| Leukemia | |||
| 13 | 0.98 | ND | UE |
| 14 | 0.62 | 6.61 | + |
| 15 | 0.98 | 2.32 | + |
| 16 | 0.36 | 0.72 | + |
| 17 | 0.41 | 0.39 | - |
| 18 | 0.24 | 0.50 | + |
| 19 | 0.09 | 0.62 | + |
| 20 | 0.16 | 0.26 | + |
| 21 | 0.19 | 1.85 | + |
| 22 | 0.30 | 0.31 | - |
| 23 | 0.19 | 0.29 | + |
| 24 | 0.22 | 0.38 | + |
| 25 | 0.37 | 0.29 | - |
| 26 | 0.11 | 0.14 | - |
Bold numbers indicate the WT1-specific CTL frequencies that significantly (≥ 1.5-fold) increased after WT1 vaccination. ND, not determined. UE, unevaluable due to lack of the measurement of WT1-specific CTL frequencies after WT1 vaccination.
The highest ones among WT1-specific CTL frequencies determined after WT1 vaccination are shown.
Table 3. A correlation between clinical and immunological responses (P = 0.0397).
| Immunological responses
|
||
|---|---|---|
| Clinical responses | + | - |
| + (n = 11) | 8/11 | 3/11 |
| - (n = 8) | 2/8 | 6/8 |
An increase in the frequencies of WT1 peptide-specific cytoplasmic IFN-γ-positive cells after WT1 vaccination was observed in neither of the patients with breast cancer, 2 of the 8 patients with lung cancer, and 6 of the 11 patients with AML (data not shown). No significant correlation was observed between increases in the frequencies of WT1 peptide-specific cytoplasmic IFN-γ-positive cells and either clinical responses or the increase in the frequencies of tetramer-positive CD8+ T cells.
Discussion
The main purpose of this phase I clinical study was to evaluate the toxicity of the WT1 vaccination. No hematopoietic damage such as leukocytopenia occurred in patients with de novo AML or patients with solid tumor with intact hematopoiesis. In the former, malignant cells were derived from leukemic transformation of myeloid progenitor cells but sufficient normal hematopoietic stem cells had been retained. In contrast, severe leukocytopenia occurred in the two patients with MDS after a single dose of 0.3 mg of the modified WT1 peptide (33). It is well known that MDS is a stem cell disease and results from leukemic transformation of hematopoietic stem cells, which means that the majority of peripheral blood cells with a normal appearance are derived from the leukemic, transformed hematopoietic stem cells (38, 39). It is, therefore, reasonable to assume that leukocytopenia was caused by the killing of the WT1-expressing leukemic, transformed hematopoietic stem cells, from which the majority of peripheral blood cells in MDS were derived, by the WT1-specific CTLs induced by the WT1 vaccination (33, 38, 39). Therefore, the leukocytopenia that occurred in these two MDS patients can be considered evidence of efficacy rather than an adverse effect (33). The adverse effect of the WT1 vaccination in cases where normal hematopoiesis is maintained can thus be limited to local erythema at the injection sites of the WT1 vaccine. Taken together, these findings lead us to conclude that the WT1 vaccination is both tolerable and safe for leukemia and solid tumor patients who retain normal hematopoiesis.
The WT1 gene is physiologically expressed in some organs such as kidney, BM, and pleura. Experimental evidence demonstrating that WT1-specific CTLs kill WT1-expressing tumor cells, but not normal cells is accumulating (25–27, 29). WT1-specific CTLs have been shown to specifically kill bcr/abl-positive leukemic, transformed stem cells, but not to damage normal hematopoietic stem cells (27). In mice immunized with MHC class I-restricted 9-mer WT1 peptides or WT1 cDNA, the WT1-specific CTLs induced killing of WT1-expressing tumor cells, but never damaged normal tissues (25, 29). There are at least four possible mechanisms by which WT1-specific CTLs can ignore physiologically WT1-expressing normal cells. First, WT1 expression levels in normal cells are lower than those in tumor cells. However, this possibility is unlikely because WT1 expression levels in CD34+ normal hematopoietic progenitor cells are similar to those in leukemic cells at the single-cell level (40). Second, expression of MHC class I molecules may be lower in normal cells than in tumor cells. Third, in WT1-expressing normal cells, WT1 peptides may not be presented on MHC class I molecules, or the presentation of WT1 peptides onto the molecules may be weak. The poor presentation of WT1 peptides could be ascribed to differences between normal and transformed cells in the processing of WT1 proteins in proteosomes or in the transport of the processed WT1 peptides onto cell surface. Fourth, compared to WT1-expressing tumor cells, WT1-expressing normal cells do not, or weakly, express cell surface costimulatory molecules needed for recognition and/or killing by WT1-specific CTLs.
Clinical responses observed in our study were satisfactory for a phase I clinical study. The frequencies of WT1-specific CTLs in one (patient 14) of the two MDS patients were as high as 0.62% before WT1 vaccination and then increased to 6.61% after a single WT1 vaccination concurrent with the clinical responses. This drastic WT1-specific CTL responses after only one WT1 vaccination may be caused by high immunogenicity of the WT1 protein, as demonstrated by a previous investigation of ours. We found that ≈78% of the patients with hematopoietic malignancies such as AML, chronic myelocytic leukemia, and MDS showed in vivo response to the WT1 protein derived from their leukemic blast cells and produced the WT1 antibody (31). The WT1-specific CTL frequencies before WT1 vaccination were significantly higher in patients with hematopoetic malignancies (mean: 0.37% vs. 0.098%; P = 0.0019) or solid tumors (mean: 0.40% vs. 0.098%; P = 0.0065) than in healthy volunteers (data not shown). These results indicated that patients with WT1-expressing tumor cells responded to the WT1 protein derived from the tumor cells and elicited WT1-specific CTLs before WT1 vaccination, suggesting that the WT1 protein was naturally immunogenic. The existence of the WT1-specific CTLs at high frequencies before WT1 vaccination may have contributed to the favorable clinical responses both in patients with hematopoietic malignancies and those with solid tumors.
Peptide or DNA vaccinations targeting tumor-associated antigens (TAAs) such as gp100, tyrosinase, NY-ESO-1, and prostate-specific antigen have been performed with clinical responses for cancer patients (41–43). Because the clinical response rate in our clinical study was comparable to or better than those in the above clinical studies, and WT1 vaccination could be expected to be effective for both patients with solid tumors in advanced stages and those with hematopoetic malignancies, WT1 is one of the most promising and universal target antigens for cancer immunotherapy.
A significant correlation between immunological and clinical responses was observed in a cohort of all the patients examined. The correlation between the increase in WT1-specific CTL frequencies and clinical responses provided us with evidence indicating that WT1-specific CTLs induced by WT1 vaccination played an important role in the clinical responses. Therefore, clinical responses to WT1 vaccination should be predictable from the degree of increase in WT1-specific CTL frequencies after one to three WT1 vaccinations. No correlation was found between clinical and immunological responses evaluated by the frequency assay of WT1-specific IFN-γ-producing CTLs. WT1-specific IFN-γ-producing CTLs, which can be expected to be functionally active, are likely to reach tumor sites and be scarce in peripheral blood.
Acknowledgments
We thank M. Yamamoto, M. Kondo, and S. Watanabe for the preparation of the manuscript and M. Mishima and K. Ishizaka for excellent technical assistance.
Abbreviations: CTL, cytotoxic T lymphocyte; AML, acute myelocytic leukemia; MDS, myelodysplastic syndrome; BM, bone marrow; PBMC, peripheral blood mononuclear cell.
References
- 1.Call, K. M., Glaser, T., Ito, C. Y., Buckler, A. J., Pelletier, J., Haber, D. A., Rose, E. A., Kral, A., Yeger, H., Lewis, W. H., et al. (1990) Cell 60, 509–520. [DOI] [PubMed] [Google Scholar]
- 2.Gessler, M., Poustka, A., Cavenee, W., Neve, R. L., Orkin, S. H. & Bruns, G. A. (1990) Nature 343, 774–778. [DOI] [PubMed] [Google Scholar]
- 3.Drummond, I. A., Madden, S. L., Rohwer-Nutter, P., Bell, G. I., Sukhatme, V. P. & Rauscher, F. J., III (1992) Science 257, 674–678. [DOI] [PubMed] [Google Scholar]
- 4.Englert, C., Hou, X., Maheswaran, S., Bennett, P., Ngwu, C., Re, G. G., Garvin, A. J., Rosner, M. R. & Haber, D. A. (1995) EMBO. J. 14, 4662–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Godyer, P., Dehbi, M., Torban, E., Bruening, W. & Pelletier, J. (1995) Oncogene 10, 1125–1129. [PubMed] [Google Scholar]
- 6.Hewitt, S. M., Hamada, S., McDonnell, T. J., Rauscher, F. J., III, & Saunders, G. F. (1995) Cancer Res. 55, 5386–5389. [PubMed] [Google Scholar]
- 7.Inoue, K., Sugiyama, H., Ogawa, H., Nakagawa, M., Yamagami, T., Miwa, H., Kita, K., Hiraoka, A., Masaoka, T., Nasu, K., et al. (1994) Blood 84, 3071–3079. [PubMed] [Google Scholar]
- 8.Brieger, J., Weidmann, E., Fenchel, K., Mitrou, P. S., Hoelzer, D. & Bergmann, L. (1994) Leukemia 8, 2138–2143. [PubMed] [Google Scholar]
- 9.Menssen, H. D., Renkl, H. J., Rodeck, U., Maurer, J., Notter, M., Schwartz, S., Reinhardt, R. & Thiel, E. (1995) Leukemia 9, 1060–1067. [PubMed] [Google Scholar]
- 10.Bergmann, L., Miething, C., Maurer, U., Brieger, J., Karakas, T., Weidmann, E. & Hoelzer, D. (1997) Blood 90, 1217–1225. [PubMed] [Google Scholar]
- 11.Menssen, H. D., Renkl, H. J., Rodeck, U., Kari, C., Schwartz, S. & Thiel, E. (1997) Int. J. Cancer 70, 518–523. [DOI] [PubMed] [Google Scholar]
- 12.Tamaki, H., Ogawa, H., Ohyashiki, K., Ohyashiki, J. H., Iwama, H., Inoue, K., Soma, T., Oka, Y., Tatekawa, T., Oji, Y., et al. (1999) Leukemia 13, 393–399. [DOI] [PubMed] [Google Scholar]
- 13.Ogawa, H., Tamaki, H., Ikegame, K., Soma, T., Kawakami, M., Tsuboi, A., Kim, E. H., Hosen, N., Murakami, M., Fujioka, T., et al. (2003) Blood 101, 1698–1704. [DOI] [PubMed] [Google Scholar]
- 14.Oji, Y., Ogawa, H., Tamaki, H., Oka, Y., Tsuboi, A., Kim, E. H., Soma, T., Tatekawa, T., Kawakami, M., Asada, M., et al. (1999) Jpn. J. Cancer Res. 90, 194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oji, Y., Miyoshi, S., Maeda, H., Hayashi, S., Tamaki, H., Nakatsuka, S., Yao, M., Takahashi, E., Nakano, Y., Hirabayashi, H., et al. (2002) Int. J. Cancer 100, 297–303. [DOI] [PubMed] [Google Scholar]
- 16.Miyoshi, Y., Ando, A., Egawa, C., Taguchi, T., Tamaki, Y., Tamaki, H., Sugiyama, H. & Noguchi, S. (2002) Clin. Cancer Res. 8, 1167–1171. [PubMed] [Google Scholar]
- 17.Oji, Y., Miyoshi, Y., Koga, S., Nakano, Y., Ando, A., Nakatsuka, S., Ikeba, A., Takahashi, E., Sakaguchi, N., Yokota, A., et al. (2003) Cancer Sci. 94, 606–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oji, Y., Yamamoto, H., Nomura, M., Nakano, Y., Ikeba, A., Nakatsuka, S., Abeno, S., Kiyotoh, E., Jomgeow, T., Sekimoto, M., et al. (2003) Cancer Sci. 94, 712–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamagami, T., Sugiyama, H., Inoue, K., Ogawa, H., Tatekawa, T., Hirata, M., Kudoh, T., Akiyama, T., Murakami, A. & Maekawa, T. (1996) Blood 87, 2878–2884. [PubMed] [Google Scholar]
- 20.Inoue, K., Tamaki, H., Ogawa, H., Oka, Y., Soma, T., Tatekawa, T., Oji, Y., Tsuboi, A., Kim, E. H., Kawakami, M., et al. (1998) Blood 91, 2969–2976. [PubMed] [Google Scholar]
- 21.Tsuboi, A., Oka, Y., Ogawa, H., Elisseeva, OA., Tamaki, H., Oji, Y., Kim, E. H., Soma, T., Tatekawa, T., Kawakami., et al. (1999) Leuk. Res. 23, 499–505. [DOI] [PubMed] [Google Scholar]
- 22.Li, H., Oka, Y., Tsuboi, A., Yamagami, T., Miyazaki, T., Yusa, S., Kawasaki, K., Kishimoto, Y., Asada, M., Nakajima, H., et al. (2003) Int. J. Hematol. 77, 463–470. [DOI] [PubMed] [Google Scholar]
- 23.Osaka, M., Koami, K. & Sugiyama, T. (1997) Int. J. Cancer 72, 696–699. [DOI] [PubMed] [Google Scholar]
- 24.Oka, Y., Elisseeva, O. A., Tsuboi, A., Ogawa, H., Tamaki, H., Li, H., Oji, Y., Kim, E. H., Soma, T. Asada, M., et al. (2000) Immunogenetics 51, 99–107. [DOI] [PubMed] [Google Scholar]
- 25.Oka, Y., Udaka, K., Tsuboi, A., Elisseeva, O. A., Ogawa, H., Aozasa, K., Kishimoto, T. & Sugiyama, H. (2000) J. Immunol. 164, 1873–1880. [DOI] [PubMed] [Google Scholar]
- 26.Ohminami, H., Yasukawa, M. & Fujita, S. (2000) Blood 95, 286–293. [PubMed] [Google Scholar]
- 27.Gao, L., Bellantuono, I., Elsasser, A., Marley, S. B., Gordon, M. Y., Goldman, J. M. & Stauss, H. J. (2000) Blood 95, 2198–2203. [PubMed] [Google Scholar]
- 28.Tsuboi, A., Oka, Y., Udaka, K., Murakami, M., Masuda, T., Nakano, A., Nakajima, H., Yasukawa, M., Hiraki, A., Oji, Y., et al. (2002) Cancer Immunol. Immunother. 51, 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsuboi, A., Oka, Y., Ogawa, H., Elisseeva, OA., Li, H., Kawasaki, K., Aozasa, K., Kishimoto, T., Udaka, K., Sugiyama, H., et al. (2000) J. Clin. Immuol. 20, 195–202. [DOI] [PubMed] [Google Scholar]
- 30.Gaiger, A., Reese, V., Disis, M. L. & Cheever, M. A. (2000) Blood 96, 1480–1489. [PubMed] [Google Scholar]
- 31.Elisseeva, O. A., Oka, Y., Tsuboi, A., Ogata, K., Wu, F., Kim, E. H., Soma, T., Tamaki, H., Kawakami, M., Oji, Y., et al. (2002) Blood 99, 3272–3279. [DOI] [PubMed] [Google Scholar]
- 32.Scheibenbogen, C., Letsch, A., Thiel, E., Schmittel, A., Mailaender, V., Baerwolf, S., Nagorsen, D. & Keilholz, U. (2002) Blood 100, 2132–2137. [DOI] [PubMed] [Google Scholar]
- 33.Oka, Y., Tsuboi, A., Murakami, M., Hirai, M., Tominaga, N., Nakajima, H., Elisseeva, O. A., Masuda, T., Nakano, A., Kawakami, M., et al. (2003) Int. J. Hematol. 78, 56–61. [DOI] [PubMed] [Google Scholar]
- 34.Tsuboi, A., Oka, Y., Osaki, T., Kumagai, T., Tachibana, I., Hayashi, S., Murakami, M., Nakajima, H., Elisseeva, O. A., Wu, F., et al. (2004) Microbiol. Immunol. 48, 175–184. [DOI] [PubMed] [Google Scholar]
- 35.Wang, F., Bade, E., Kuniyoshi, C., Spears, L., Jeffery, G., Marty, V., Groshen, S. & Weber, J. (1999) Clin. Cancer Res. 5, 2756–2765. [PubMed] [Google Scholar]
- 36.Monsurro, V., Nagorsen, D., Wang, E., Provenzano, M., Dudley, M. E., Rosenberg, S. A. & Marincola, F. M. (2002) J. Immunol. 168, 5933–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ikuta, Y., Okugawa, T., Furugen, R., Nagata, Y., Takahashi, Y., Wang, L., Ikeda, H., Watanabe, M., Imai, S. & Shiku, H. (2000) Int. J. Cancer 87, 553–558. [DOI] [PubMed] [Google Scholar]
- 38.Nilsson, L., Astrand-Grundstrom, I., Arvidsson, I., Jacobsson, B., Hellstrom-Lindberg, E., Hast, R. & Jacobsen, S. E. (2000) Blood 96, 2012–2021. [PubMed] [Google Scholar]
- 39.van Lom, K., Hagemeijer, A., Smit, E., Hahlen, K., Groeneveld, K. & Lowenberg, B. (1995) Leukemia 9, 1818–1821. [PubMed] [Google Scholar]
- 40.Hosen, N., Sonoda, Y., Oji, Y., Kimura, T., Minamiguchi, H., Tamaki, H., Kawakami, M., Asada, M., Kanato, K., Motomura, M., et al. (2002) Br. J. Haematol. 16, 409–420. [DOI] [PubMed] [Google Scholar]
- 41.Jager, E., Gnjatic, S., Nagata, Y., Stockert, E., Jager, D., Karbach, J., Neumann, A., Rieckenberg, J., Chen, Y. T., Ritter, G., et al. (2000) Proc. Natl. Acad. Sci. USA 97, 12198–12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Slingluff, C. L., Jr., Petroni, G. R., Yamshchikov, G. V., Barnd, D. L., Eastham, S., Galavotti, H., Patterson, J. W., Deacon, D. H., Hibbitts, S., Teates, D., et al. (2003) J. Clin. Oncol. 21, 4016–4026. [DOI] [PubMed] [Google Scholar]
- 43.Kaufman, H. L., Wang, W., Manola, J., DiPaola, R. S., Ko, Y.-J., Sweeney, C., Whiteside, T. L., Schlom, J., Wilding, G. & Weiner, L. M. (2004) J. Clin. Oncol. 22, 2122–2132. [DOI] [PubMed] [Google Scholar]