

Abstract
Aryl hydrocarbon receptor-interacting protein-like 1 (AIPL1) is a member of the FK-506-binding protein family expressed specifically in retinal photoreceptors. Mutations in AIPL1 cause Leber congenital amaurosis, a severe early-onset retinopathy that leads to visual impairment in infants. Here we show that knockdown of AIPL1 expression in mice also produces a retinopathy but over a more extended time course. Before any noticeable pathology, there was a reduction in the level of rod cGMP phosphodiesterase (PDE) proportional to the decrease in AIPL1 expression, whereas other photoreceptor proteins were unaffected. Consistent with less PDE in rods, flash responses had a delayed onset, a reduced gain, and a slower recovery of flash responses. We suggest that AIPL1 is a specialized chaperone required for rod PDE biosynthesis. Thus loss of AIPL1 would result in a condition that phenocopies retinal degenerations in the rd mouse and in a subgroup of human patients.
Mutations in several genes, including aryl hydrocarbon receptor-interacting protein-like 1 (AIPL1), cause Leber congenital amaurosis (LCA), a severe early-onset retinopathy (1–4). AIPL1 is expressed specifically in adult rod photoreceptors (1, 5), where its function is essential but not understood. AIPL1 possesses one peptidyl-prolyl isomerase (PPI) domain and three consecutive tetratricopeptide repeats (TPR). Sequence comparison places AIPL1 in the FK-506-binding protein (FKBP) family. FKBPs and cyclophilins comprise the immunophilin superfamily of proteins (6), many of which function as molecular chaperones (7–9). Thus AIPL1 might fulfill a molecular chaperone function for retinal protein folding (10). The presence of TPR motifs downstream from a PPIase domain in AIPL1 makes it a close relative of the larger members of the FKBP family such as FKBP52 and AIP, which function in the maturation/translocation of dioxin and steroid receptors, respectively (11, 12). The chaperone functions of this protein family typically do not act at the step of initial polypeptide folding. Rather, these proteins are “specialized chaperones” that assist specific client proteins in later stages of maturation, subunit assembly, transport, and degradation (11, 13–15). Many such client proteins are components of signal transduction pathways. Therefore, the primary sequence of AIPL1 suggests a possible role as a specialized accessory chaperone important for photoreceptor protein(s). In another study, AIPL1 was found to interact with and aid in the processing of farnesylated proteins in vitro, suggesting a role for AIPL1 in the processing of farnesylated proteins (16).
To investigate the function of AIPL1 in vivo, we created a murine mutant model by genetic manipulations. Based on the human disease, we postulated that conventional gene knockout would produce an AIPL1 null mutant with photoreceptors that never fully mature or that degenerate too rapidly for detailed cell biological and physiological studies. To circumvent this problem, we used a knockdown approach to produce a mutant in which AIPL1 expression was reduced but not extinguished. Our analyses of this hypomorphic mutant suggest that AIPL1 functions as a chaperone specific for rod cGMP phosphodiesterase (PDE) biosynthesis.
Materials and Methods
Generation of the AIPL1 Hypomorphic (h) Mutant Allele. Genomic fragments spanning exons 1–2 and exon 3 were amplified by PCR from 129/Sv mouse genomic DNA. These fragments were cloned into the vector pGT-N29 (New England Biolabs, Beverly, MA) flanking the neor gene, including the bovine polyadenylation signal, to generate the AIPL1 targeting vector. Linearized vector DNA was electroporated into J1 embryonic stem cells, and neomycin-resistant colonies were selected. The AIPL1 mutant allele carrying the neor gene insertion in intron 2 was identified by PCR and confirmed by DNA sequencing (Fig. 1A). A targeted clone was microinjected into C57BL/6 blastocysts, and male chimeras were crossed with C57BL/6 mice. Mice homozygous for the mutant allele (h/h) were identified by PCR (Fig. 1B). The primers used for genomic PCR were A5 (5′-GTACGGGTATACATGTGTGTATCTATGAG), C3 (5′-AGCCTGTTGCCTTCTCCATGAAG) and B5 (5′-CGAGATCAGCAGCCTCTGTTCCAC). All animal experimentation conformed to institutional guidelines.
Fig. 1.

Generation of an AIPL1 hypomorphic (h) mutant. (A) Targeting strategy for the insertion of a neor marker into intron 2 of the AIPL1 gene. PCR primers for identification of the mutant and WT alleles are shown as arrowheads. (B) Identification of the mutant allele by genomic PCR. Primer combinations are shown (Upper). (C) Analysis of AIPL1 transcript by semiquantitative PCR. GAPDH was used as an internal standard. (D) Reduced AIPL1 immunofluorescence (yellow) in mutant photoreceptor inner segment. Cell nuclei (blue) were counterstained with Hoechst dye 33342. OS, outer segment; IS, inner segment; ONL, outer (photoreceptor) nuclear layer; INL, inner nuclear layer. (E) Decreased AIPL1 level on immunoblots (Left). Actin served as a general loading control. RDS was a control for amounts of photoreceptor proteins. Data from triplet experiments were plotted as percent of the control (Right).
mRNA Assays. Total RNA was isolated by using the TRIzol reagent (GIBCO). First-strand cDNA synthesis was primed with oligo(dT)20 by using the ThermoScript RT-PCR System (Invitrogen). GAPDH was amplified together as an internal standard for quantification. PCR was carried out for different cycle numbers to ensure that amplification had not yet reached a plateau. PCR products were separated on agarose gels and quantified by using the Fluor-S MultiImager (Bio-Rad). PCR primers for GAPDH mRNA assays were PG5 (5′-TGAAGGTCGGTGTGAACGGATTTGGC) and PG3 (5′-CATGTAGGCCATGAGGTCCACCAC). Primers for AIPL1 mRNA assays were PA5 (5′-ATGGACGTCTCTCTACTCCTCAATG and PA3 (5′-CTCCAGCTTCAGCCACTCAAC). Primers for PDEα mRNA assays were PDEα 5 (5′-ATGGGTGAGGTGACAGCAGAG) and PDEα3 (5′-TACTGGATGCAACAGGACTTAG). Primers for PDEβ mRNA assays were PDEβ 5 (5′-ATGAGCCTCAGTGAGGAACAGGTAC) and PDEβ 3 (5′-TTATAGGATACAGCAGGTCGAG). Primers for PDEγ mRNA assays were PDEγ5 (5′-TGACAGAGTCCAGAAGCTAAGG) and PDEγ3(5′-TAAATGATGCCATACTGGGCCAG).
Antibodies, Immunoblotting, and Immunofluorescence. A His-tagged fusion protein encompassing the full-length mouse AIPL1 was expressed in Escherichia coli, purified, and used to generate polyclonal antibodies in rabbit. The antibodies were affinity-purified. Immunoblotting and immunofluorescence were performed as described (17). Rod PDE holoenzyme is composed of αβγ2 (18, 19). PDEα- and β-specific antibodies were purchased from Affinity BioReagents (Neshanic Station, NJ). PDE-PatB was raised against the PDE holoenzyme (20). PDE-NC and -CAT were raised against the noncatalytic and catalytic regions of the PDE but did not distinguish between PDEα- and β-subunits (21). Retinal histology was performed as described (22).
Electroretinographic Recording. Electroretinograms (ERGs) were recorded as described (23, 24). The initial segments of the a waves from dark-adapted full-field ERGs in response to bright white flashes were fitted with a mathematical “component” model that estimates parameters involved in rod phototransduction (22, 25).
Single Cell Recording. AIPL1 mutant and WT mice at 7–10 weeks were dark-adapted for a minimum of 12 h. Retinas were dissected under infrared light and put into oxygenated Leibovitz's L-15 medium (GIBCO) with 10% (wt/vol) BSA and 10 mM glucose and stored on ice until use. A small aliquot was chopped finely in L-15 containing DNase type IV-S (Sigma) and loaded into an experimental chamber. The tissue was perfused with 139 mM Na+/3.6 mM K+/2.4 mM Mg2+/1.2 mM Ca2+/123.3 mM Cl–/20 mM 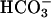 mM Hepes/0.02 mM EDTA 10 mM glucose/3 mM succinate/0.5 mM l-glutamate/MEM amino acids (GIBCO)/BME vitamins (Sigma), equilibrated with 95% O2/5% CO2 and heated to 36.5–37.5°C, pH 7.4. The outer segment membrane current of a rod was recorded with a suction pipette containing Locke's medium 140 mM Na+/3.6 mM K+/2.4 mM Mg2+/1.2 mM Ca2+/145.8 mM Cl–/10 mM Hepes/0.02 mM EDTA/10 mM glucose, and a current-to-voltage converter (Axopatch 200A, Axon Instruments, Union City, CA). Signals were filtered at 30 Hz (–3 dB, 8-pole Bessel, Frequency Devices, Haverhill, MA) and digitized at 400 Hz (Pulse 8.07, HEKA Electronics, Lambrecht/Pfalz, Germany). Corrections were not made for the delay introduced by filtering. Rods were stimulated with light passing through an interference filter (500 nm, Omega Optical, Brattleboro, VT) and neutral density filters.
mM Hepes/0.02 mM EDTA 10 mM glucose/3 mM succinate/0.5 mM l-glutamate/MEM amino acids (GIBCO)/BME vitamins (Sigma), equilibrated with 95% O2/5% CO2 and heated to 36.5–37.5°C, pH 7.4. The outer segment membrane current of a rod was recorded with a suction pipette containing Locke's medium 140 mM Na+/3.6 mM K+/2.4 mM Mg2+/1.2 mM Ca2+/145.8 mM Cl–/10 mM Hepes/0.02 mM EDTA/10 mM glucose, and a current-to-voltage converter (Axopatch 200A, Axon Instruments, Union City, CA). Signals were filtered at 30 Hz (–3 dB, 8-pole Bessel, Frequency Devices, Haverhill, MA) and digitized at 400 Hz (Pulse 8.07, HEKA Electronics, Lambrecht/Pfalz, Germany). Corrections were not made for the delay introduced by filtering. Rods were stimulated with light passing through an interference filter (500 nm, Omega Optical, Brattleboro, VT) and neutral density filters.
cGMP Assay. Total cGMP was measured under dark- and light-adapted conditions from WT and mutant mice by using the competitive enzyme immunoassay kit from Cayman Chemical (Ann Arbor, MI) and following the manufacturer's protocols. Dissected retinas from three animals were used in each experimental group.
Calcium Determinations. The free [Ca2+]i of single outer segments was measured with a fluorescent indicator dye (26). Mice were dark-adapted for at least 3 h and then killed under dim red light. The eyes were removed and placed in Locke's solution with 3 mM Hepes and 5 mM Na ascorbate, pH 7.4. Retinas were dissected in infrared illumination and incubated at room temperature with 10 μM fluo-5F acetoxymethyl ester (Molecular Probes). After 30 min, the retinas were perfused with 35–38°C bicarbonate-buffered DMEM (D-2902, Sigma) supplemented with 15 mM NaHCO3/2 mM Na succinate/0.5 mM Na glutamate/2 mM Na gluconate/5 mM NaCl. For improved collection of fluorescence, we used only rods on the floor of the perfusion chamber. Fluorescence was excited with 488-nm light from an argon ion laser (American Laser, Salt Lake City), focused to a 10-μm diameter spot on the rod outer segment. To prevent dye bleaching, the intensity of the laser was attenuated to 2–5 × 1010 photons μm–2·s–1 with reflective neutral density filters (Newport, Fountain Valley, CA). Fluorescence was detected with a photomultiplier tube of low dark count with a restricted photocathode (Model 9130/100A, Electron Tubes, Ruislip, U.K.). The current from the tube was amplified by a low-noise current-to-voltage converter (PDA-700, Terahertz Technology, Oriskany, NY), filtered at 1 kHz with a low-pass 8-pole Bessel filter (BenchMaster VBF8, Kemo, Kent, U.K.) and acquired at 2 kHz by using Digidata 1200 and software from Axon Instruments. The temperature close to the rod was recorded with a miniature thermocouple (0.05-mm diameter, T type, Cu-CuNi; Omega Engineering, Stamford, CT) and a digital thermometer (Model HH-25T, Omega Engineering).
The [Ca2+]i in darkness and after illumination were estimated from the initial fluorescence after laser exposure and the steady-state fluorescence measured 30–60 sec later, according to [Ca2+] = Kd[(F–Fmin)/(Fmax–F)], where Kd is the temperature-adjusted dissociation constant, F is the measured fluorescence, and Fmin and Fmax are the fluorescence minimum (at low Ca2+) and maximum (at high Ca2+). Fmin and Fmax were estimated for each rod by exposure first to a low Ca2+-ionomycin solution 140 mM NaCl/3.6 mM KCl/3.08 mM MgCl2/2 mM EGTA/0.01 mM ionomycin/3 mM Hepes, pH 7.4; and then to a high Ca2+-ionomycin solution 50 mM CaCl2/3.6 mM KCl/0.005 mM ionomycin/3 mM Hepes/140 mM sucrose, pH 7.4. This procedure corrects for any differences in dye loading and fluorescence collection across rods.
Results
Reduced AIPL1 Level in the Mutant Photoreceptors. PCR and DNA sequencing analyses showed that the AIPL1 mutant allele had the neomycin resistance gene inserted between exons 2 and 3 in the same orientation as the AIPL1 gene (Fig. 1 A). The neomycin cassette was terminated by a polyadenylation signal of the bovine growth hormone gene (Fig. 1 A). Analysis of retinal mRNA confirmed that the AIPL1 transcript level was severely reduced (Fig. 1C). A lower abundance of AIPL1 protein in the mutant photoreceptors was apparent by immunofluorescence, and the protein was correctly localized in the inner segments (Fig. 1D). Quantification on immunoblots revealed that the level of AIPL1 protein in the mutant was decreased to 20–25% of that for WT (Fig. 1E). Thus the mutant allele was a strong hypomorph (h).
Progressive Photoreceptor Degeneration in the AIPL1 Hypomorphic Mutant. By morphological criteria, photoreceptors in the AIPL1 mutant retinas developed normally, although the AIPL1 deficiency eventually gave rise to a retinal degeneration. Until 3 months of age, light microscopy showed that the photoreceptor layer thickness was not significantly reduced in the mutant retinas (Fig. 2A), but some disorganization of the outer segments and increased pycnotic nuclei were apparent by electron microscopy (not shown). Expression of glial fibrillary acidic protein, a marker of retinal degeneration, was markedly up-regulated (Fig. 2B). By 8 months of age, more than half of the photoreceptors were lost, and the photoreceptor inner and outer segments were shortened (Fig. 2 A). We therefore restricted biochemical and functional studies to ages younger than 10 weeks. We found no evidence of a cone defect by immunofluorescence for cone opsin up to 11 months of age (not shown).
Fig. 2.

Age-dependent retinal degeneration in the AIPL1 mutant. (A) Thinning of outer segment and outer nuclear layers at 8 months in mutant retinas. (B) Glial fibrillary acidic protein (yellow) up-regulation at 3 months of age in the mutant retina. Nuclei (blue) were counterstained with Hoechst dye.
Lower Abundance of Rod PDE, but Not of Other Photoreceptor Proteins, in the Mutant. To test the hypotheses that AIPL1 serves as a specialized chaperone for photoreceptor-specific proteins or is required for protein farnesylation, we surveyed a large number of proteins for their levels of expression and electrophoretic mobilities by immunoblotting and subcellular distributions by immunofluorescence. Rhodopsin, peripherin/RDS; PDEα-, β-, and γ-subunits; PDEδ; transducin α-, β-, and γ-subunits; retinal guanylyl cyclase 1; cGMP-gated cationic channel, Tulp1 (Tubby-like protein 1); arrestin; rhodopsin kinase; and RPGRIP (retinitis pigmentosa GTPase regulator-interacting protein) were selected for analysis, because they either are dedicated to phototransduction, specifically expressed in photoreceptors, isoprenylated, involved in the pathogenesis of LCA, or meet a combination of these criteria.
Immunoblotting of retinal homogenates from WT and mutant mice showed that, of all of the proteins tested, only PDE was reduced in abundance (Fig. 3A). Quantification of the immunoblotting results indicated that PDE was reduced to ≈20% of the WT level (Fig. 3B). Further analyses on low crosslinking acrylamide gels and by subunit-specific antibodies showed that all three subunits of PDE were similarly reduced (Fig. 3C). Consistent with these findings, immunofluorescence showed a marked reduction of PDE subunits in the rod outer segments (Fig. 3D). Analyses of rod PDE by immunoblotting and immunofluorescence were repeated by using five separate antibodies specific for the catalytic subunits of PDE. All gave results similar to those shown in Fig. 3 B–D. The subcellular distributions of all other proteins were normal when examined by immunofluorescence (data not shown).
Fig. 3.

Identification of rod cGMP PDE as the only protein affected by the loss of AIPL1. (A) Immunoblotting of photoreceptor proteins. (Lower) Actin as a general loading control. Channel, cGMP-gated channel; Rho kinase, rhodopsin kinase; Ret-GC, retinal guanylyl cyclase 1; RDS, peripherin/RDS. PDE was significantly reduced, whereas all other photoreceptor proteins were unaffected. (B) Quantification of PDE from three pairs of independent samples shown as mean ± SEM. (C) Proportional reduction in all three subunits of PDE. (Top) The two catalytic subunits of PDE were separated on low-crosslinking SDS/PAGE and detected with an antibody that recognized both subunits. (Middle) A PDEγ-specific antibody revealed a decrease of this subunit as well. (Bottom) Actin is shown as a loading control. (D) Reduced immunofluorescence for PDE in the mutant rods. Cell nuclei were stained blue with Hoechst dye. (E) Comparable mRNA levels for all three PDE subunits in WT and mutant retinas shown by RT-PCR.
If AIPL1 were to function as a chaperone or to assist in protein farnesylation, it should modulate PDE levels at a posttranslational stage, rather than through transcriptional regulation. Nevertheless, to rule out the latter possibility, we analyzed the mRNA levels of each PDE subunit by semiquantitative RT-PCR. There was no substantial decrease in the mRNA levels of any of the PDE subunits (Fig. 3E), confirming that the defect in PDE biosynthesis was posttranscriptional.
Because PDEα, transducin γ (Gγt), and rhodopsin kinase are normally farnesylated, it was notable that neither Gγt nor rhodopsin kinase was reduced in abundance, shifted in mobility on SDS/PAGE (Fig. 3A), or mislocalized by immunofluorescence (not shown). The finding is particularly significant for Gγt. Due to its low molecular weight of ≈6 kDa, a disruption in its farnesylation would unambiguously shift its electrophoretic mobility (16, 27). These observations rule out a generalized defect in protein farnesylation in the mutant photoreceptors.
Delayed Photoresponse Onset and Recovery in Mutant Rods. In visual transduction, photoexcited rhodopsin activates many transducins. Each transducin activates a PDE catalytic subunit, which hydrolyzes cGMP. cGMP-gated channels close, and the rod hyperpolarizes. Because PDE is central to the transduction process, we investigated the physiological consequences of a PDE reduction in the mutant photoreceptors.
In a cohort of young mice (5–6 weeks old) tested by ERG, the mutant mice (n = 8) showed a 0.44-msec increase in mean latency (P = 0.007) and a 31% reduction in the geometric mean gain of phototransduction (P = 0.007) compared with WT mice (n = 9) (Fig. 4). The increased latency was consistent with a delayed diffusional encounter of activated transducin with PDE (28), due to the lower concentration of the latter. The reduction in the gain of phototransduction suggests that PDE was activated at a lower rate in mutant rods. The amplitudes of the a and b waves were normal at 6–7 weeks but became progressively lower with age (6–8 months; not shown), as expected simply from the decrease in the number of photoreceptors and shortening of outer segments (Fig. 2 A).
Fig. 4.

Dark-adapted ERG a waves in response to a bright flash of white light (28 foot Lambert-seconds, presented at time = 0 msec) from WT and AIPL1 mutant mice. Recordings (dotted lines) were fit according to the component model of the activation phase of rod phototransduction (continuous lines).
Photoresponses were also recorded from single rods (Fig. 5A). Although the maximal amplitude of flash responses in mutant rods (11.3 ± 0.5 pA, mean ± SEM, n = 47) was not different from WT rods (10.8 ± 0.5 pA, n = 21), mutant rods showed delayed onset and slower initial rate of rise of the single photon response (Fig. 5B), corroborating the ERG results based on the mass photoreceptor response. However, the mutant quantal response continued to rise for a longer period, leading to a larger amplitude and a greater time to peak. Recovery was slowed, prolonging integration time, defined as integral of the response divided by response amplitude. Mutant rods were more sensitive than normal (Fig. 5C). The half-saturating flash strengths from 47 mutant rods and 21 WT rods were 29 ± 1 and 51 ± 3 photons μm–2, respectively (P < 8e–13). The increased flash sensitivity and integration time combined to make mutant rods 2.4-fold more sensitive than WT rods to steps of light (not shown). An intensity of 77 ± 11 photons μm–2·s–1 gave rise to a half-maximal response in mutant rods (n = 6), whereas for WT (n = 10) 182 ± 18 photons μm–2·s–1 were required.
Fig. 5.

Photoresponses of single rods. (A) Averaged normalized flash responses of a mutant rod (thick traces) and a WT rod (thin traces). The maximum response amplitude was 11.4 pA for the mutant rod and 15.9 pA for the WT rod. (B) Averaged single-photon responses of mutant (thick trace) and WT (thin trace) rods. Averaged dim flash responses from mutant and WT rods were scaled to the average ratio of the ensemble variance to mean amplitude for rods of each type. Flashes generating responses with mean amplitudes <0.2 rmax were considered to be dim. The times to peak were 215 and 130 msec, and the integration times were 545 and 259 msec for the mutant and WT rods, respectively. (C) Stimulus-response relations of cells in A. Results were fit with: r/rmax = 1 – exp(–ki), where i is the flash strength, k is ln (2)/i1/2, and i1/2 is the flash strength producing a half-maximal response. i1/2 values were 28 and 50 photons μm–2 for the mutant and WT rods, respectively.
Changes in cGMP and Ca2+. A reduction in basal PDE activity should elevate the free cGMP concentration and support a higher Ca2+ influx by increasing the number of open cGMP-gated channels in darkness (29). Because intracellular Ca2+ is a key regulator of the phototransduction cascade and appears to be critical for rod viability (30), these changes were explored. A priori, it was not clear whether the total cGMP concentration would be increased in the mutant, because PDE both degrades free cGMP and sequesters a large pool of tightly bound cGMP at noncatalytic sites (31). We found that total cGMP levels were lower in the AIPL1 mutant both under dark- and light-adapted conditions (Fig. 6). This would mean that the increase in free cGMP was not sufficiently large to compensate for the 80% loss of PDE and hence the bound cGMP pool. In mice heterozygous for the rd allele (32, 33), which have a milder loss of PDE, retinal cGMP is also lower (34, 35). The ratio of cGMP levels in WT vs. heterozygous rd is the same in darkness and after light exposure. In contrast, the fractional decrease in cGMP in the AIPL1 mutant retinas was significantly greater upon light exposure than in darkness (Fig. 6), supporting the idea that the lowered basal PDE activity in the dark-adapted AIPL1 mutant retinas may have resulted in an elevated level of free cGMP.
Fig. 6.

Measurement of cGMP levels from WT and mutant retinas. Both dark- and light-adapted cGMP levels were lower in the mutant than in the WT retinas, and the fractional reduction of cGMP level in the mutant retinas was more pronounced under light adaptation (P < 0.001).
Higher free cGMP would increase channel opening and enhance Ca2+ influx. We therefore measured the free Ca2+ in WT and mutant rod outer segments. Fig. 7A shows the averaged decrease in fluorescence after laser exposure for a sample of WT and mutant rods. The level of Ca2+ in the dark-adapted rods was derived from the initial fluorescence. The steady-state plateau fluorescence reflects the decreased Ca2+ concentration caused by closure of channels after laser exposure and efflux of Ca2+ by the Na+/K+-Ca2+ exchanger. Fluorescence was greater under both conditions for the mutant rods. After calibration of the fluorescence (see Materials and Methods), the dark-adapted Ca2+ concentrations were 322 ± 64 nM (n = 12) for the mutant and 240 ± 17 nM (n = 56) for the WT controls, a difference in the mean that was not statistically significant. However, if we define a “high” concentration of calcium as the 90th percentile of the WT distribution, then one-third of mutant rods exceeded the high concentration of 392 nM found for dark-adapted WT rods, i.e., a higher proportion than WT (Fisher's exact test, P = 0.04). Unexpectedly, closure of cGMP-gated channels in the mutant by light exposure did not reduce the Ca2+ concentration to the same fraction as in WT rods (Fig. 7B; P = 0.004). The light-exposed Ca2+ concentration was therefore significantly higher in the mutant (96 ± 16 nM, n = 12) than in the WT rods (42 ± 3 nM, n = 56).
Fig. 7.

Enhanced Ca2+ concentration in mutant rods. (A) Mean fluorescence from seven WT (black trace) and seven mutant (gray trace) rods, detected as photomultiplier tube current. The decreases in fluorescence reflect the light-dependent fall in Ca2+ concentration after laser exposure (see text). The kinetics of [Ca2+] change was similar for mutant and WT rods. (B) Ca2+ concentration in WT and mutant rods. WT values in the present experiments did not differ from those reported previously (26, 49) and were combined to give mean values (±SEM) of 240 ± 17 nM (dark) and 42 ± 3 nM (light, n = 56). Mutant values were 322 ± 64 nM (dark) and 96 ± 16 nM (light, n = 12). After converting Ca2+ concentrations to logarithms to better normalize the distributions, the mean dark values were not significantly different. However, the proportion of mutant rods with “high” [Ca2+] was significantly greater than that of WT rods (see Results). The difference between the WT and mutant under light adapted conditions was highly significant (P = 0.0007).
Discussion
In this study, we investigated the in vivo function of AIPL1 by analyzing a murine mutant model in which AIPL1 expression was too low to sustain photoreceptor survival over the long term. This provided us with an opportunity to examine morphologically normal photoreceptors at an early age with a variety of approaches. Because AIPL1 mutations cause LCA, and this protein is likely to act as a chaperone, it follows that retinal proteins involved in the pathogenesis of LCA or severe recessive RP are candidate client proteins for AIPL1. Along this line of reasoning, we included the cGMP-gated channel (36), RPGRIP (37), and guanylyl cyclase (38) in our analyses. A putative role for AIPL1 in processing farnesylated proteins (16) implicated PDEα, rhodopsin kinase, and Gγt. Careful analyses of an extensive array of photoreceptor-enriched proteins for which antibodies were available identified PDE as the only client protein of AIPL1 in rods. Our study does not support a general role for AIPL1 in protein farnesylation but strongly suggests that AIPL1 is a specialized chaperone for rod PDE. It remains to be seen whether this is the only role of AIPL1. Transient expression of AIPL1 in cone precursors during embryonic development (39) and the greater disease severity in LCA (AIPL1 mutations) (4) than recessive retinitis pigmentosa (PDE mutations) (40) appear to imply a role for AIPL1 in developing cones. Further study is needed to clarify this question.
It has long been recognized that functional PDE is difficult to express in cell cultures (41–43). One possible reason is the lack of an essential chaperone, namely AIPL1. Yet, repeated efforts to coexpress in COS cells the three subunits of PDE together with AIPL1 or with a homologous protein (FKBP8) as a control failed to increase the yield of either the total or soluble fraction of PDE. Barring technical issues, additional factors, absent in COS cells, may be required to promote PDE synthesis. Alternatively, AIPL1 may subserve a quality-control mechanism whereby only properly folded, fully assembled PDE holoenzyme is given facilitated transport to the outer segment, whereas misfolded or partially assembled PDE is rapidly degraded. Indeed, AIPL1 was reported to interact with NUB1 (44), which targets substrate proteins to the degradative pathway (45). Consistent with this view, mutant mouse photoreceptors lacking PDEγ produce little PDE activity or PDEα/β-polypeptides (46).
The availability of mutant rods with a reduced PDE concentration in an otherwise normal-appearing cell allowed us to test certain predictions about phototransduction. In darkness, hydrolysis of cGMP by PDE balances its synthesis by guanylyl cyclases (30). Because basal PDE activity consists of thermal activation of PDE (47), it decreases with a fall in PDE concentration. Free cGMP would accumulate, cGMP-gated channels open, and intracellular Ca2+ increase, which would then decrease guanylyl cyclase activity, bringing cGMP and Ca2+ to a somewhat elevated steady-state level. Yet any increase in dark current or Ca2+ in our mutant rods must have been small, because they were not resolved by the suction electrode method, by ERG or by direct measurements of dark-adapted Ca2+ levels. We did observe a slower falling phase in dim flash responses and a higher sensitivity to light for mutant rods, consistent with a reduction in basal PDE activity (48). Furthermore, the initiation of the photoresponse was delayed, showing that normally, a significant portion of the effective latency for the activation of phototransduction is attributable to the collision time between transducin and PDE (28).
It is remarkable that the retinas of the mutant mice slowly degenerated, even though the effect of decreased PDE on the phototransduction apparatus seemed relatively mild. A similar phenomenon was described recently for a gain-of-function GCAP1 mutant (Y99C) expressed in transgenic mice (49). In that case, the mutant altered cGMP synthesis rather than hydrolysis. Nonetheless, this mutant also produced a slowing in the falling phase of the dim flash response and higher sensitivity, as well as an increase in Ca2+ that was small and not statistically significant for a slowly degenerating line but large and significant for a line that degenerated rapidly. LCA patients with AIPL1 mutations add yet another example for which disturbances in cGMP metabolism give rise to retinal degeneration. How these disturbances trigger photoreceptor cell death and the role of Ca2+ in this process remain important questions for future investigations.
Acknowledgments
We thank Drs. V. Arshavsky (Harvard Medical School, Boston), J. Chen (University of Southern California, Los Angeles), R. Cote (University of New Hampshire, Durham), A. Dizhoor (Pennsylvania College of Optometry, Elkins Park), R. Lee (University of California, Los Angeles), R. Molday (University of British Columbia, Vancouver), M. Simon (California Institute of Technology, Pasadena), and G. Travis (University of California, Los Angeles) for antibodies; Norman Michaud and Akella Sreedevi for histology; and Scott Adam for DNA sequencing. This work was supported by grants from the Macular Vision Research Foundation, National Institutes of Health (Grants EY10309, EY11358, EY01844, and Core Grant EY14104); the Knights Templar Eye Foundation, the Foundation Fighting Blindness; and the Massachusetts Lions Eye Research Fund.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: AIP, aryl hydrocarbon receptor-interacting protein; AIPL1, AIP-like 1; FKBP, FK-506-binding protein; LCA, Leber congenital amaurosis; PDE, cGMP phosphodiesterase; ERG, electroretinogram.
References
- 1.Sohocki, M. M., Bowne, S. J., Sullivan, L. S., Blackshaw, S., Cepko, C. L., Payne, A. M., Bhattacharya, S. S., Khaliq, S., Qasim Mehdi, S., Birch, D. G., et al.. (2000) Nat. Genet. 24, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanein, S., Perrault, I., Gerber, S., Tanguy, G., Barbet, F., Ducroq, D., Calvas, P., Dollfus, H., Hamel, C., Lopponen, T., et al. (2004) Hum. Mutat. 23, 306–317. [DOI] [PubMed] [Google Scholar]
- 3.Koenekoop, R. K. (2004) Surv. Ophthalmol. 49, 379–398. [DOI] [PubMed] [Google Scholar]
- 4.Dharmaraj, S., Leroy, B. P., Sohocki, M. M., Koenekoop, R. K., Perrault, I., Anwar, K., Khaliq, S., Devi, R. S., Birch, D. G., De Pool, E., et al. (2004) Arch. Ophthalmol. 122, 1029–1037. [DOI] [PubMed] [Google Scholar]
- 5.van der Spuy, J., Chapple, J. P., Clark, B. J., Luthert, P. J., Sethi, C. S. & Cheetham, M. E. (2002) Hum. Mol. Genet. 11, 823–831. [DOI] [PubMed] [Google Scholar]
- 6.Snyder, S. H., Lai, M. M. & Burnett, P. E. (1998) Neuron 21, 283–294. [DOI] [PubMed] [Google Scholar]
- 7.Gothel, S. F. & Marahiel, M. A. (1999) Cell. Mol. Life Sci. 55, 423–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colley, N. J., Baker, E. K., Stamnes, M. A. & Zuker, C. S. (1991) Cell 67, 255–263. [DOI] [PubMed] [Google Scholar]
- 9.Pratt, W. B., Galigniana, M. D., Harrell, J. M. & DeFranco, D. B. (2004) Cell. Signalling 16, 857–872. [DOI] [PubMed] [Google Scholar]
- 10.Chapple, J. P., Grayson, C., Hardcastle, A. J., Saliba, R. S., van der Spuy, J. & Cheetham, M. E. (2001) Trends Mol. Med. 7, 414–421. [DOI] [PubMed] [Google Scholar]
- 11.Petrulis, J. R. & Perdew, G. H. (2002) Chem. Biol. Interact. 141, 25–40. [DOI] [PubMed] [Google Scholar]
- 12.Kimmins, S. & MacRae, T. H. (2000) Cell Stress Chaperones 5, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Young, J. C., Barral, J. M. & Ulrich Hartl, F. (2003) Trends Biochem. Sci. 28, 541–547. [DOI] [PubMed] [Google Scholar]
- 14.Carver, L. A. & Bradfield, C. A. (1997) J. Biol. Chem. 272, 11452–11456. [DOI] [PubMed] [Google Scholar]
- 15.Blatch, G. L. & Lassle, M. (1999) BioEssays 21, 932–939. [DOI] [PubMed] [Google Scholar]
- 16.Ramamurthy, V., Roberts, M., van den Akker, F., Niemi, G., Reh, T. A. & Hurley, J. B. (2003) Proc. Natl. Acad. Sci. USA 100, 12630–12635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hong, D. H., Yue, G., Adamian, M. & Li, T. (2001) J. Biol. Chem. 276, 12091–12099. [DOI] [PubMed] [Google Scholar]
- 18.Baehr, W., Devlin, M. J. & Applebury, M. L. (1979) J. Biol. Chem. 254, 11669–11677. [PubMed] [Google Scholar]
- 19.Deterre, P., Bigay, J., Forquet, F., Robert, M. & Chabre, M. (1988) Proc. Natl. Acad. Sci. USA 85, 2424–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee, R. H., Navon, S. E., Brown, B. M., Fung, B. K.-K. & Lolley, R. N. (1988) Invest. Ophthalmol. Visual Sci. 29, 1021–1027. [PubMed] [Google Scholar]
- 21.Mou, H. & Cote, R. H. (2001) J. Biol. Chem. 276, 27527–27534. [DOI] [PubMed] [Google Scholar]
- 22.Hong, D. H., Pawlyk, B. S., Shang, J., Sandberg, M. A., Berson, E. L. & Li, T. (2000) Proc. Natl. Acad. Sci. USA 97, 3649–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li, T., Sandberg, M. A., Pawlyk, B. S., Rosner, B., Hayes, K. C., Dryja, T. P. & Berson, E. L. (1998) Proc. Natl. Acad. Sci. USA 95, 11933–11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sandberg, M. A., Lee, H., Matthews, G. P. & Gaudio, A. R. (1991) Invest. Ophthalmol. Visual Sci. 32, 1508–1516. [PubMed] [Google Scholar]
- 25.Hood, D. C. & Birch, D. G. (1994) Invest. Ophthalmol. Visual Sci. 35, 2948–2961. [PubMed] [Google Scholar]
- 26.Woodruff, M. L., Sampath, A. P., Matthews, H. R., Krasnoperova, N. V., Lem, J. & Fain, G. L. (2002) J. Physiol. 542, 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terada, K. & Mori, M. (2000) J. Biol. Chem. 275, 24728–24734. [DOI] [PubMed] [Google Scholar]
- 28.Lamb, T. D. & Pugh, E. N., Jr. (1992) J. Physiol. 449, 719–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arshavsky, V. Y., Lamb, T. D. & Pugh, E. N., Jr. (2002) Annu. Rev. Physiol. 64, 153–187. [DOI] [PubMed] [Google Scholar]
- 30.Fain, G. L., Matthews, H. R., Cornwall, M. C. & Koutalos, Y. (2001) Physiol. Rev. 81, 117–151. [DOI] [PubMed] [Google Scholar]
- 31.Gillespie, P. G. & Beavo, J. A. (1989) Proc. Natl. Acad. Sci. USA 86, 4311–4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bowes, C., Li, T., Danciger, M., Baxter, L. C., Applebury, M. L. & Farber, D. B. (1990) Nature 347, 677–680. [DOI] [PubMed] [Google Scholar]
- 33.Pittler, S. J. & Baehr, W. (1991) Proc. Natl. Acad. Sci. USA 88, 8322–8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferrendelli, J. A. & Cohen, A. I. (1976) Biochem. Biophys. Res. Commun. 73, 421–427. [DOI] [PubMed] [Google Scholar]
- 35.Doshi, M., Voaden, M. J. & Arden, G. B. (1985) Exp. Eye Res. 41, 61–65. [DOI] [PubMed] [Google Scholar]
- 36.Dryja, T. P., Finn, J. T., Peng, Y.-W., McGee, T. L., Berson, E. L. & Yau, K.-W. (1995) Proc. Natl. Acad. Sci. USA 92, 10177–10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dryja, T. P., Adams, S. M., Grimsby, J. L., McGee, T. L., Hong, D. H., Li, T., Andreasson, S. & Berson, E. L. (2001) Am. J. Hum. Genet. 68, 1295–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perrault, I., Rozet, J. M., Calvas, P., Gerber, S., Camuzat, A., Dollfus, H., Chatelin, S., Souied, E., Ghazi, I., Leowski, C., et al. (1996) Nat. Genet. 14, 461–464. [DOI] [PubMed] [Google Scholar]
- 39.van der Spuy, J., Kim, J. H., Yu, Y. S., Szel, A., Luthert, P. J., Clark, B. J. & Cheetham, M. E. (2003) Invest. Ophthalmol. Visual Sci. 44, 5396–5403. [DOI] [PubMed] [Google Scholar]
- 40.McLaughlin, M. E., Sandberg, M. A., Berson, E. L. & Dryja, T. P. (1993) Nat. Genet. 4, 130–134. [DOI] [PubMed] [Google Scholar]
- 41.Piriev, N. I., Yamashita, C., Samuel, G. & Farber, D. B. (1993) Proc. Natl. Acad. Sci. USA 90, 9340–9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin, N. & Baehr, W. (1994) J. Biol. Chem. 269, 3265–3271. [PubMed] [Google Scholar]
- 43.Granovsky, A. E., Natochin, M., McEntaffer, R. L., Haik, T. L., Francis, S. H., Corbin, J. D. & Artemyev, N. O. (1998) J. Biol. Chem. 273, 24485–24490. [DOI] [PubMed] [Google Scholar]
- 44.Akey, D. T., Zhu, X., Dyer, M., Li, A., Sorensen, A., Blackshaw, S., Fukuda-Kamitani, T., Daiger, S. P., Craft, C. M., Kamitani, T. & Sohocki, M. M. (2002) Hum. Mol. Genet. 11, 2723–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hipp, M. S., Raasi, S., Groettrup, M. & Schmidtke, G. (2004) J. Biol. Chem. 279, 16503–16510. [DOI] [PubMed] [Google Scholar]
- 46.Tsang, S. H., Gouras, P., Yamashita, C. K., Kjeldbye, H., Fisher, J., Farber, D. B. & Goff, S. P. (1996) Science 272, 1026–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rieke, F. & Baylor, D. A. (1996) Biophys. J. 71, 2553–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nikonov, S., Lamb, T. D. & Pugh, E. N., Jr. (2000) J. Gen. Physiol. 116, 795–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olshevskaya, E. V., Calvert, P. D., Woodruff, M. L., Peshenko, I. V., Savchenko, A. B., Makino, C. L., Ho, Y. S., Fain, G. L. & Dizhoor, A. M. (2004) J. Neurosci. 24, 6078–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]