

Abstract
In cyanobacteria, KaiC is an essential hexameric clock protein that forms the core of a circadian protein complex. KaiC can be phosphorylated, and the ratio of phospho-KaiC to non-phospho-KaiC is correlated with circadian period. Structural analyses of KaiC crystals identify three potential phosphorylation sites within a 10-Å radius of the ATP binding regions that are at the T432, S431, and T426 residues in the KaiCII domains. When these residues are mutated by alanine substitution singly or in combination, KaiC phosphorylation is altered, and circadian rhythmicity is abolished. These alanine substitutions do not prevent KaiC from hexamerizing. Intriguingly, the ability of KaiC overexpression to repress its own promoter is also not prevented by alanine substitutions at these sites, implying that the capability of KaiC to repress its promoter is not sufficient to allow the clockwork to oscillate. The KaiC structure and the mutational analysis suggest that S431 and T426 may share a phosphate that can shuttle between these two residues. Because the phosphorylation status of KaiC oscillates over the daily cycle, and KaiC phosphorylation is essential for clock function as shown here, daily modulations of KaiC activity by phosphorylation at T432 and S431/T426 seem to be key components of the circadian clockwork in cyanobacteria.
Circadian clocks are self-sustained biochemical oscillators whose properties include temperature compensation and a highly precise time constant of ≈24 h. These properties are difficult to explain by known biochemical reactions. Understanding the mechanism of these unusual oscillators will require characterizing the structures, interactions, modifications, and activities of the molecular components of circadian clocks. The simplest cells that are known to exhibit circadian phenomena are the prokaryotic cyanobacteria (1, 2), where considerable progress has been recently achieved in the identification of essential clock proteins and their structures (3).
A mutational analysis discovered that this system is regulated by at least three essential clock genes, kaiA, kaiB, and kaiC, which form a cluster on the chromosome (4). The three-dimensional structures of the proteins encoded by the kai genes have been determined (3, 5–10). The Kai proteins interact with each other (11, 12) to form large complexes in vivo in which KaiC is the core (13). The KaiC core protein can exist in both phosphorylated and nonphosphorylated forms in vivo (14–16). The ratio of phospho-KaiC to non-phospho-KaiC is correlated with the period of the oscillation (16). However, it is not known whether this phosphorylation is essential for proper clock function.
The addition of ATP to purified KaiC in vitro initiates a KaiC autophosphorylation (5, 14, 15), and it also stimulates the formation of hexameric KaiC ring complexes (17, 18), which is consistent with the observation of KaiC in high molecular weight complexes in vivo (13). KaiC autophosphorylation is stimulated in vitro by KaiA, whereas KaiB antagonizes the effects of KaiA on KaiC autophosphorylation (5, 15, 16, 19). KaiC will also dephosphorylate in vitro, implying that KaiC will continuously turn over its phosphorylation status in the presence of ATP (16). We found that dephosphorylation of KaiC in vitro is inhibited by KaiA, and this effect of KaiA is also antagonized by KaiB; therefore, the apparent KaiA stimulation of KaiC autophosphorylation might be due to an inhibition of dephosphorylation (16). The in vitro results are relevant to events happening within cells: in vivo, KaiA stabilizes KaiC in the phosphorylated form, and KaiB antagonizes the effect of KaiA (15, 16). Not only is KaiC phosphorylated in vivo, but the proportion of phospho-KaiC to non-phospho-KaiC oscillates over the circadian cycle (15, 16, 20).
KaiC seems to be predominantly phosphorylated on threonine and serine residues (15), but identification of the specific residues involved has not yet been reported. Nishiwaki et al. (21) state in a companion article published in this issue of PNAS that a mass spectrometric assay of KaiC indicates that the S431 and T432 residues within KaiC are phosphorylated and that mutation of these residues reduces KaiC phosphorylation and causes arrhythmicity. We found by analyses of the KaiC crystal structure these two phosphorylation sites and in addition we found another putative phosphorylation site at T426. We report here that mutation of these threonine and serine residues to alanine residues alters the KaiC phosphorylation profile and abolishes rhythmicity. Therefore, we conclude that T432 and the S431/T426 doublet are phosphorylation sites that are essential to clock function in cyanobacteria.
Materials and Methods
Crystallographic Refinement of Phospho-KaiC. Coordinates of dephospho-KaiC (Protein Data Bank code 1TF7; ref. 10) in combination with native KaiC diffraction data were used to calculate difference (Fo – Fc) Fourier electron density maps that were displayed at various thresholds (2.5σ to 5σ range). Difference electron densities around T and S residues of all six subunits, and in particular those located within a sphere of a 10-Å radius around the γ-phosphate group of ATP molecules in the CI and CII domains, were then visually inspected. A strong difference electron density peak (5σ level) was observed at <2 Å from the γ-OH of T432 residues in all six subunits, and a somewhat weaker peak (4σ level) was present at <2 Å from the γ-OH groups of S431 residues in four subunits (A, B, E, and F). In addition, these second peaks were located at ≈3.5 Å from the γ-oxygens of T426 residues.
Subsequently, phosphate groups were placed at 1.6 Å from the γ-oxygens of all six T432 and the four S431 residues, the topology and parameter dictionary files were updated, and several cycles of positional and B factor refinement were carried out with the program cns (22). In addition to phosphates at T432 and S431 from six and four subunits, respectively, a single Mg2+ ion per KaiCII domain was included in the refinement. The final Rwork and Rfree (for 5% randomly selected reflections) for all 79,746 data in the 30.0- to 2.8-Å resolution range are 0.255 and 0.289, respectively. The root mean square deviations from standard values for bond lengths and bond angles are 0.009 Å and 1.5°, respectively.
Experimental Strains, KaiC Mutagenesis, and Rhythm Assay. Growth conditions of cyanobacterial strains have been described (16). The QuikChange XL Site-Directed Mutagenesis system (Stratagene) was applied to mutagenize single, double, or triple residues at T426, S431, or T432 of KaiC from threonine or serine to alanine, as shown in Table 1. In cyanobacteria, the expression of different KaiCs was driven by the trc promoter (trcp) in a kaiC-null strain with a kaiBCp::luxAB reporter (16); in Escherichia coli (BL21/DE3), the expression of various KaiCs with a His-6 tag fused to the C terminus was driven by T7p.
Table 1. Mutagenized KaiCs and cyanobacterial strains.
| KaiC mutagenesis*
|
|||
|---|---|---|---|
| Strain | KaiC version | Residue | Nucleotide† |
| WT | KaiCWT | none | none |
| T426A | KaiCT426A | Thr426→Ala | 1276ACT1278→GCT |
| S431A | KaiCS431A | Ser431→Ala | 1291TCA1293→GCA |
| T432A | KaiCT432A | Thr432→Ala | 1294ACA1296→GCA |
| TSa | KaiCTSa | Thr426→Ala | 1276ACT1278→GCT |
| Ser431→Ala | 1291TCA1293→GCA | ||
| Thr426→Ala | 1276ACT1278→GCT | ||
| TSTa | KaiCTSTa | Ser431→Ala | 1291TCA1293→GCA |
| Thr432→Ala | 1294ACA1296→GCA | ||
Residue and nucleotide positions are those relative to the translation start site of KaiC.
Altered nucleotides are in bold.
For rhythm measurement, colonies grown on agar plates for 5 days at 30°C in constant light (LL) were synchronized with a 12-h dark treatment before releasing the cultures into free-running LL conditions to monitor the luminescence of a kaiBCp::luxAB reporter strain (16). For observation of the effect of trcp-driven KaiC on rhythmicity, isopropyl β-d-thiogalactoside (IPTG) was applied under the agar medium on the third day of the rhythm assay (final IPTG concentration of 0, 2, or 32 μM) and the assay was continued for 4 more days.
KaiC Phosphorylation Assay. Cyanobacterial cultures were grown to an OD750 of 0.2. After a 12-h dark pulse, the cultures were treated with 10 μM IPTG for 3 h at 30°C in light with air bubbling, and ≈40 ml of the cultures were collected for preparation of total extracts. Immunoblot analysis for KaiC was performed on 5 μg of total proteins per lane according to the previous description (20), except that we used a highly specific mouse polyclonal antibody to KaiC and a SuperSignal West Pico Chemiluminescent Substrate (Pierce). The phosphorylated KaiC signals were quantified as before (16) and analyzed by Student's t test.
In Vivo KaiC Hexamer Detection. The in vivo hexamerization of wild-type or mutant KaiCs was assayed by native-gel immunoblots in the presence of ATP. Extracts of cyanobacterial strains expressing wild-type or mutant KaiCs collected at circadian time 3 (CT3) were prepared in the following buffer: 50 mM Hepes (pH 7.4), 137 mM KCl, 10% glycerol, 1 mM EDTA, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 5 μg/ml pepstatin A, 1 mM phenylmethylsulfonyl fluoride, and 5 mM ATP. Five micrograms of total protein was resolved in 7.5% nondenaturing PAGE with standard running buffer but without SDS, and 1 mM ATP was added to both gel and running buffers. The gels were immunoblotted and analyzed as described above to detect the oligomer forms of the various KaiCs.
Results and Discussion
Crystal Structure of KaiC from Synechococcus elongatus. We recently reported the crystal structure of the full-length core protein from the cyanobacterial circadian clock system, the KaiC homohexamer, at 2.8-Å resolution (10). The structure assumes the shape of a double doughnut with approximate dimensions of 100 by 100 Å (Fig. 1A). Twelve ATP molecules are bound at the interfaces between the subunits in both KaiCI and KaiCII domains (Fig. 1 A and B). The central channel has a diameter of ≈22 Å but is somewhat constricted at the C-terminal end and partly sealed by arginine residues from six subunits that protrude into the cavity. The narrower waist region demarcates the junction between the KaiCI and KaiCII halves from individual subunits. The 245-residue core domains of CI and CII exhibit a very similar fold that resembles the central portion of RecA from E. coli (23). This finding is consistent with the earlier observation that the kaiC gene seems to be an internally duplicated version of a recA/dnaB-like gene (11, 24). The overall fold of the core of the KaiCI and KaiCII domains consists of a mostly parallel-stranded twisted β-sheet with seven strands that is surrounded by α-helices (Fig. 1C).
Fig. 1.

Structure of KaiC and phosphorylation sites. (A) Ribbon diagram of the overall structure of the KaiC homo-hexamer with bound ATP molecules shown in a van der Waals representation. The CI and CII domains are labeled, and carbon, oxygen, nitrogen, and phosphorus atoms of ATPs are colored gray, red, blue, and yellow, respectively. Phosphate groups of residues T432 in all subunits are highlighted as orange spheres, and phosphates of residues S431 in subunits A, B, E, and F are highlighted as black spheres. (B) The intersubunit ATP binding site. Portions of the KaiCII domains from adjacent subunits A and F (in parentheses) are shown in a transparent surface representation (colored gray and cyan, respectively). Side chains of T426, S431, and T432, as well as residues interacting (directly or by means of Mg2+) with the triphosphate moiety of ATP, are shown in a stick representation and are labeled. The Mg2+ ion is depicted as a red sphere, and its coordination sphere is indicated with thin solid lines. Distances between the phosphorus atoms of ATP (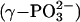 ) and the phosphates of T432 and S431 are highlighted. (C) Secondary and tertiary structure of the KaiCII monomer with the bound ATP molecule. Residues T426, S431, and T432 are located in an extended loop region (gray ribbon) that links the β7 and β8 strands and features a single α-helical turn.
) and the phosphates of T432 and S431 are highlighted. (C) Secondary and tertiary structure of the KaiCII monomer with the bound ATP molecule. Residues T426, S431, and T432 are located in an extended loop region (gray ribbon) that links the β7 and β8 strands and features a single α-helical turn.
Both KaiCI and KaiCII contain shared regions that include Walker A (P loop) and B motifs that are often involved in ATP/GTP binding and hydrolysis (Fig. 2A). Indeed, when the Walker A motifs in KaiC are mutated, ATP nucleotide binding is eliminated and rhythmicity is severely disrupted or abolished (14, 25). KaiC contains a conserved glutamate (E78/E79 and E318/E319 in the CI and CII halves, respectively; Fig. 2 A) that has been proposed, based on the structure of RecA, to activate the water nucleophile for in-line attack of the ATP γ-phosphate (23, 24). RecA itself forms helical filaments (23, 26), but also hexameric ring structures of unknown function (27).
Fig. 2.

Consequences of phosphorylation for KaiCII intersubunit organization. (A) Sequence of KaiC from S. elongatus with the KaiCI (top line) and KaiCII (bottom line) domains aligned. Residues 1–13 and 498–519 including the C-terminal His-6 tag (residues 520–525) are disordered in the crystal structure. Amino acids that form part of the Walker A and B motifs are red/blue and boxed; serines and threonines that lie within a 10-Å sphere from the ATP γ-phosphates in the KaiCI and KaiCII domains (P··· Cα distance) are green and are numbered. Serines and threonines contained in the Walker A and B motifs and located within this sphere are blue, and amino acids forming the linker between the CI and CII domains are underlined. (B) Example of the final Fourier 2Fo – Fc sum electron density (blue; 1.5σ level) with the super-imposed Fo – Fc difference electron density (red; 3.5σ level before incorporation of phosphate groups) for S431 and T432 in the F subunit. Only residues 425–433 of the loop region connecting the KaiCII β7 and β8 strands are shown, and selected residues are labeled. (C) Intersubunit interactions as a result of phosphorylation of T432 and S431. Carbon atoms of residues from the A and F subunits are colored gray and cyan, respectively, selected residues and secondary structural elements are labeled, and hydrogen bonds are drawn as thin dashed lines.
The crystal structure also revealed differences between the ATP binding modes in the KaiCI and KaiCII halves (10). At the KaiCI domain subunit interfaces, the nucleobase of ATP is contacted by the side chains of three amino acids and is thus in a tight grip. By comparison, the triphosphate portion is relatively loosely bound, and the γ-phosphate exhibited a disorder between two main conformations. In contrast, no direct hydrogen bonding contacts are established between amino acid side chains and the ATP nucleobase at the subunit interface between KaiCII domains. However, in KaiCII, all three phosphate groups are firmly held in place by extensive interactions to amino acid side chains from both subunits at the interface.
Structure-Based Identification of Phosphorylation Sites. For the crystallographic structure analysis, KaiC protein from S. elongatus was expressed in E. coli and purified in the presence of ATP. Before the crystallization trials, ATP was removed and replaced by ATPγS (10). The protein was found to be a mixture of the dephospho- and phospho-forms by SDS/PAGE analysis, so it was quite reasonable to expect that the crystal structure might disclose potential phosphorylation sites. Indeed, difference Fourier density potentially accounting for a phosphate group appeared in the vicinity of residues T432 in all six subunits during the final cycles of refinement (10), but no additional refinement with phosphorylated threonines was carried out.
We have now carried out a detailed analysis of the (Fo – Fc) difference electron density present in the final dephospho-KaiC structure (see Materials and Methods). In addition to T432, the structural data also identify S431 as a putative phosphorylation site in four subunits. Residues S431 and T426 face each other in a loop region that connects the β7 and β8 strands (Fig. 1C). Although it is clear from the difference electron density that it is the S431 side chain that is covalently bound to a phosphate group in the crystal (Fig. 2B), inserting the phosphate at 1.6 Å from the γ-oxygen of S431 brings the γ-oxygen of T426 in hydrogen bonding distance from the phosphate (Fig. 2C and Table 2). We speculated that this phosphate group could possibly shuttle between the two residues and therefore also included T426 in the mutational analysis (see below). The crystallographic refinement was continued with phosphorylated T432 (in all subunits) and S431 residues (in four subunits). An example of the final Fourier (2Fo – Fc) sum electron density in the region of the three threonine and serine residues is depicted in Fig. 2B. The difference density maps also showed a strong peak near the β- and γ-phosphate groups of ATP. Because additional potentially inner-sphere contacts between the peak and the side chains of residues T295, E318, E319, and D378 could be established, it was interpreted as a Mg2+ ion and subsequently included in the refinement of all six subunits. The coordination geometry of the Mg2+ ions at the subunit interface is shown in Fig. 1B. It is noteworthy that inspection of difference electron density maps around the KaiCI halves did not reveal any strong peaks that could be taken as evidence for either phosphorylated threonine or serine residues.
Table 2. Hydrogen bonding and van der Waals interactions at subunit interfaces as a result of phosphorylation.
| Residue* | Atom | Residue and atom* | Distance, Å |
|---|---|---|---|
| pT432 (A) | O1P | S381 (F) O | 4.1 |
| S381 (F) OG | 4.5 | ||
| O2P | R385 (F)NH2 | 3.0 | |
| O3P | R385 (F)NH2 | 3.1 | |
| E318 (F) OE2 | 2.7 | ||
| S379 (F) OG | 4.0 | ||
| pS341 (A) | O1P | D417 (F) OC | 3.8 |
| O2P | T426 (A)OG1 | 2.6 | |
| O3P | T426 (A)OG1 | 3.0 | |
| H429(A)ND1 | 3.5 | ||
| Wat78 | O | T432 (A) O1P | 3.6 |
| T431 (A) O3P | 3.6 | ||
| D417 (F)OD1 | 2.8 |
Distances vary somewhat depending on the particular subunit pair.
Letters in parentheses designate the subunit.
Consequences of S431 and T432 Phosphorylation for the Subunit Interface in KaiC Hexamers. The presence of phosphate groups at S431 and in particular at T432 leads to additional interactions between amino acids from adjacent subunits (Fig. 2C). In the case of T432, intersubunit contacts are newly established to R385 and E318 by means of the phosphate group (Table 2). The effect is somewhat subtler for S431 where the addition of a phosphate creates a hydrogen bond to H429 (Fig. 2C). This histidine crosses the interface and interacts with D427 from the adjacent subunit. A further interaction of the S431 phosphate is the aforementioned hydrogen bond to the γ-hydroxyl group of T426. These two residues are not in hydrogen bonding contact in the dephospho-KaiC structure. The newly established contacts as a result of phosphorylation of S431, although not directly linking residues across the subunit interface, are likely to exert a stabilizing effect on existing interactions by tying them into a network of hydrogen bonds. Moreover, in some subunits, a water molecule is observed to bridge the phosphate groups of residues T432 and S431 (Table 2). A reasonable conclusion with regard to the observed changes at the subunit interface as a result of S431 and T432 phosphorylation is that the presence of phosphates results in tighter binding between adjacent KaiCII domains. This effect is particularly evident in the case of T432 residues where phosphorylation creates an intersubunit salt bridge (Fig. 2C).
Alanine Substitutions at T432, S431, and T426 Alter KaiC Phosphorylation in Vivo. To test whether the putative phosphorylation sites at T432, S431, and T426 are essential for circadian rhythmicity, mutations by alanine substitution were made at these sites as individual, double (S431/T426), and triple (T432/S431/T426) replacements (Table 1). KaiC's phosphorylation status in vivo can be determined from SDS/PAGE and immunoblotting. As has been shown previously, nonphosphorylated KaiC has a faster mobility in SDS/PAGE than do the various forms of phosphorylated KaiC (15, 16). This characteristic results in upward band shifts as KaiC is phosphorylated. Fig. 3 shows that the KaiC mutations described in Table 1 affect KaiC phosphorylation. Cyanobacterial extracts collected from the KaiC-null strain (ΔKaiC) do not react with our antibody. Approximately 20% of wild-type KaiC (KaiCWT) is present in the upper bands (phosphorylated forms) by the KaiC expression assay in cyanobacteria we described previously (16). The T432A and T426A mutations significantly reduce the amount of phosphorylated KaiC, but the S431A mutation seems to increase the ratio of phospho-KaiC/nonphospho-KaiC. The S431A/T426A double mutation has an amount of phosphorylation that is roughly equivalent to that of wild-type KaiC, whereas the triple T432A/S431A/T426A mutant is essentially nonphosphorylated.
Fig. 3.

Mutation of key phosphorylation sites alters the phosphorylation status of KaiC. (A) In vivo KaiC phosphorylation profiles in the kaiC-null cyanobacteria (ΔKaiC) expressing trcp-driven wild-type or mutagenized KaiCs (Table 1). After a 3-h pulse of the inducer IPTG in light starting at circadian time 0 (CT0), the expression patterns of KaiC proteins were determined by immunoblotting with mouse anti-KaiC antibody. Filled arrows indicate phosphorylated KaiC bands, and the open arrow denotes nonphosphorylated KaiC bands. (B) Densitometry of the phospho-KaiC levels in various strains, normalized to the density of the phospho-KaiCS431A band. The values are mean ± SD; asterisks denote significant differences vs. wild type (*, P < 0.05; **, P < 0.01).
These SDS/PAGE profiles clearly indicate that mutating these three residues will significantly alter phosphorylation of KaiC that is expressed in cyanobacteria. We do not believe that these three residues are the only residues that can be phosphorylated on KaiC; there is very likely to be phosphorylation at a variety of KaiC residues. However, based on the structural analyses of crystalline KaiC, these are the only three residues that seem to be reproducibly autophosphorylated in vitro. Moreover, when T432, S431, and T426 are all mutated to alanine, KaiC expressed in cyanobacteria seems incapable of phosphorylation anywhere. T432 stands out as a key site, because mutation at this single site abolishes most of the phosphorylation. The T426A mutation reduces phosphorylation significantly, but less than the alanine mutation at T432. Intriguingly, the S431A mutation significantly increases KaiC phosphorylation, implying that the presence of serine at this site might inhibit the phosphorylation of KaiC at other sites.
Alanine Substitutions at T432, S431, or T426 Abolish Circadian Rhythmicity. Cyanobacteria are arrhythmic when the kaiC gene is deleted (4). When wild-type KaiC (KaiCWT) is expressed at an appropriate level in a kaiC-null strain of cyanobacteria under the control of an IPTG-inducible promoter (trcp), rhythmicity is restored (16). For example, in ΔkaiC/trcp::kaiC strains, rhythmicity is evident when IPTG is absent because trcp is slightly leaky (16). At low concentrations (0–8 μM) of IPTG that allow appropriate levels of KaiCWT expression, rhythmicity is allowed, as can be assessed with a luminescence reporter of kaiBC promoter (kaiBCp) activity (16) (Fig. 4). At higher levels of KaiCWT expression, the activity of kaiBCp is repressed and arrhythmic, indicative of negative feedback regulation (4). Expression of KaiCs mutated at T432, S431, or T426 will not, however, allow rhythmicity to be restored at any concentration of IPTG (0, 2, and 32 μM IPTG are shown in Fig. 4). This finding means that the capacity to be phosphorylated at each of these three sites is essential for proper clock function in cyanobacteria. The S431A mutation seems to enhance KaiC phosphorylation (Fig. 3), but it also disrupts rhythmicity (Fig. 4), implying the importance of the proper balance of phosphorylation. The double (S431A/T426A) and triple (T432A/S431A/T426A) mutants are also arrhythmic (data not shown).
Fig. 4.

Mutation of potential phosphorylation residues of KaiC abolishes circadian rhythmicity. After a 12-h synchronizing dark pulse, colonies of kaiC-null cyanobacteria harboring the constructs for trcp-driven wild-type KaiC (WT) or mutagenized KaiCs (T426A, S431A, or T432A) were released to constant light (LL) for measurement of kaiBCp-driven luminescence. After 72 h in LL, the inducer IPTG (0, 2, or 32 μM) was added as shown by the orange dashed lines. Heavy solid lines indicate the average luminescence traces, and red bars denote the SD (n = 3 for each trace).
Surprisingly, however, these mutant KaiCs can still function to repress the overall activity of the kaiBC promoter, as shown by the dramatic decrease of luminescence after 32 μM IPTG administration (Fig. 4). This result implies that feedback repression of KaiC onto its promoter is not sufficient to allow the clockwork to oscillate in cyanobacteria. Apparently the capability of KaiC to repress kaiBCp activity is dissociable from more subtle effects of KaiC that are enabled by phosphorylation on T432 and S431 (and T426; see below). Finally, because the T432/S431/T426 phosphorylations were found in the crystal structure after expression of KaiCWT in E. coli and autophosphorylation of purified KaiCWT in vitro, the observations that alanine mutations at these sites reduce phosphorylation in cyanobacteria (Fig. 3) and prevent an oscillating clock (Fig. 4) mean that these phosphorylations are not artifacts of in vitro autophosphorylation, but are also important in cyanobacteria in situ.
Alanine Substitutions at T432, S431, and T426 Do Not Hinder Hexamerization of KaiC. As shown previously (17), KaiC hexamerization in the presence of ATP can be determined by electrophoresis on native gels. We expressed KaiCWT and the alanine mutants of KaiC in cyanobacterial cells and analyzed their ability to form hexamers by electrophoresis of these cells' extracts in native gels containing ATP. As depicted in Fig. 5, all of the alanine mutants of KaiC have the capability to form hexamers in cyanobacterial cells. Two different forms of hexamers are distinguished on these native gels that are probably due to varying degrees of phosphorylation, with the upper hexamer band (HU) being more highly phosphorylated. The relative level of these forms is quantified in Fig. 5B as the relative density of the lower hexamer band (HL) as a percentage of the total KaiC hexamers (HU + HL).
Fig. 5.

Mutations of key phosphorylation sites do not affect the hexamerization of KaiC but alter the profiles of the KaiC hexamers. (A) Native-gel immunoblotting assay for wild-type or mutagenized KaiCs in cyanobacterial extracts collected at circadian time 3 (CT3). No KaiC monomer was detected in any strain under the conditions described in Materials and Methods. The predicted approximate positions of the KaiC monomer (M) and two distinct hexamer bands (upper = HU, lower = HL) are indicated by arrows. (B) Ratio of the lower hexamer band (HL) to the total hexamer population (HU + HL) as determined by densitometry. Data are mean ± SD.
Phosphorylations Within KaiC Affect Subunit Interactions and Circadian Rhythmicity. Intersubunit interactions of KaiC are favorable from an electrostatic perspective (10). From our current analyses of the KaiC structure, phosphorylation at the T432 and S431 residues will be expected to enhance binding between adjacent KaiCII domains. This interpretation is especially likely in the case of the phosphorylation at T432 that adds an intersubunit salt bridge. The alanine substitutions that we tested do not prevent hexamerization (Fig. 5). Therefore, phosphorylation at T432 and S431 is essential for circadian oscillations, but is not necessary for KaiC hexamerization or its ability to repress its own promoter's activity (Fig. 4).
What about T426? It is unlikely that the arrhythmicity evoked by the T426A mutation (Fig. 4) is simply due to the inability of the alanine at position 426 to form a hydrogen bond to the phosphate group of S431. Rather, T426 may constitute an alternative phosphorylation site to S431. Our structural analysis is based on the state of KaiC as it was trapped in the crystal structure, and it might be true that crystallization favored the attachment of phosphate to S431. If so, our crystal structure might be merely one possible phospho-KaiC form among several others. When the S431 residue was mutated to alanine, the overall phosphorylation of KaiC seemed to increase (Fig. 3). This result suggests that phosphorylation at S431 inhibits phosphorylation at other sites, possibly at T426 and elsewhere. Perhaps phosphate can shuttle between S431 and T426. Even more speculatively, maybe the phosphate oscillates between these two residues over the circadian cycle, thereby contributing to the rhythmic modulation of KaiC activity.
The phosphorylation status of KaiC is known to oscillate over the daily cycle, with more phospho-KaiC in the night phase (15). Moreover, the ratio of phospho-KaiC/nonphospho-KaiC is correlated with the period of the cyanobacterial clock (16). Our study shows that KaiC phosphorylation is essential to clock function, because mutations of residues identified from structural analyses that alter or abolish phosphorylation also eliminate rhythmicity (but intriguingly, not kaiBCp repression). We previously suggested a model wherein the activity of a KaiC-containing complex is modulated by KaiC phosphorylation mediated by KaiA and KaiB (2, 16). Because the phosphorylation status of KaiC oscillates over the daily cycle, and KaiC phosphorylation is essential for clock function (this study), daily changes in the phosphorylation status of T432 and S431/T426 seem to be key components of the circadian clockwork in cyanobacteria (2, 15, 16).
Acknowledgments
We thank Drs. H. Yan and R. Mernaugh for their assistance with the anti-KaiC antibody, and Drs. Wayne Anderson, Kathleen Gould, Ronald Laskey, and D. Martin Watterson for discussions. This work was supported by National Institutes of Health (NIH) Grant GM67152 and National Science Foundation Grant MCB-9874371 (to C.H.J.), in part by NIH Grant GM55237 (to M.E.), and by a Vanderbilt University Medical Center Discovery grant (to M.E. and C.H.J.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: trcp, trc promoter; IPTG, isopropyl β-d-thiogalactoside.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 1U9I).
See Commentary on page 13697.
References
- 1.Ditty, J. L., Williams, S. B. & Golden, S. S. (2003) Annu. Rev. Genet. 37, 513–543. [DOI] [PubMed] [Google Scholar]
- 2.Johnson, C. H. (2004) Curr. Issues Mol. Biol. 6, 103–110. [PubMed] [Google Scholar]
- 3.Johnson, C. H. & Egli, M. (2004) Nat. Struct. Mol. Biol. 11, 584–585. [DOI] [PubMed] [Google Scholar]
- 4.Ishiura, M., Kutsuna, S., Aoki, S., Iwasaki, H., Andersson, C. R., Tanabe, A., Golden, S. S., Johnson C. H. & Kondo T. (1998) Science 281, 1519–1523. [DOI] [PubMed] [Google Scholar]
- 5.Williams, S. B., Vakonakis, I., Golden, S. S. & LiWang, A. C. (2002) Proc. Natl. Acad. Sci. USA 99, 15357–15362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vakonakis, I., Sun, J., Wu, T., Holzenburg, A., Golden, S. S. & LiWang, A. C. (2004) Proc. Natl. Acad. Sci. USA 101, 1479–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye, S., Vakonakis, I., Ioerger, T. R., LiWang, A. C. & Sacchettini, J. C. (2004) J. Biol. Chem. 279, 20511–20518. [DOI] [PubMed] [Google Scholar]
- 8.Garces, R. G., Wu, N., Gillon, W. & Pai, E. F. (2004) EMBO J. 23, 1688–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uzumaki, T., Fujita, M., Nakatsu, T., Hayashi, F., Shibata, H., Itoh, N., Kato, H. & Ishiura, M. (2004) Nat. Struct. Mol. Biol. 11, 623–631. [DOI] [PubMed] [Google Scholar]
- 10.Pattanayek, R., Wang, J., Mori, T., Xu, Y., Johnson, C. H. & Egli, M. (2004) Mol. Cell 15, 375–388. [DOI] [PubMed] [Google Scholar]
- 11.Iwasaki, H., Taniguchi, Y., Kondo, T. & Ishiura, M. (1999) EMBO J. 18, 1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taniguchi, Y., Yamaguchi, A., Hijikata, A, Iwasaki, H., Kamagata, K., Ishiura, M., Go, M. & Kondo, T. (2001) FEBS Lett. 496, 86–90. [DOI] [PubMed] [Google Scholar]
- 13.Kageyama, H., Kondo, T. & Iwasaki, H. (2003) J. Biol. Chem. 278, 2388–2395. [DOI] [PubMed] [Google Scholar]
- 14.Nishiwaki, T., Iwasaki, H., Ishiura, M. & Kondo, T. (2000) Proc. Natl. Acad. Sci. USA 97, 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwasaki, H., Nishiwaki, T., Kitayama, Y., Nakajima, M. & Kondo, T. (2002) Proc. Natl. Acad. Sci. USA 99, 15788–15793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu, Y., Mori, T. & Johnson, C. H. (2003) EMBO J. 22, 2117–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mori, T., Saveliev, S. V., Xu, Y., Stafford, W. F., Cox, M. M., Inman, R. B. & Johnson, C. H. (2002) Proc. Natl. Acad. Sci. USA 99, 17203–17208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayashi, F., Suzuki, H., Iwase, R., Uzumaki, T., Miyake, A., Shen, J.-R., Imada, K., Furukawa, Y., Yonekura, K., Namba, K., et al. (2003) Genes Cells 8, 287–296. [DOI] [PubMed] [Google Scholar]
- 19.Kitayama, Y., Iwasaki, H., Nishiwaki, T. & Kondo, T. (2003) EMBO J. 22, 1–8.12505979 [Google Scholar]
- 20.Xu, Y., Mori, T. & Johnson, C. H. (2000) EMBO J. 19, 3349–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishiwaki, T., Satomi, Y., Nakajima, M., Lee, C., Kiyohara, R., Kageyama, H., Kitayama, Y., Temamoto, M., Yamaguchi, A., Hijikata, A., et al. (2004) Proc. Natl. Acad. Sci. USA 101, 13927–13932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M. & Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 23.Story, R. M., Weber, I. T. & Steitz, T. A. (1992) Nature 355, 318–325. [DOI] [PubMed] [Google Scholar]
- 24.Leipe, D. D., Aravind, L., Grishin, N. V. & Koonin, E. V. (2000) Genome Res. 10, 5–16. [PubMed] [Google Scholar]
- 25.Mori, T. & Johnson, C. H. (2001) Sem. Cell Dev. Biol. 12, 271–278. [DOI] [PubMed] [Google Scholar]
- 26.VanLoock, M. S., Yu, X., Yang, S., Lai, A. L., Low, C., Campbell, M. J. & Egelman, E. H. (2003) Structure 11, 1–20. [DOI] [PubMed] [Google Scholar]
- 27.Yu, X. & Egelman, E. H. (1997) Nat. Struct. Mol. Biol. 4, 101–104. [DOI] [PubMed] [Google Scholar]