

Abstract
Change in redox status has long been known to link light to the posttranslational regulation of chloroplast enzymes. So far, studies have been conducted primarily with thioredoxin-linked members of the stroma that function in a broad array of biosynthetic and degradatory processes. Consequently, little is known about the role of redox in regulating the growing number of enzymes found to occur in the lumen, the site of oxygen evolution in thylakoid membranes. To help fill this gap, we have studied AtFKBP13, an FKBP-type immunophilin earlier shown to interact with a redox-active protein of the lumen, and found the enzyme to contain a pair of disulfide bonds in x-ray structural studies. These disulfides, which in protein mutagenesis experiments were shown to be essential for the associated peptidyl-prolyl isomerase activity, are unique to chloroplast FKBPs and are absent in animal and yeast counterparts. Both disulfide bonds were redox-active and were reduced by thioredoxin from either chloroplast or bacterial sources in a reaction that led to loss of enzyme activity. The results suggest a previously unrecognized paradigm for redox regulation in chloroplasts in which activation by light is achieved in concert with oxygen evolution by the oxidation of sulfhydryl groups (conversion of SH to S—S). Such a mechanism, occurring in the thylakoid lumen, is in direct contrast to regulation of enzymes in the stroma, where reduction of disulfides targeted by thioredoxin (S—S converted to SH) leads to an increase in activity in the light.
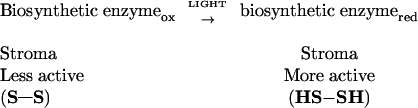
Evidence accumulated during the past three decades has shown that chloroplast enzymes are differentially regulated by light in a manner compatible with their function (1, 2). Thus, biosynthetic enzymes, such as regulated members of the Calvin cycle, are posttranslationally converted from a less active disulfide (S—S) state in the dark to a more active sulfhydryl (SH) state by light (Eq. 1). By contrast, other such enzymes, e.g., those linked to carbohydrate degradation, behave in the opposite manner, i.e., they are converted from a more active (S—S) state in the dark to a less active (SH) state in the light (Eq. 2).
 |
[1] |
 |
[2] |
In this way, chloroplasts use light to maximize available resources by directing biochemical processes diurnally in an effective manner. The ferredoxin/thioredoxin system, composed of ferredoxin, ferredoxin-thioredoxin reductase, and a thioredoxin, facilitates these redox changes in target enzymes that so far are either located in, or exposed to, the stroma.
Recent advances in proteomics and genomics have led to the unexpected finding that the lumen, the site of oxygen evolution in chloroplast thylakoids, contains numerous enzymes in addition to those directly associated with the light reactions (3–7). Prominent among the lumen inhabitants are more than a dozen immunophilins (4–6, 8, 9), proteins originally defined as receptors for immunosuppressive drugs (10) that were later found to have peptidyl-prolyl cis-trans isomerase (PPIase) activity (11, 12). Our recent study on AtFKBP13 (Protein Data Bank ID code 1U79), a representative of the FKBP immunophilin group and a resident of the chloroplast thylakoid lumen, demonstrated an interaction between the precursor form and the Rieske protein, a luminal participant in photosynthetic electron transfer (4). Coupled with evidence that a stromal cyclophilin ROC4, now referred to as AtCYP20-3 (8, 13), is activated by thioredoxin (14), this finding raised the question of whether the regulation of AtFKBP13 could be linked to redox. We have addressed the question by determining first the structure of AtFKBP13 and then its response to redox agents.
The 3D structure of AtFKBP13 revealed a unique pair of disulfide bonds that are absent in animal homologues (Cys-5,17 and Cys-106,111). Further, thioredoxins from both chloroplasts (m-type) and Escherichia coli specifically reduced both disulfides in a reaction that was accompanied by the loss of PPIase activity. That evidence, together with similar results obtained with derivatives harboring cysteine → serine mutations, suggests that luminal AtFKPB13 resembles a stromal cyclophilin counterpart, AtCYP20-3, in undergoing thiol redox modulation (14). The lumen compartment appears to differ from the stroma, however, in one fundamental respect: activation of a resident immunophilin is achieved by oxidation (conversion of 2SH to S—S) rather than reduction (S—S to 2SH) as for AtCYP20-3 and many other biosynthetic enzymes. The results support the view that enzymes of the lumen, such as AtFKPB13, are activated by oxidation in the light (compare Eqs. 1 and 3). The evidence further suggests that the response of a regulatory enzyme to redox change depends on the redox milieu of the host compartment.
 |
[3] |
Materials and Methods
Protein Expression and Purification. The AtFKBP13 gene was cloned into the pGEX-KG vector and the GST-fusion protein was overexpressed in E. coli BL21 (DE3) cells as described (4). In brief, the cells were grown in LB medium to an OD600 of 0.6 at 37°C and expression of the recombinant protein was induced for 6 h with 0.5 mM isopropyl-d-thiogalactopyranoside at 30°C. Cells were harvested by centrifugation at 4,200 × g for 10 min at 4°C and were resuspended in ice-cold lysis buffer (20 mM Tris·HCl, pH 7.5/0.5 M NaCl/1 mM DTT). After sonication, the crude lysate was centrifuged at 42,400 × g for 1 h at 4°C and the supernatant fraction was applied to a GST-affinity column. The GST-AtFKBP13 fusion protein was eluted with 50 mM Tris·HCl (pH 7.5) containing 10 mM reduced glutathione. The fusion protein was cleaved by thrombin and dialyzed in 20 mM Tris·HCl (pH 7.5)/0.5 M NaCl with a 3,500 molecular weight cutoff dialysis tube (Spectra/Por, Spectrum Laboratories, Houston). Thrombin and the cleaved GST were removed by passing the preparation sequentially through 5-ml HiTrap benzamidine FF (Pharmacia) and 5-ml GSTrap FF columns. The final purification step was achieved by gel filtration on a HiLoad 16/60 Superdex-75 column (Pharmacia) previously equilibrated with buffer solution containing 50 mM Tris·HCl (pH 7.5) and 150 mM NaCl. The purified protein was analyzed by SDS/PAGE, native PAGE, and matrix-assisted laser desorption ionization–time-of-flight MS. The molecular weight was determined to be 13,656 ± 1.07 and the protein used for crystallography was >95% pure. Dynamic light-scattering data showed the protein to be a monomer.
Crystallization, Data Collection, and Structure Determination. The AtFKBP13 protein was crystallized by the hanging-drop vapordiffusion method (15) at 20°C. Single crystals were grown in 8–11% PEG 550MME/2.5 M ammonium sulfate/100 mM Tris (pH 7.9) within 2 weeks. Single crystals were rapidly swept through mother liquor containing 20% (vol/vol) glycerol as a cryoprotectant and were flash-cooled in liquid nitrogen at –80°C. The diffraction data were collected by using synchrotron radiation at the X12-B beam line, National Synchrotron Light Source, Brookhaven National Laboratory (Upton, NY) with an ADSC Q315 charge-coupled device detector (Area Detector Systems Corporation, Poway, CA). All diffraction intensities were integrated and scaled by using the hkl software package (16). The crystal belongs to the orthorhombic space group C2221 with unit-cell parameters a = 89.02, b = 126.60, c = 119.40 Å. The Matthews coefficient (17) suggested that the asymmetric unit contained between four and six molecules (Mr = 14,000). The structure was determined by the molecular replacement method with yeast FKBP (Protein Data Bank ID code 1YAT) as the search model (18, 19). Structure refinements were carried out with cns (20) and model building was performed with the program o (21). No residue of AtFKBP13 in the final model is present in the disallowed region of the Ramachandran plot. All figures were produced by using molscript (22). The data collection and refinement statistics are presented in Table 1.
Table 1. Crystal parameters, data collection, processing, and refinement statistics.
| Unit-cell parameters | |
| Space group | C2221 |
| a, Å | 89.026 |
| b, Å | 126.606 |
| c, Å | 119.404 |
| Matthews coefficient | 2.5 |
| Percentage solvent | 50.3 |
| No. of molecules in ASU* | 5 |
| Data collection | |
| X-ray source | NSLS, BNL (X12B)† |
| Detector | ADSC Q315 CCD‡ |
| Wavelength, Å | 0.979 |
| Resolution, Å | 1.85 |
| Total observations | 44,839 |
| Unique reflections | 20,740 |
| Completeness, % | 93.6 |
| Redundancy | 3.7 |
| Rsym§ | 0.080 |
| Refinement | |
| R factor/Rfree¶ | 0.21/0.23 |
| Data | |Fo| > 3.0σ (|Fo|) |
| Final model | |
| Nonhydrogen atoms | 4,630 |
| Waters | 463 |
| Average B factors, Å2 | |
| Protein | 22.24 |
| Water | 32.82 |
| rms deviation in bond length, Å | 0.007 |
| rms deviation in bond angles, ° | 1.7 |
ASU, asymmetric unit.
National Synchrotron Light Source, Brookhaven National Laboratory.
Area Detector Systems Corporation, charge-coupled device.
Rsym = ΣhklΣi[|Ii(hkl) — 〈I(hkl)〉|]/ΣhklI(hkl).
R factor = Σhkl|Fo(hkl)| — |Fc(hkl)|/Σhkl|Fo(hkl)|.
FKBP13 Reduction by Thioredoxin. Reduction experiments were carried out with recombinant AtFKBP13 purified after cleavage by thrombin. Reduction by the NADP/thioredoxin system of E. coli was performed as described (23). Recombinant AtFKBP13 (1.5 μg) was incubated with 0.25 mM NADPH, 0.3 μg of NADP-dependent thioredoxin reductase (NTR), and 0.3 μg of thioredoxin in 50 mM Tris·HCl (pH 7.5) at 25°C for 20 min in a final volume of 20 μl. Newly exposed cysteines resulting from disulfide reduction were labeled with addition of the thiol-specific fluorescent probe monobromobimane to 2 mM. After labeling, the protein sample was separated by SDS/PAGE (24) and the fluorescence recorded by using a Gel Doc-1000 fitted with a UV 365-nm Transilluminator and the quantity one analysis program (Bio-Rad). Subsequently, the protein pattern was revealed by staining with Coomassie blue G-250 and captured by using a scanner.
PPIase Assay. All assays were carried out by using the GST-AtFKBP13 fusion protein. GST alone showed no PPIase activity, and the fusion protein showed no reduction of PPIase activity compared with thrombin-cleaved pure protein (data not shown). PPIase assay was performed by using the protocol of Kofron et al. (25), with minor modifications. Enzyme, 45 nM, was incubated with 1.5 mg of α-chymotrypsin in reaction buffer in a final volume of 0.5 ml (50 mM Hepes/100 mM NaCl, pH 8), and the reaction was allowed to stabilize to 10°C. AtFKBP13 was reduced by incubation with 0.5 μM chloroplast m-type [Arabidopsis Genome Initiative (AGI) no. At3g15300] or E. coli thioredoxin and 500 μM DTT for 20 min at 25°C. The synthetic peptide Suc-Ala-Ala-Pro-Phe-paranitroanilide was dissolved in 470 mM LiCl in trifluoroethanol to maximize the amount of peptide present as the cis-isomer. The reaction was started by adding peptide substrate to a final concentration of 60 μM, catalysis was monitored at 390 nm in a Cary 3E UV/visible spectrometer (Varian), and data were obtained with the kinetics application. kcat/Km values were calculated as kobs – k0/[PPIase], where k0 represents the first-order rate constant for spontaneous cis-trans isomerization (26).
Results and Discussion
Structure of AtFKBP13 Reveals Unique Disulfide Bridges. The overall structure of AtFKBP13 molecule was found to consist of six β-strands and two α-helices (Fig. 1A). The β-strands form an integral antiparallel β-sheet that constitutes the core of the protein. The secondary structures are arranged in the following order: β1, β4, β5a, α2, β5b, α1, β2, β6a, β6b, and β3. The β5-strand of AtFKBP13 is split into two fragments, β5a and β5b, as in Methanococcus thermolithotrophicus MtFKB17 (27) and Homo sapiens HsFKB12 (28). Between β5a and β5b is the inserted α2-helix, similar to that of MtFKB17 (27). The β6-strand, which is unique to AtFKBP13, is formed by strands β6a and β6b connected by a short loop. Alignment between AtFKBP13 and representatives of other FKBPs, namely human HsFKB12 (29) and the macrophage infectivity potentiator protein from Legionella pneumophila, LpMIP (30), through the Dali server (31), gives the rms deviation values of 1.3 Å (with a Z-value of 17.5) and 1.4 Å (with a Z-value of 17.2), respectively (Fig. 1B). Significant similarities exist between the core regions of these proteins. The best matches are found in the catalytic region that contains the highly conserved residues, including Tyr-37, Phe-47, Asp-48, Phe-59, Val-66, Ile-67, Trp-72, Tyr-99, Ile-113, Leu-119, and Phe-121 involved in binding and maintaining the hydrophobic core of FK506 (32).
Fig. 1.

Structures of AtFKBP13. The active-site disulfides are shown as a ball-and-stick representation and are yellow. (A) Three-dimensional structure of the AtFKBP13 monomer. (B) Comparison of the Cα atom backbones of AtFKBP13 with human (Hs) and L. pneumophila (Lp) counterparts. AtFKBP13 (residues 5–129), HsFKBP12 (residues 1–107), and LpFKBP25 (residues 496–612) are magenta, blue, and green, respectively.
The tertiary structure of AtFKBP13 is highly unique in showing the presence of two disulfide bonds formed by Cys-5–Cys-17 and Cys-106–Cys-111 (Fig. 1 A). These disulfides, which are not present in FKBPs from animals or yeast, are consistent with the presence of conserved cysteine residues in the amino acid sequence of AtFKBP13 (data not shown). Structural comparison with human and L. pneumophila FKBPs indicates that the PPIase domain in AtFKBP13 has an additional strand (β6a and β6b) inserted at the C terminus, the position occupied by Cys-106 and Cys-111 (Fig. 1B). The disulfide bond between Cys-5 and Cys-17 at the N terminus is located at the β1-strand (Fig. 1 A). The disulfide bonds form additional secondary structures that are located on either sides of the central β-sheets and reflect the intrinsic versatility and flexibility of these regions. The cysteine residues exist in their oxidized state in the crystal structure as shown by their clearly defined electron density maps (see Fig. 2 for a detailed map of Cys-106–Cys-111 at the C terminus). Both disulfides appear to be stable, with low conformational strain based on their calculated theoretical dihedral energies of 3.60 kcal·mol–1 and 4.00 kcal·mol–1 for the N- and C-terminal bonds, respectively (33). The corresponding value varies from 0.5 to 4.7 for most protein disulfide bonds (34) and rarely reaches stability values >5.0. These highly stable disulfides suggest that the reduced form of the cysteines could act as reductants.
Fig. 2.

Stereoview of the 2Fo – Fc maps contoured at the 1.2σ level at the region of the C-terminal disulfide. The amino acid residues in the region are numbered.
In AtFKBP13, the residues around the two disulfides form two grooves on the protein surface, designated N and C (Fig. 3A). Groove C is built exclusively by residues in the vicinity of Cys-106, including Gly-105, Pro-114, Ser-110, Leu-112, and Ile-113 and is essentially hydrophobic. The Sγ atoms of Cys-106 and Cys-111 are exposed on the protein surface (Fig. 3B). Unlike groove C, the formation of groove N involves residues Phe-7, Phe 16, Lys-19, Thr-90, and Asp-122. The residue Cys-5 is solvent-exposed, whereas the sulfur atom of Cys-17 is buried (Fig. 3C). The two grooves are adjacent to each other on the protein surface (27.6 Å between atoms Sγ of Cys-5 and Cys-106), but they appear to have different accessibility and probably function independently to some extent. The structural features of AtFKBP13 support the idea that this protein could be regulated by redox and possibly act as a regulatory link to other proteins in the thylakoid lumen.
Fig. 3.

Further representations of AtFKBP13 structure. (A) Surface charge distributions (blue for positive and red for negative charge) of AtFKBP13 monomer. (B) Space-filling representation in which the sulfur atoms of Cys-106 and Cys-111 are exposed on the surface of the molecule. (C) Space-filling representation in which the Sγ atom of Cys-5 is fully exposed on the surface, and the sulfur atom of Cys-17 is buried.
FKBP13 Is a Target for Reduction by Thioredoxin. The possibility of a thioredoxin-linked reduction of AtFKBP13 arose from the finding of two solvent-exposed disulfides in its structure and from recent evidence that AtCYP20-3, a functionally related protein present in the chloroplast stroma, has been found to be a thioredoxin target (14, 35). We tested this possibility by applying the NADP/thioredoxin-reducing system of E. coli combined with fluorescence gel analysis with monobromobimane as a thiol-specific probe. In this assay, AtFKBP13 was incubated with thioredoxin and NTR in the presence of NADPH, a source of reducing equivalents. The newly formed —SH groups were labeled with monobromobimane, the proteins were separated by SDS/PAGE, and the fluorescence was recorded.
The reduction of disulfide(s) by the NADP/thioredoxin system was reflected by an increase in the fluorescence of the treated protein (Fig. 4). Owing to the absence of additional free cysteines in the sequence of AtFKBP13, the protein alone did not react with monobromobimane (Fig. 4, lane 1, control treatment). By contrast, incubation of AtFKPB13 with the NADP/thioredoxin system for 20 min resulted in marked fluorescent labeling, indicating disulfide reduction (Fig. 4, lane 2, complete treatment). Reduction was not observed when any one of the components of the system was omitted, i.e., thioredoxin, NTR, or NADPH (Fig. 4, lanes 3–5). A similar experiment performed with mutant forms of AtFKBP13 in which either the N- or C-terminal disulfide (Cys-5/17 or Cys-106/111) was replaced by serine established that both S—S bonds are fully reduced by thioredoxin (data not shown). Additional assays using a low concentration of DTT (0.5 mM) as hydrogen donor and different types of thioredoxin (E. coli, chloroplast f and m, and extraplastidic h) indicated the chloroplast m-type to be the most efficient. DTT alone was a poor reducing agent under these conditions.
Fig. 4.

Reduction of AtFKBP13 by the NADP/thioredoxin system from E. coli. Ctrl, control (FKBP13 alone); Cpl, complete thioredoxin system (NADPH + NTR + Trx) plus FKBP13; Trx, thioredoxin. -Trx, -NTR, and -NADPH indicate the complete thioredoxin system in which each of these components was individually omitted. Protein refers to the complete treatment in which the gel was stained with Coomassie blue.
AtFKPB13 PPIase Activity Is Regulated by Redox Status. Having structurally resolved the position of the disulfide bridges and established their reducibility by thioredoxin, we sought to investigate the effect of redox status on catalysis of the PPIase reaction by AtFKBP13. In contrast to AtCYP20-3, the isolated, oxidized form of AtFKBP13 efficiently catalyzed the peptidylprolyl cis-trans isomerization of a chromogenic synthetic pentapeptide in a standard PPIase assay (Table 2). To ascertain the importance of each disulfide bridge to total PPIase activity, mutant forms of AtFKBP13 were constructed which lacked either one or both cysteine pairs. AtFKBP13 proteins containing mutations in the amino (C5S/C17S) or carboxy (C106S/C111S) terminal cysteine pairs showed, respectively, ≈30% and ≈55% reduction in catalytic efficiency (kcat/Km). This result suggests that the surface-exposed C-terminal disulfide bridge may be more important for catalysis and may constitute the major site of regulation by thioredoxin. Activity was reduced almost 80% in the quadruple cysteine mutant (C5S/C17S, C106S/C111S). After incubation and reduction of AtFKBP13 with an Arabidopsis thioredoxin m reduced by DTT, PPIase activity was reduced by 55%, in keeping with the conclusion that AtFKBP13 PPIase activity can be modulated by redox state in vitro. DTT alone had a minimal effect. Unlike certain enzymes of the Calvin cycle (e.g., fructose bisphosphatase), neither pH nor Mg2+ appeared to be a factor in determining the extent of activation by thioredoxin (data not shown) (1). AtFKBP13 was found to remain catalytically active down to pH 5 (data not shown), indicating that this enzyme is functionally adapted to the acidic conditions of the thylakoid lumen.
Table 2. Requirement for both disulfide (S-S) groups for PPIase activity of FKBP13.
| FKBP13 | kcat/Km, s-1 μM-1 |
|---|---|
| WT | 11.6 |
| C5S/C17S | 8.3 |
| C106S/C111S | 5.4 |
| No Cys | 3.1 |
| + DTT | 10.2 |
| + Trx m | 9.2 |
| DTT + Trx m | 5.3 |
Trx, thioredoxin.
Concluding Remarks. The present results extend the capacity for redox regulation, long known for chloroplast stromal enzymes, to AtFKBP13, a resident of the thylakoid lumen. The results suggest that the regulatory response of an enzyme to redox change depends not only on its function, but also on its location within the cell. Further, although specifics are not known, the experiments above raise the possibility that the observed oxidative activation reflects a pattern of regulation operative in the lumen. According to this view, AtFKBP13 and perhaps other redox-regulated proteins of the lumen are photoreduced by thioredoxin m by photosystem I on entering and traversing the stroma. Once imported into the lumen, the reduced, inactive form of AtFKBP13 is converted to the active form by photooxidation by a photosystem II reaction, either directly by molecular oxygen or, indirectly, by way of a redox-active protein (Fig. 5). Such a mode of activation, in which an enzyme of the lumen is rendered active when oxidized in the light, contrasts with the mechanism functional in the stroma. The relation of electron transfer proteins of the lumen (36) [HCF164 (37), thylakoid luminal protein (38), oxygen-evolving enhancer protein (39), and CcdA (40)] to the scheme depicted in Fig. 5 requires further work. The mechanism presented appears to be independent of the previously demonstrated interaction of the precursor form of FKPB13 with the Rieske protein (4).
Fig. 5.

Proposed mechanism for the light-mediated regulation of AtFKBP13 in the thylakoid lumen. Trx, thioredoxin; PSI, photosystem I; PSII, photosystem II; Fd, ferredoxin. Rp is an undefined redox protein.
Acknowledgments
We thank Scott Luzzi (Michael Marletta laboratory, University of California, Berkeley) for assisting with the spectrophotometer and Nick Cai and Heather Lee for preparing the illustrations. This work was supported by the Academic Research Fund of the National University of Singapore (to K.S.), a U.S. Department of Energy grant (to S.L.), and funds from the California Agricultural Experiment Station (to B.B.B.). P.R. was supported by a Biotechnology and Biological Sciences Research Council Wain Fellowship.
Abbreviations: PPIase, peptidyl-prolyl cis-trans isomerase; NTR, NADP-dependent thioredoxin reductase.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 1U79).
References
- 1.Buchanan, B. B. (1980) Annu. Rev. Plant Physiol. 31, 341–374. [Google Scholar]
- 2.Schürmann, P. & Jacquot, J.-P. (2000) Annu. Rev. Plant Physiol. Plant Mol. Biol. 51, 371–400. [DOI] [PubMed] [Google Scholar]
- 3.Gupta, R., He, Z. Y. & Luan, S. (2002) Nature 417, 567–571. [DOI] [PubMed] [Google Scholar]
- 4.Gupta, R., Mould, R. M., He, Z. & Luan, S. (2002) Proc. Natl. Acad. Sci. USA 99, 15806–15811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peltier, J. B., Emanuelsson, O., Kalume, D. E., Ytterberg, J., Friso, G., Rudella, A., Liberles, D. A., Söderberg, L., Roepstorff, P., von Heijne, G. & Van Wijk, K. J. (2002) Plant Cell 14, 211–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schubert, M., Petersson, U. A., Haas, B. J., Funk, C., Schröder, W. P. & Kieselbach, T. (2002) J. Biol. Chem. 277, 8354–8365. [DOI] [PubMed] [Google Scholar]
- 7.Spetea, C., Hundal, T., Lundin, B., Heddad, M., Adamska, I. & Andersson, B. (2004) Proc. Natl. Acad. Sci. USA 101, 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He, Z. Y., Li, L. & Luan, S. (2004) Plant Physiol. 134, 1248–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Romano, P. G. N., Edvardsson, A., Ruban, A. V., Andersson, B., Vener, A. V., Gray, J. E. & Horton, P. (2004) Plant Physiol. 134, 1244–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Handschumacher, R. E., Harding, M. W., Rice, J., Drugge, R. J. & Speicher, D. W. (1984) Science 226, 544–547. [DOI] [PubMed] [Google Scholar]
- 11.Fischer, G., Wittmann-Liebold, B., Lang, K., Kiefhaber, T. & Schmid, F. X. (1989) Nature 337, 476–478. [DOI] [PubMed] [Google Scholar]
- 12.Schreiber, S. L. (1991) Science 251, 283–287. [DOI] [PubMed] [Google Scholar]
- 13.Romano, P. G. N., Horton, P. & Grey, J. E. (2004) Plant Physiol. 134, 1268–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Motohashi, K., Kondoh, A., Stumpp, M. T. & Hisabori, T. (2001) Proc. Natl. Acad. Sci. USA. 98, 11224–11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McPherson, A. (1990) Eur. J. Biochem. 101, 387–396. [Google Scholar]
- 16.Otwinowski, Z. & Minor, W. (1997) Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 17.Matthews, B. (1968) J. Mol. Biol. 33, 491–497. [DOI] [PubMed] [Google Scholar]
- 18.Rossmann, M. G. (1990) Acta Crystallogr. A 46, 73–82. [DOI] [PubMed] [Google Scholar]
- 19.Navaza, J. (1994) Acta Crystallogr. A 50, 157–163. [Google Scholar]
- 20.Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 21.Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991) Acta Crystallogr. A 47, 110–119. [DOI] [PubMed] [Google Scholar]
- 22.Kraulis, P. J. (1991) J. Appl. Crystallogr. 24, 946–950. [Google Scholar]
- 23.Wong, J. H., Balmer, Y., Cai, N., Tanaka, C. K., Vensel, W. H., Hurkman, W. J. & Buchanan, B. B. (2003) FEBS Lett. 547, 151–156. [DOI] [PubMed] [Google Scholar]
- 24.Laemmli, U. K. (1970) Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 25.Kofron, J. L., Kuzmic, P., Kishore, V., Colon-Bonilla, E. & Rich, D. H. (1991) Biochemistry 30, 6127–6134. [DOI] [PubMed] [Google Scholar]
- 26.Liu, J., Albers, M. W., Chen, C. M., Schreiber, S. L. & Walsh, C. T. (1990) Proc. Natl. Acad. Sci. USA 87, 2304–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki, R., Nagata, K., Yumoto, F., Kawakami, M., Nemoto, N., Furutani, M., Adachi, K., Maruyama, T. & Tanokura, M. (2003) J. Mol. Biol. 328, 1149–1160. [DOI] [PubMed] [Google Scholar]
- 28.Meadows, R. P., Nettesheim, D. G., Xu, R. X., Olejniczak, E. T., Petros, A. M., Holzman, T. F., Severin, J., Gubbins, E., Smith, H. & Fesik, S.W. (1993) Biochemistry 32, 754–765. [DOI] [PubMed] [Google Scholar]
- 29.Wilson, K. P., Yamashita, M. M., Sintchak, M. D., Rotstein, S. H., Murcko, M. A., Boger, J., Thomson, J. A., Fitzgibbon, M. J., Black, J. R. & Navia, M. A. (1995) Acta Crystallogr. D 51, 511–521. [DOI] [PubMed] [Google Scholar]
- 30.Riboldi-Tunnicliffe, A., König, B., Jessen, S., Weiss, M. S., Rahfeld, J., Hacker, J., Fischer, G. & Hilgenfeld, R. (2001) Nat. Struct. Biol. 8, 779–783. [DOI] [PubMed] [Google Scholar]
- 31.Holm, L. & Sander, C. (1993) J. Mol. Biol. 233, 123–138. [DOI] [PubMed] [Google Scholar]
- 32.Radzicka, A., Acheson, S. A. & Wolfenden, R. (1992) Bioorg. Chem. 20, 382–386. [Google Scholar]
- 33.Weiner, S. J., Kollman, P. A., Case, D. A., Singh, U. C., Ghio, C., Alagona, G., Profeta, S., Jr., & Weiner, P. K. (1984) J. Am. Chem. Soc. 106, 765–784. [Google Scholar]
- 34.Darby, N. T. & Creighton, T. E. (1995) Biochemistry 34, 11725–11735. [DOI] [PubMed] [Google Scholar]
- 35.Motohashi, K., Koyama, F., Nakanishi, Y., Ueoka-Nakanishi, H. & Hisabori, T. (2003) J. Biol. Chem. 278, 31848–31852. [DOI] [PubMed] [Google Scholar]
- 36.Friso, G., Giacomelli, L., Ytterberg, A. J., Peltier, J.-B., Rudella, A., Sun, Q. & van Wijk, K. J. (2004) Plant Cell 16, 478–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lennartz, K., Plucken, H., Seidler, A., Westhoff, P., Bechtold, N. & Meierhoff, K. (2001) Plant Cell 13, 2539–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee, K., Lee, J., Kim, Y., Bae, D., Kang, K. Y., Yoon, S. C. & Lim, D. (2004) Electrophoresis 25, 532–541. [DOI] [PubMed] [Google Scholar]
- 39.Heide, H., Kalisz, H. M. & Follmann, H. (2004) J. Plant Physiol. 161, 139–149. [DOI] [PubMed] [Google Scholar]
- 40.Page, M. L., Hamel, P. P., Gabilly, S. T., Zegzouti, H., Perea, J. V., Alonso, J. M., Ecker, J. R., Theg, S. M., Christensen, S. K. & Merchant, S. (2004) J. Biol. Chem. 279, 32474–32482. [DOI] [PubMed] [Google Scholar]