

Abstract
Autologous T cells genetically modified to express a chimeric antibody receptor (CAR) against carboxy-anhydrase-IX (CAIX) were administered to 12 patients with CAIX-expressing metastatic renal cell carcinoma (RCC). Patients were treated in three cohorts with a maximum of 10 infusions of a total of 0.2 to 2.1 × 109 CAR T cells. CTC grade 2–4 liver enzyme disturbances occurred at the lowest CAR T cell doses, necessitating cessation of treatment in four out of eight patients in cohorts 1 and 2. Examination of liver biopsies revealed CAIX expression on bile duct epithelium with infiltration of T cells, including CAR T cells. Subsequently four patients were pre-treated with CAIX monoclonal antibody (mAb) G250 to prevent CAR-specific toxicity and showed no liver toxicities and indications for enhanced peripheral T cell persistence. No clinical responses were recorded. This report shows that CAIX-targeting CAR T cells exerted antigen-specific effects in vivo and induced liver toxicity at the lowest dose of 0.2 × 109 T cells applied, illustrating the potency of receptor-modified T cells. We provide in-patient proof that the observed “on-target” toxicity is antigen-directed and can be prevented by blocking antigenic sites in off-tumor organs and allowing higher T cell doses.
Introduction
Adoptive transfer of antigen-specific T cells has shown therapeutic successes in the treatment of viral infections and tumors.1,2,3,4,5 Treatment of patients with gene-engineered T cells equipped with either chimeric antigen receptors (CARs) or T cell receptors (TCRs) provides an attractive strategy to provide therapeutic immunity. Despite some marked successes,6,7 gene-engineered T cells failed to yield antitumor responses in a substantial number of patients.8,9,10,11,12 One of the main challenges in the field of T cell engineering is receptor specificity as engineered T cells endowed with high-affinity receptors proved significantly toxic when tumor-associated antigens were targeted that are also expressed, even at low level, on normal tissue,8,11,12,13,14,15 so-called “on-target” toxicity. We have designed a CAR-directed against carboxy-anhydrase-IX (CAIX) and treated patients with CAIX-expressing metastatic renal cell carcinoma (RCC).16 In a previous report on the first three patients treated in this clinical trial, we reported (i) limiting liver enzyme disturbances in two patients, most likely caused by on-target toxicity; (ii) a limited peripheral persistence of transferred CAR T cells; and (iii) immunogenicity of the CAIX CAR receptor.8
We have now extended our observations based on an amended clinical protocol in nine additional patients in which we addressed two therapy-related questions related to on-target toxicity. First, can on-target toxicity be prevented or diminished when treating patients with lower doses of CAR T cells and can a maximum tolerated dose (MTD) of CAR T cells be determined? Second, can on-target toxicity be prevented or diminished by shielding the CAIX sites in the liver but not tumor by applying a parental CAIX monoclonal antibody (mAb) before T cell treatment and will such a pre-treatment enhance the MTD of CAR T cells? Previously, administration of CAIX mAb was shown to be well tolerated,17 and to saturate liver uptake of further CAIX mAb at a single low dose of 5 mg and leaving CAIX antigen in RCC metastasis accessible.18,19,20
Here, we provide in-patient proof that the observed on-target toxicity is antigen-directed and that effective blocking of a CAR-specific antigen expressed on normal (off-tumor) tissue resulted in an improved toxicity profile and allowed higher T cell doses.
Results
In vitro characteristics of CAIX CAR T cells for patient treatment
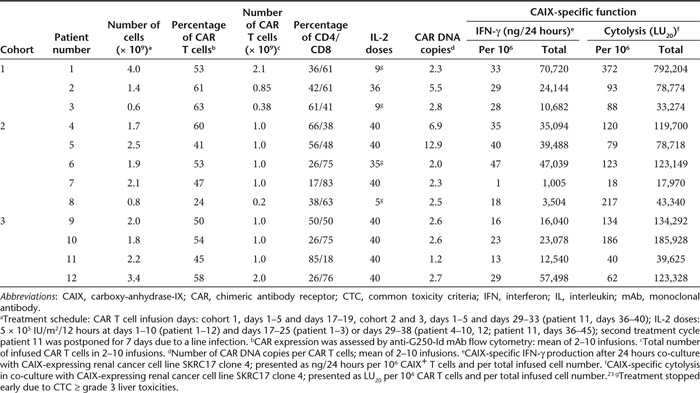
Detailed pre-infusion characteristics of CAIX CAR T cells for patient treatment are summarized in Table 1. Of the administered T cells to the 12 patients, a median of 61% were CD8+ (range, 18–83%) and 53% (range, 24–65%) expressed the CAIX CAR, with similar expression on both CD4 and CD8 T cell subsets. The CAR T cells had incorporated a median of 2.6 copies of the CAR transgene in their DNA (range, 1.2–12.9). We report a median CAR-mediated net cytolytic activity of 107 LU20/106 CAR T cells (range, 18–372) and a CAR-mediated net interferon-γ (IFN-γ) production of 29 ng/24 hours/106 CAR T cells (range, 1–47). Specific IFN-γ production by samples from therapeutic infusions was at least 20-fold higher than production of interleukin-5 (IL-5), tumor necrosis factor-α, and IL-4.
Table 1. Characteristics of pre-infusion CAIX CAR T cells.
Patients and treatment
Between March 2002 and December 2010, 12 metastatic RCC patients were treated, Table 2 provides patient characteristics and disease history, and consort diagram (Figure 1) showing compliance to eligibility criteria and protocol treatment. Patients had their diseased kidney removed and presented with metastasis, primarily in lung and a wide range of anatomical sites and were refractory to prior treatment with IFN-α and/or tyrosine kinase inhibitors. Patients were treated with CAIX CAR T cells in three cohorts, as further illustrated in Supplementary Figure S1. In cohort 1, an in-patient uptitration scheme was applied of a maximum of eight infusions at days 1–5 and days 17–19 with 2 × 107 to 2 × 109 CAR T cells per infusion; three patients were treated in this cohort with an actual cumulative dose of 0.38 to 2.1 × 109 CAR T cells (Table 1). Two out of three patients developed dose-limiting toxicities of liver enzymes (see below) and the treatment protocol was amended (see Supplementary Figures S1 and S2). In cohort 2, patients were treated according a conventional 3 × 3 phase I approach, starting at a dose of 1 × 108 CAR T cells per infusion and applying a maximum of 10 infusions at days 1–5 and days 29–33. At the starting dose, a liver enzyme dose-limiting toxicity was obtained in the third and a second in the fifth patient (i.e., patient 6 and 8, Table 3); patients received an actual cumulative dose of 0.2 to 1.0 × 109 CAR T cells (Table 1). Therefore, cohort 2 was closed without applying a dose escalation and assessment of a MTD of CAIX CAR T cells. In cohort 3, patients were treated as in cohort 2, but with pre-treatment of an intravenous infusion of 5 mg of the anti-CAIX mAb G250 3 days before start of each series of CAR T cell infusions, in order to block CAIX in liver and prevent liver enzyme toxicity. Three patients were treated at the starting (cumulative) dose of 1 × 109 CAR T cells without toxicity and CAR T cell dose was escalated to 2 × 109 CAR T cells (cumulative) for next patients.
Table 2. Patient characteristics and disease history before CAIX CAR T cell therapy.
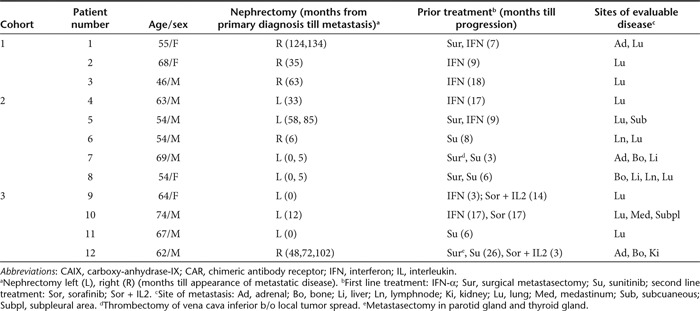
Figure 1.

Consort diagram showing compliance to eligibility criteria and protocol treatment. CAIX, carboxy-anhydrase-IX; CAR, chimeric antibody receptor; mAb, monoclonal antibody; MTD, maximum tolerated dose.
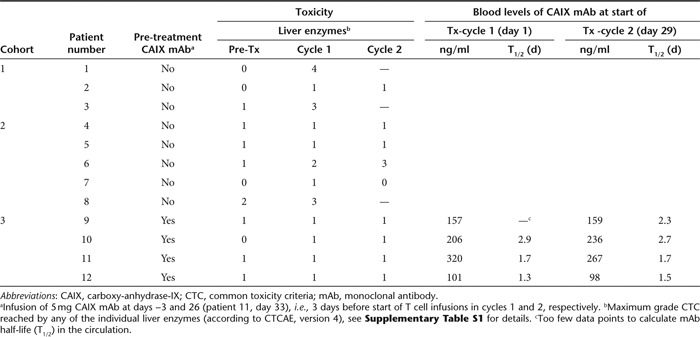
Table 3. Toxicities and blocking of CAIX antigen sites by CAIX mAb during T cell therapy.
From 2005 onwards several new, highly active treatment options were introduced for RCC.21 Consequently, patient accrual gradually diminished as patients were in a too poor condition to meet eligibility criteria after these multilineage treatments. The study was terminated after one patient in the second dose level of cohort 3 was treated.
No clinical responses were obtained and the median overall survival was 9.5 months (range: 3–33 months) for cohorts 1 (n = 3 patients) and 2 (n = 5 patients), and 12.5 months (6–24 months) for cohort 3 (n = 4 patients).
Evaluation of on-target toxicity
We have previously reported8 that two out of three patients in cohort 1 developed transient liver enzyme disturbances (CTC grades 3–4). This observation necessitated cessation of treatment in patients 1 and 3, corticosteroid treatment in patient 1 and reduction of maximal T cell dose to 2 × 108 T cells in patients 2 and 3.8 The toxicity was likely to be caused by an antigen-specific encounter of the CAR T cells with CAIX expressing bile duct epithelial cells, so-called “on-target toxicity”. Here, we add to these data our observations in nine more patients treated according to an amended protocol according a conventional phase I strategy without (cohort 2) and with (cohort 3) pre-treatment with anti-CAIX mAb (see Supplementary Figures S1 and S2).
In cohort 2, the patients 6 and 8 developed transient CTC grade 3 liver enzyme disturbances after 10 and 2 T cell infusions, respectively. Again, treatment was halted and corticosteroids were applied. Of note, patient 8 presented with CTC grade 2 liver enzyme levels before treatment, but was eligible because of the presence of liver metastasis (Tables 2 and 3, and Supplementary Table S1). Liver biopsies were taken from patient 6 at day 40 and from patient 8 at day 4, i.e., 7 and 2 days after the last T cell infusion, respectively. Pathological examination showed intact liver architecture, minor (patient 6) to purulent cholangitis (patient 8), limited (patient 6) to massive (patient 8) T cell infiltrates around the bile ducts with sporadic bile duct epithelium infiltration, and clear CAIX antigen expression on the bile duct epithelial cells (Figure 2). Remarkably, in patient 8, the portal T cell infiltrate consisted predominantly of CD4 T cells (CD4:CD8 ratio, 4.0) while the CAR T cell infusions consisted predominantly of CD8 T cells (ratio, 0.6; Table 1). Flow cytometry analysis confirmed presence of CAR T cells in this biopsy and revealed that 0.07% of total T cells were in fact CAR T cells, whereas the percentage of CAR T cells in a corresponding blood sample was 0.15 (Supplementary Figure S3). In addition, staining of cryosections using G250 anti-idiotype mAb NuH82 suggested the presence of CAR T cells, but poor morphology prohibited the assessment of their location.
Figure 2.

T cell infiltration in liver. Immunohistochemistry of biopsy of liver taken from patient 8 at day 4, i.e., 2 days after last CAR T cell infusion (original magnification ×200, all sections), showing liver parenchyma (L), portal triangle (P) with bile duct (B) and stained for expression of CAIX antigen and for CD3, CD4, and CD8 T cells. CD8 T cells were lining at the basal side (arrowheads) or infiltrating (arrow) the epithelium of the bile duct. CAIX, carboxy-anhydrase-IX; CAR, chimeric antibody receptor.
In cohort 3, patients were pre-treated with 5 mg CAIX mAb followed by a cumulative dose of 1 × 109 (three patients) or 2 × 109 CAR T cells (one patient). Dose-limiting toxicities or higher than grade 1 liver toxicity were not observed (Table 3 and Supplementary Table S1).
CAIX mAb blood levels
Blood concentrations of CAIX mAb at start of CAR T cell infusions were similar for treatment cycles 1 and 2 (i.e., days 1 and 29) of individual patients and ranged from 98 to 320 ng/ml with a T1/2 of 1.7 days (median; range 1.3–2.9), Table 3. As a result, CAIX mAb was detectable in blood up to 6–10 days after start of CAR T cell infusions.
CAR T cell blood levels
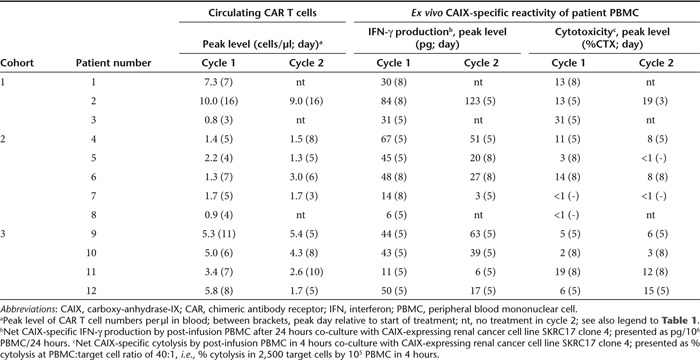
CAR T cells were detectable in peripheral blood by flow cytometry during a median of 39 days (range, 12–74; n = 12 patients).22 The number of circulating CAR T cells peaked at a median of 2.8/µl (range, 0.8–10.0) and 2.6/µl (range, 1.3–9.0) for treatment cycles 1 and 2, respectively, at 6 days (range, 3–16) after start of infusions in each treatment (Table 4). We analyzed CAR T cell persistence, defined as the last day of flow cytometry CAR T cell detection relative to start of treatment cycle 2, in patients receiving an equal cumulative dose of 1 × 109 CAR T cells in cohort 2 (patients 4–7) versus cohort 3 (patients 9–11). Persistence of CAR T cells tend to be somewhat longer in cohort 3 (21, 23, 37 days) when compared with cohort 2 (10, 5, 14, 21 days; P = 0.06 using Student's t-test). Patient 12, treated in cohort 3 with a total of 2 × 109 CAR T cells developed a fast and vigorous anti-CAIX CAR T cell response, and CAR T cells were detected for only 6 days relative to start of treatment cycle 2 (Supplementary Data).
Table 4. Characteristics of post-infusion CAIX CAR T cells.
Post-treatment peripheral blood mononuclear cell show CAIX-specific T cell functions
CAIX-specific IFN-γ production by post-treatment peripheral blood mononuclear cell (PBMC), expressed as pg IFN-γ/106 PBMC/24 hours, is presented in Table 4 and further specified in Supplementary Table S2a. CAIX-specific IFN-γ production peaked at 5 days (median, range: 5–8) after start of each treatment cycle, with a median IFN-γ production of 43 pg/106 PBMC/24 hours (range 3–123 pg). The IFN-γ production by post-infusion PBMC correlates with pre-infusion CAIX CAR T cell IFN-γ production potency (Pearson's correlation r = 0.45, P = 0.04; Supplementary Table S2b and Figure 3).
Figure 3.

CAR-specific IFN-γ production by post-treatment PBMC correlates with pre-infusion CAR T cell IFN-γ production potency. Maximum IFN-γ production by CAR T cells in 1 × 106 PBMC per 24 hours is calculated from the IFN-γ production of pre-infusion CAR T cells and the proportion of CAR T cells in PBMC and is expressed as pg IFN-γ/106 PBMC/24 hours, see Supplementary Table S3b. The measured IFN-γ production in post-treatment PBMC correlates with the calculated maximum IFN-γ production by CAR T cells in PBMC; this correlation is significant (Pearson's correlation analysis: r = 0.452; P = 0.04; regression line: y = 23 + 0.32x). CAR, chimeric antibody receptor; IFN, interferon; PBMC, peripheral blood mononuclear cell.
Post-treatment PBMC exerted CAIX-specific cytolysis in 17 out of 21 treatment cycles, peaking again at 5 days (range, 3–8) after start of treatment are presented in Table 4 and further specified in Supplemental Table S3a. CAIX-specific cytolysis by post-treatment PBMC did not correlate with pre-infusion CAIX CAR T cell cytolytic (Supplementary Table S3b).
Pre-treatment with CAIX mAb does not seem to affect the ex vivo activity of patients' PBMC in cohort 3 when compared with cohort 2.
Blood cytokine levels
Blood cytokine levels were assessed in plasma samples obtained at regular intervals before, during, and after CAR T cell infusions and are presented in Figure 4 for individual patients and treatment cycles at day 1 (pre-treatment) and at the days that levels peaked. Blood cytokine peak levels occurred at 5 days (median, range: 5–8) after start of CAR T cell infusions and IL-2 injections in cycle 1. For treatment cycle 2, levels peaked 2 days (range, 1–5) earlier and peak cytokine levels were slightly lower than those for cycle 1. Most prominent were the expected elevations (greater than twofold over pre-treatment) of IL-2 levels, in 17 out of 21 treatment cycles, and the putative IL-2 induced IL-5 elevations23 in 19 out of 21 treatment cycles. IFN-γ elevations, possibly produced by in vivo-activated CAR T cells,23 were seen in only 9 out of 21 treatment cycles, and IL-10 elevations were observed in 13 out of 21 treatment cycles.
Figure 4.

Blood cytokine levels. Blood cytokine levels were assessed in plasma samples obtained before, during, and following CAIX CAR T cell therapy by cytokine bead arrays. For both treatment cycles, pre-treatment (day 1) and peak levels are shown for individual patients. Peak day is presented as median and range. CAIX, carboxy-anhydrase-IX; CAR, chimeric antibody receptor; IFN, interferon; IL, interleukin; TNF, tumor necrosis factor.
Discussion
Adoptive T cell therapy in advanced metastatic malignancies is showing increasingly encouraging clinical data, using both unmodified and receptor gene-modified T cells.24 Early clinical trials using CAR or TCR gene-modified T cells provided, although often showing limited clinical success, important lessons for the next generation trials,14,25 regarding on-target toxicity and limited peripheral persistence of transferred CAR T cells, and immunogenicity of the CAR receptor.8,22 On-target toxicity was first reported for the CAIX CAR, used to treat patients with metastatic RCC and consisted of limiting liver enzyme elevations that were most likely caused by CAR T cells that recognized CAIX antigen expressed at low levels on bile duct epithelial cells.8 In our current studies, we have extended our observations and assessed whether this on-target toxicity also occurred at lower doses of CAR-transduced T cells, examined the underlying mechanism of on-target toxicity in more detail, and explored a strategy to attenuate activity of CAR T cells against bile duct epithelial cells. The latter was done by pretreating patients with a low dose of the CAIX mAb G250, from which the CAR was constructed, in an attempt to block access of engineered T cells to CAIX expressed in bile ducts.
Already at the lowest dose explored, 1 × 108 CAR T cells per infusion with a maximum cumulative dose of 1 × 109 CAR T cells, two out of five patients experienced dose-limiting liver enzyme disturbances after 2 and 10 infusions, respectively. Thus, the MTD of CAR T cells given according to this treatment protocol is lower than 1 × 109 CAR T cells (cumulative dose). Liver biopsies revealed presence of CAR T cells in the liver and confirmed previous observations of CAIX expression on bile duct epithelial cells, as well as inflammation and T cell infiltration in the portal areas around bile ducts.8 The expression of CAIX in normal tissues is restricted to scattered areas in the upper gastrointestinal mucosa and gastrointestinal-related structures (bile ducts and exocrine ducts of pancreas).26,27 Damage of the most vulnerable of these structures, i.e., bile ducts, will result in immediate release of liver enzymes in blood. Despite that pre-existing liver enzyme disturbances (Supplementary Table S1) will have lowered the threshold for the particular liver enzyme(s) to reach for a higher toxicity grade, all patients showed a clear increase in liver enzyme values following the infusion of anti-CAIX CAR-transduced T cells. This finding, together with the presence of CAIX on bile ducts and the presence of CAR T cells infiltrating the bile ducts, makes it unlikely that other causes contribute to the observed liver toxicity. The massive portal area CD4 T cell infiltrate in patient 8 bears resemblance with findings in a receptor gene therapy tumor transplantation model in immune-deficient mice28 that suggested dominant reactivity of CD4 T cells at the border of tumor antigen-positive tissue.
In an attempt to circumvent on-target liver toxicity, the next four patients received CAIX mAb pre-treatment before infusions of a total of 1 × 109 (three patients) or 2 × 109 (one patient) CAR T cells. This blocking strategy was based on an extensive set of clinical radio-immunotherapy data; (i) a 131I-labeled CAIX mAb cG250 dose escalation study revealed that 2–5 mg CAIX mAb saturated the CAIX liver sites;29 (ii) 131I-labeled CAIX mAb (5 mg) given 4 days after an infusion with 5 mg of 125I-labeled CAIX mAb did not accumulate in the liver, whereas an equal distribution of both radio-isotopes in the tumor metastasis was recorded;19,20 (iii) in subsequent clinical radio-immunotherapy studies, 5 mg of the radiolabeled CAIX mAb was applied as “scout” or diagnostic dose of radiolabeled CAIX mAb;30,31 these studies warranted the use of 5 mg CAIX mAb to block CAIX sites in the liver. It is noteworthy that actual CAIX mAb blood levels at start of CAR T cell infusions (98–320 ng/ml) do not saturate CAIX-binding sites on RCC cell lines in vitro and do not prevent CAIX CAR T cells to kill the RCC cell lines. Effective in vitro blocking of CAR–CAIX interactions using RCC cell lines is only observed at concentrations above 10 µg/ml CAIX mAb.
Since none of the patients pre-treated with CAIX mAb experienced any sign of liver enzyme disturbance above pre-treatment levels, it can be concluded that shielding CAIX antigen sites in the liver by CAIX mAb attenuates the toxicity of CAR T cells and is anticipated to result in a higher MTD of CAR T cells. Unfortunately, neither the MTD nor the T cell dose that would yield effective antitumor activity of a combined treatment with CAR T cells and CAIX mAb have formally been established. CAIX mAb was detectable in blood up to 6–10 days after start of CAR T cell infusions (days 1–5) potentially resulting in effective shielding of CAIX antigen sites in liver during T cell therapy. Direct evidence to substantiate shielding of these sites cannot be given as no liver biopsies are available of these patients.
We observed effective blocking of CAIX CAR T cell-induced toxicity of liver in metastatic RCC patients by pre-treatment with a high-affinity CAIX mAb (Ka = 4 × 109 l/mol). Key to successful application of this approach may be the administration of a high affinity mAb with excellent homing to the tumor and ability to selectively saturate CAIX-binding sites outside the tumor.19,20,29 Whether this approach would be applicable for shielding of other CAR-related on-target toxicities needs to be further explored.
On-target toxicity is a serious drawback of receptor gene therapy and has also been reported for CARs directed against Her2/Neu and CD19 and TCRs directed against the HLA-A2–restricted antigens MARTI, gp100, and CEA.11,12,13,15,32 Together with our results, these findings illustrate the great potency of receptor gene-modified T cells and underscore the need to select “safer” T cell target antigens that are strictly expressed on malignant cells.
Cancer/testis (CT) antigens are a class of shared antigens expressed in a variety of tumor types, but silenced in healthy cells, except for germ line cells and thymic medullary epithelial cells. Preclinical and clinical studies have shown the immunogenic nature of these antigens.33 Clinical proof of the value of targeting CT antigens came from the treatment of metastatic melanoma and synovial cell carcinoma with NY-ESO-1 TCR retargeted T cells, which resulted in significant and durable clinical responses without signs of toxicities.7 Along the same line, we and others currently explore specific members of the “CT” family of MAGE antigens because of their restricted expression in multiple tumors and their pivotal role in tumor pathogenesis.34,35,36
In this clinical trial, patients were treated with so-called “first generation” CAR T cells and without lymphodepletion preconditioning. In these immunocompetent patients, CAR T cells elicited vigorous humoral and cellular anti-CAIX CAR immune responses limiting T cell persistence up to 4 weeks after the first treatment cycle and up to 1–3 weeks after the second treatment cycle.22 Remarkably, following CAIX mAb pre-treatment, no anti-CAR immune responses developed and T cell persistence recovered up to 3–5 weeks following both first and second treatment cycle in patients treated at the total dose of 1 × 109 CAR T cells. Although, the inter-relationship between shielding CAIX antigen sites in the liver and lack of immunity directed against CAR T cells needs to be further explored, inflammation in liver induced by tissue damage by CAR T cells may take part in these observations. In patients treated with receptor-engineered T cells with lymphodepleting preconditioning, only limited anti-receptor immune responses have been documented.37 Of note, these studies mostly applied only a single infusion of receptor-engineered T cells in contrast to the multi-dosing strategy and two treatment cycles in present study, which in itself is more prone to elicit immune responses. Taken together, it is noteworthy that blocking antigen exposure in an off-tumor organ not only abrogates on-target toxicity but apparently also enhances the availability of peripheral CAR T cells.
We demonstrated low to moderate CAIX-specific functions in post-treatment patients' PBMC, mostly at times when numbers of circulating CAR T cells peaked. Notably, pre-infusion CAR T cell IFN-γ production but not cytolytic activity correlated with these activities in post-treatment PBMC. In addition, we observed increased IFN-γ levels post-treatment in the blood of 6 out of 12 patients. Thus, we confirm previous observations that gene-modified T cells maintain their transgene-specific function in vivo.6,7,23 Importantly, these ex vivo functions of PBMC seem not to be affected by the in vivo CAIX mAb pre-treatment in cohort 3.
Clinical antitumor responses of T cells irrespective of gene-modification appear to be directly related to T cell persistence,38 which in turn is reported to be associated with differentiation state and replicative capacity of transferred T cells.6,39 Improving T cell persistence is yet another important goal of receptor gene therapy.24,25,40 Here, we may have prevented on-target toxicity through CAIX mAb pre-treatment, which at the lowest T cell dose (total 1 × 109 CAR T cells) coincided with reduced humoral and cellular immunity directed against CAR T cells and an apparent improvement of the peripheral persistence. Future strategies to improve T cell persistence should include pre-selection and controlled in vitro propagation of T cells to generate T cells with preferred less differentiated phenotype, in combination with a patient lymphodepleting preconditioning regimen.14,24,25,40
Materials and Methods
Patients. Patients had disseminated clear cell RCC, measurable progressive disease not amenable for curative surgery and for whom no standard treatments exist, and a primary tumor expressing CAIX (Table 2). The clinical protocol (DDHK97-29/P00.0040C) and amendments referred to the Declaration of Helsinki protocols and were approved by the governmental regulatory authorities and the institutional medical ethical review board. Patients were included after written informed consent was obtained. The inclusion and exclusion criteria for patients participating in this study have been reported previously.8
Retroviral supernatant and preparation of autologous CAIX CAR T-lymphocytes. The clinical CAIX CAR vector batch, produced in May 2002 (batch no. M4.50086; BioReliance, Sterling, UK),41 was approved for clinical ex vivo gene therapy by the Dutch Inspectorate for Public Health and proved stable till protocol closure (December 2010).42,43 Patient autologous T-lymphocytes were transduced with the SFG-CAIX CAR retrovirus as described.41,44,45
Patient treatment schedule and evaluation. Following written informed consent, patients were treated with CAIX CAR expressing autologous T-lymphocytes in three different cohorts (Figure 1 and Supplementary Figure S1).
In cohort 1, we aimed to evaluate potential safety concerns of CAR T cells. We have treated patients with CAR T cells in an in-patient dose escalation scheme, as described.46 Treatment consisted of intravenous doses of 2 × 107 T cells at day 1; 2 × 108 T cells at day 2; 2 × 109 T cells at days 3–5 (treatment cycle 1) and days 17–19 (treatment cycle 2), in combination with subcutaneous injections of 5 × 105 IU/m2 human recombinant interleukin-2 (IL-2, Chiron, Amsterdam, The Netherlands) twice daily at days 1–10 and days 17–26. Adverse events were graded according the National Cancer Institute Common Toxicity Criteria (CTC), version 2.0. Tumor response was evaluated by RECIST.47
In cohort 2, we aimed to assess the MTD of CAR T cells. Patients were treated with CAR T cells in a conventional 3 × 3 phase I design, starting at 1 × 108 CAR T cells per infusion and extending to 2, 4, 8, 16, 20, 25, and 30 × 108 cells, and applying a maximum of 10 T cell infusions at days 1–5 and days 29–33 combined with IL-2, subcutaneously, 5 × 105 IU/m2 twice daily at days 1–10 and days 29–38. Dose-limiting toxicity was defined as a CTC grade 4 event, and CTC grade 2–3 events persisting longer than 22 days from onset, Supplementary Figure S2.
In cohort 3, we aimed to prevent on-target toxicity and allow safe administration of CAR T cells. We included an extra intravenous infusion of 5 mg of the anti-CAIX mAb G250 (IgG1; Ka: 4 × 109 l/mol, kindly provided by Lloyd J. Old and Eric Hoffman; LIRC, New York, NY),29 3 days before start of each series of CAR T cell infusions, which blocks CAIX antigen in the liver and leaving accessible CAIX antigen at RCC tumor sites.19,29,48
Histology and immunohistochemistry. Biopsies of liver were formalin-fixed and paraffin-embedded. Routine pathological examination and immunohistochemistry were performed on 4 µm tissue sections using the mAbs M75, CD3, CD4, and CD8 and a polymer-based detection system (Dako Envision; Dakocytomation, Glostrup, Denmark). The IgG2b mAb M75 recognizes a formalin-resistant epitope on CAIX different from that recognized by the G250 mAb.26,27,49 Part of the biopsy taken from patient 8 was frozen and cryosections stained with the G250 anti-idiotype mAb NuH82.50 In addition, single cells were prepared from another part of this biopsy for flow cytometric analysis (Supplementary Figure S3).
Flow cytometric analysis of transduced T cell infusions and patient blood samples. Expression of the CAIX CAR receptor on transduced T cell cultures was determined by indirect immunofluorescence.41,44 The absolute numbers of lymphocyte subsets, the percentages, and the absolute numbers of CAR T cells in the blood of patients before and following infusions of transduced T cells were assessed as described.51
Assessment of transgene copy number by quantitative real-time PCR. Genomic DNA was isolated from aliquots of the transduced T cell infusions and from blood taken before, during, and after therapy using the QIAamp DNA mini kit (Qiagen, Hilden, Germany). The quantitative real-time PCR was performed as described.51
Assessment of CAIX CAR-mediated T cell functions. T cells harvested from the transduction cultures and PBMC obtained before, during, and after treatment were monitored for their level of CAIX CAR-mediated T cell cytotoxicity and cytokine production in response to CAIX+ and CAIX− RCC cell lines.23 The antigen specificity of the CAR-mediated T cell effector functions was verified by blocking CAIX antigen on these cells by the CAIX mAb.44
Assessment of cytokine levels. The concentrations of IFN-γ, tumor necrosis factor-α, IL-2, IL-4, IL-5, and IL-10 in plasma samples were determined by cytokine bead arrays (BD Biosciences, San Jose, CA), according to suppliers' specifications, and expressed in pg/ml. The detection limits of the assay were 2.6 pg/ml for IL-2 and IL-4, 2.5 pg/ml for IL-5, 2.8 pg/ml for IL-10 and tumor necrosis factor-α, and 7.1 pg/ml for IFN-γ.
SUPPLEMENTARY MATERIAL Figure S1. Treatment schemes. Figure S1. Diagram showing design of phase I treatment (cohorts 2 and 3). Figure S1. Flow cytometric assessment of CAR T cells in blood sample and liver tissue obtained from patient 8 at treatment day 4. Table S1. Liver enzyme abnormalities in patients following CAIX CAR T cell therapy. Table S2a. CAIX CAR-specific IFN-γ production by post-treatment PBMC. Table S2b. CAIX CAR-specific IFN-γ production by post-treatment PBMC reflects pre-infusion CAIX CAR T cell IFN-γ production potency. Table S3a. CAIX CAR-specific cytolysis by post-treatment PBMC. Table S3b. CAIX CAR-specific cytolysis by post-treatment PBMC does not reflect pre-infusion CAIX CAR T cell cytolytic potency. Data
Acknowledgments
This work was funded in part by the Dutch Cancer Foundation (grant DDHK99-1865), the European Commission grant QLK3-1999-01262, and the Cancer Research Institute, New York, NY (clinical investigation grant “Immuno-gene therapy of metastatic renal cell cancer patients”). This article was presented in part in JCO 2006.8 The authors thank Barbara Luider and Jeannette Oosterwijk-Wakka for their technical assistance. The authors declared no conflict of interest.
Supplementary Material
References
- Butler, MO, Friedlander, P, Milstein, MI, Mooney, MM, Metzler, G, Murray, AP et al. (2011). Establishment of antitumor memory in humans using in vitro-educated CD8+ T cells. Sci Transl Med 3: 80ra34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savoldo, B, Goss, JA, Hammer, MM, Zhang, L, Lopez, T, Gee, AP et al. (2006). Treatment of solid organ transplant recipients with autologous Epstein Barr virus-specific cytotoxic T lymphocytes (CTLs). Blood 108: 2942–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee, C, Thompson, JA, Byrd, D, Riddell, SR, Roche, P, Celis, E et al. (2002). Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA 99: 16168–16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, EA, Greenberg, PD, Gilbert, MJ, Finch, RJ, Watanabe, KS, Thomas, ED et al. (1995). Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med 333: 1038–1044. [DOI] [PubMed] [Google Scholar]
- Dudley, ME, Wunderlich, JR, Yang, JC, Sherry, RM, Topalian, SL, Restifo, NP et al. (2005). Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 23: 2346–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos, M, Levine, BL, Porter, DL, Katz, S, Grupp, SA, Bagg, A et al. (2011). T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 3: 95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, PF, Morgan, RA, Feldman, SA, Yang, JC, Sherry, RM, Dudley, ME et al. (2011). Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 29: 917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers, CH, Sleijfer, S, Vulto, AG, Kruit, WH, Kliffen, M, Debets, R et al. (2006). Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol 24: e20–e22. [DOI] [PubMed] [Google Scholar]
- Till, BG, Jensen, MC, Wang, J, Chen, EY, Wood, BL, Greisman, HA et al. (2008). Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 112: 2261–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, RA, Dudley, ME, Wunderlich, JR, Hughes, MS, Yang, JC, Sherry, RM et al. (2006). Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314: 126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, LA, Morgan, RA, Dudley, ME, Cassard, L, Yang, JC, Hughes, MS et al. (2009). Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114: 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst, MR, Yang, JC, Langan, RC, Dudley, ME, Nathan, DA, Feldman, SA et al. (2011). T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 19: 620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, RA, Yang, JC, Kitano, M, Dudley, ME, Laurencot, CM and Rosenberg, SA (2010). Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 18: 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilham, DE, Debets, R, Pule, M, Hawkins, RE and Abken, H (2012). CAR-T cells and solid tumors: tuning T cells to challenge an inveterate foe. Trends Mol Med 18: 377–384. [DOI] [PubMed] [Google Scholar]
- Kochenderfer, JN, Dudley, ME, Feldman, SA, Wilson, WH, Spaner, DE, Maric, I et al. (2012). B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 119: 2709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weijtens, ME, Willemsen, RA, Valerio, D, Stam, K and Bolhuis, RL (1996). Single chain Ig/gamma gene-redirected human T lymphocytes produce cytokines, specifically lyse tumor cells, and recycle lytic capacity. J Immunol 157: 836–843. [PubMed] [Google Scholar]
- Bleumer, I, Knuth, A, Oosterwijk, E, Hofmann, R, Varga, Z, Lamers, C et al. (2004). A phase II trial of chimeric monoclonal antibody G250 for advanced renal cell carcinoma patients. Br J Cancer 90: 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwers, AH, Mulders, PF, de Mulder, PH, van den Broek, WJ, Buijs, WC, Mala, C et al. (2005). Lack of efficacy of two consecutive treatments of radioimmunotherapy with 131I-cG250 in patients with metastasized clear cell renal cell carcinoma. J Clin Oncol 23: 6540–6548. [DOI] [PubMed] [Google Scholar]
- Steffens, MG, Boerman, OC, Oyen, WJ, Kniest, PH, Witjes, JA, Oosterhof, GO et al. (1999). Intratumoral distribution of two consecutive injections of chimeric antibody G250 in primary renal cell carcinoma: implications for fractionated dose radioimmunotherapy. Cancer Res 59: 1615–1619. [PubMed] [Google Scholar]
- Steffens, MG, Oosterwijk-Wakka, JC, Zegwaart-Hagemeier, NE, Boerman, OC, Debruyne, FM, Corstens, FH et al. (1999). Immunohistochemical analysis of tumor antigen saturation following injection of monoclonal antibody G250. Anticancer Res 19(2A): 1197–1200. [PubMed] [Google Scholar]
- Escudier, B, Szczylik, C, Porta, C and Gore, M (2012). Treatment selection in metastatic renal cell carcinoma: expert consensus. Nat Rev Clin Oncol 9: 327–337. [DOI] [PubMed] [Google Scholar]
- Lamers, CH, Willemsen, R, van Elzakker, P, van Steenbergen-Langeveld, S, Broertjes, M, Oosterwijk-Wakka, J et al. (2011). Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 117: 72–82. [DOI] [PubMed] [Google Scholar]
- Lamers, CH, Langeveld, SC, Groot-van Ruijven, CM, Debets, R, Sleijfer, S and Gratama, JW (2007). Gene-modified T cells for adoptive immunotherapy of renal cell cancer maintain transgene-specific immune functions in vivo. Cancer Immunol Immunother 56: 1875–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June, C, Rosenberg, SA, Sadelain, M and Weber, JS (2012). T-cell therapy at the threshold. Nat Biotechnol 30: 611–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coccoris, M, Straetemans, T, Govers, C, Lamers, C, Sleijfer, S and Debets, R (2010). T cell receptor (TCR) gene therapy to treat melanoma: lessons from clinical and preclinical studies. Expert Opin Biol Ther 10: 547–562. [DOI] [PubMed] [Google Scholar]
- Oosterwijk, E, Ruiter, DJ, Hoedemaeker, PJ, Pauwels, EK, Jonas, U, Zwartendijk, J et al. (1986). Monoclonal antibody G 250 recognizes a determinant present in renal-cell carcinoma and absent from normal kidney. Int J Cancer 38: 489–494. [DOI] [PubMed] [Google Scholar]
- Pastoreková, S, Parkkila, S, Parkkila, AK, Opavský, R, Zelník, V, Saarnio, J et al. (1997). Carbonic anhydrase IX, MN/CA IX: analysis of stomach complementary DNA sequence and expression in human and rat alimentary tracts. Gastroenterology 112: 398–408. [DOI] [PubMed] [Google Scholar]
- Straetemans, T, Coccoris, M, Berrevoets, C, Treffers-Westerlaken, E, Scholten, CE, Schipper, D et al. (2012). T-cell receptor gene therapy in human melanoma-bearing immune-deficient mice: human but not mouse T cells recapitulate outcome of clinical studies. Hum Gene Ther 23: 187–201. [DOI] [PubMed] [Google Scholar]
- Steffens, MG, Boerman, OC, Oosterwijk-Wakka, JC, Oosterhof, GO, Witjes, JA, Koenders, EB et al. (1997). Targeting of renal cell carcinoma with iodine-131-labeled chimeric monoclonal antibody G250. J Clin Oncol 15: 1529–1537. [DOI] [PubMed] [Google Scholar]
- Brouwers, AH, Buijs, WC, Mulders, PF, de Mulder, PH, van den Broek, WJ, Mala, C et al. (2005). Radioimmunotherapy with [131I]cG250 in patients with metastasized renal cell cancer: dosimetric analysis and immunologic response. Clin Cancer Res 11(19 Pt 2): 7178s–7186s. [DOI] [PubMed] [Google Scholar]
- Divgi, CR, O'Donoghue, JA, Welt, S, O'Neel, J, Finn, R, Motzer, RJ et al. (2004). Phase I clinical trial with fractionated radioimmunotherapy using 131I-labeled chimeric G250 in metastatic renal cancer. J Nucl Med 45: 1412–1421. [PubMed] [Google Scholar]
- Brentjens, R, Yeh, R, Bernal, Y, Riviere, I and Sadelain, M (2010). Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther 18: 666–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero, OL and Chen, YT (2009). Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci 100: 2014–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas, S, De Plaen, E and Boon, T (2000). MAGE-B5, MAGE-B6, MAGE-C2, and MAGE-C3: four new members of the MAGE family with tumor-specific expression. Int J Cancer 87: 55–60. [PubMed] [Google Scholar]
- Yang, B, O'Herrin, SM, Wu, J, Reagan-Shaw, S, Ma, Y, Bhat, KM et al. (2007). MAGE-A, mMage-b, and MAGE-C proteins form complexes with KAP1 and suppress p53-dependent apoptosis in MAGE-positive cell lines. Cancer Res 67: 9954–9962. [DOI] [PubMed] [Google Scholar]
- Straetemans, T, van Brakel, M, van Steenbergen, S, Broertjes, M, Drexhage, J, Hegmans, J et al. (2012). TCR gene transfer: MAGE-C2/HLA-A2 and MAGE-A3/HLA-DP4 epitopes as melanoma-specific immune targets. Clin Dev Immunol 2012: 586314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, JL, Theoret, MR, Zheng, Z, Lamers, CH, Rosenberg, SA and Morgan, RA (2010). Development of human anti-murine T-cell receptor antibodies in both responding and nonresponding patients enrolled in TCR gene therapy trials. Clin Cancer Res 16: 5852–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, PF, Dudley, ME, Wunderlich, J, El-Gamil, M, Li, YF, Zhou, J et al. (2004). Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol 173: 7125–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, J, Khong, HT, Dudley, ME, El-Gamil, M, Li, YF, Rosenberg, SA et al. (2005). Survival, persistence, and progressive differentiation of adoptively transferred tumor-reactive T cells associated with tumor regression. J Immunother 28: 258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins, RE, Gilham, DE, Debets, R, Eshhar, Z, Taylor, N, Abken, H et al. (2010). Development of adoptive cell therapy for cancer: a clinical perspective. Hum Gene Ther 21: 665–672. [DOI] [PubMed] [Google Scholar]
- Lamers, CH, van Elzakker, P, Langeveld, SC, Sleijfer, S and Gratama, JW (2006). Process validation and clinical evaluation of a protocol to generate gene-modified T lymphocytes for imunogene therapy for metastatic renal cell carcinoma: GMP-controlled transduction and expansion of patient's T lymphocytes using a carboxy anhydrase IX-specific scFv transgene. Cytotherapy 8: 542–553. [DOI] [PubMed] [Google Scholar]
- Lamers, CH, van Elzakker, P, Luider, BA, van Steenbergen, SC, Sleijfer, S, Debets, R et al. (2008). Retroviral vectors for clinical immunogene therapy are stable for up to 9 years. Cancer Gene Ther 15: 268–274. [DOI] [PubMed] [Google Scholar]
- Lamers, CH, van Elzakker, P, van Steenbergen, SC, Luider, BA, Groot, C, van Krimpen, BA et al. (2013). Long-term stability of T cell activation and transduction components critical to the processing of clinical batches of gene-engineered T cells. Cytotherapy, in press (doi:10.1016/j.jcyt.2012.12.006). [DOI] [PubMed]
- Lamers, CH, Willemsen, RA, Luider, BA, Debets, R and Bolhuis, RL (2002). Protocol for gene transduction and expansion of human T lymphocytes for clinical immunogene therapy of cancer. Cancer Gene Ther 9: 613–623. [DOI] [PubMed] [Google Scholar]
- Lamers, CH, Willemsen, RA, van Elzakker, P, van Krimpen, BA, Gratama, JW and Debets, R (2006). Phoenix-ampho outperforms PG13 as retroviral packaging cells to transduce human T cells with tumor-specific receptors: implications for clinical immunogene therapy of cancer. Cancer Gene Ther 13: 503–509. [DOI] [PubMed] [Google Scholar]
- Canevari, S, Stoter, G, Arienti, F, Bolis, G, Colnaghi, MI, Di Re, EM et al. (1995). Regression of advanced ovarian carcinoma by intraperitoneal treatment with autologous T lymphocytes retargeted by a bispecific monoclonal antibody. J Natl Cancer Inst 87: 1463–1469. [DOI] [PubMed] [Google Scholar]
- Eisenhauer, EA, Therasse, P, Bogaerts, J, Schwartz, LH, Sargent, D, Ford, R et al. (2009). New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45: 228–247. [DOI] [PubMed] [Google Scholar]
- Steffens, MG, Boerman, OC, de Mulder, PH, Oyen, WJ, Buijs, WC, Witjes, JA et al. (1999). Phase I radioimmunotherapy of metastatic renal cell carcinoma with 131I-labeled chimeric monoclonal antibody G250. Clin Cancer Res 5 (suppl. 10): 3268s–3274s. [PubMed] [Google Scholar]
- Grabmaier, K, Vissers, JL, De Weijert, MC, Oosterwijk-Wakka, JC, Van Bokhoven, A, Brakenhoff, RH et al. (2000). Molecular cloning and immunogenicity of renal cell carcinoma-associated antigen G250. Int J Cancer 85: 865–870. [DOI] [PubMed] [Google Scholar]
- Uemura, H, Okajima, E, Debruyne, FMJ and Oosterwijk, E (1994). Internal image antiidiotype antibodies related to renal-cell carcinoma-associated antigen G250. Int J Cancer 56: 609-614. [DOI] [PubMed] [Google Scholar]
- Lamers, CH, Gratama, JW, Pouw, NM, Langeveld, SC, Krimpen, BA, Kraan, J et al. (2005). Parallel detection of transduced T lymphocytes after immunogene therapy of renal cell cancer by flow cytometry and real-time polymerase chain reaction: implications for loss of transgene expression. Hum Gene Ther 16: 1452–1462. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.