

Abstract
The Scianna system was named in 1974 when it was appreciated that two antibodies described in 1962 in fact identified antithetical antigens. However, it was not until 2003 that the protein on which antigens of this system are found and the first molecular variants were described. Scianna was the last, previously serologically defined, protein-based blood group system to be characterized at the molecular level, marking the end of an era in immunohematology. This story highlights the critical role that availability of laboratory reagents for serologic testing has played in the initial characterization of a blood group and sets the stage for the development of new reagents, such as recombinant proteins, to assist in this process. The central role that genetics has played, both by classic pedigree analysis and by molecular techniques, in the discovery and characterization of this blood group is reviewed.
The high- and low-prevalence antigens that constitute the Scianna (SC) blood group system are caused by variants in the erythroid membrane-associated protein (ERMAP).1 Scianna was initially identified by serologic methods; the clinical significance of antibodies specific to SC is uncertain, although case reports demonstrating rare cases of hemolytic disease attributed to SC variants exist. Genetic analyses, both in the classic and molecular approaches, have been central to the discovery and elaboration of the SC system. This article reviews the story of the SC blood group from a genetic point of view, emphasizing the way it has been brought into focus thanks to genetic tools ranging from pedigree analysis to physical mapping.
History
Nomenclature: Sc1, Sc2, Sc3, and Sc4
The story of the 13th International Society of Blood Transfusion (ISBT) blood group system began in 1962, when a new high-prevalence antigen was reported alongside a coexisting anti-D in a 25-year-old, multiparous woman of Italian descent in Miami, Florida, who experienced several fetal deaths as a result of hemolytic disease of the fetus and newborn (HDFN).2 She came to clinical attention because of difficulty obtaining compatible blood. Her ABO and Rh typings were O ccddee, and her husband’s were O CCDee. After an unremarkable first pregnancy and birth, she experienced three subsequent and progressively earlier fetal demises at term and at 7 and 6 months gestation in the late 1950s. After her second fetal death, her anti-D titer was demonstrated at 256, and the new antibody to a high-prevalence antigen, originally named anti-Sm, was demonstrated at a titer of 16. An informative family study revealed three antigen-negative siblings with a likely autosomal dominant mode of antigen inheritance, and no unrelated antigen-negative specimens were identified in a population survey of 600 D– random individuals. A clue to the genetic position of the responsible locus was present even in this defining family: based on the pedigree, it could not be determined whether the new antigen was part of the Rh system as it was in linkage disequilibrium with cc in that kindred.
In spite of this very dramatic introduction, the clinical importance of the new antigen was uncertain, as the concurrent anti-D clearly could account for the proband’s unfortunate obstetric history. While the work of the Miami group was in the pipeline for publication, the Winnipeg Rh Laboratory, in Manitoba, reported an antibody to a new low-prevalence antigen arising in a 50-year-old man with stomach cancer.3 In this patient, the antibody originally named anti-Bua found in serum Char. was identified during a routine pretransfusion crossmatch. As the patient had been transfused with three units of blood 14 days earlier, this delayed serologic transfusion reaction was investigated, which revealed that although his serum was crossmatch-compatible with all three donor samples before transfusion, it reacted with one of the three samples after transfusion. A follow-up survey of 18 panel red blood cells (RBCs) demonstrated one reactive cell, suggesting a relatively high prevalence for this new antigen; however, this proved not to be the case, as only one of the next 1,000 donors was positive. The families of all three of these probands took part in pedigree analysis, one of which was extremely informative with a kindred of both parents and nine offspring. These studies in classic genetics demonstrated that the new locus segregated independently from ABO, MNSs, P, Rh, Kell, Kidd, Duffy, and X-chromosome.
Genetics and Inheritance
It did not take long for the relationship between the Sm and Bua to be postulated, tested, and proven. In 1964, the anti-Bua serum was used to type the available members of the index Sm family (Fig. 1). The importance of using this serum as a typing reagent is underscored by the fact that it was required to demonstrate that the parent generation consists of a mating of two Sm/Bua heterozygotes (parents PM Sr. and RM): the F1 generation consists of four Sm– homozygotes, one Sm/Bua heterozygote (individual AM), and one Bua/Bua homozygote (individual CS). Without it, the zygosities of AM and CS could not be determined. This is the only outbred family in which both parents are Sm/Bua heterozygotes.
Fig. 1.
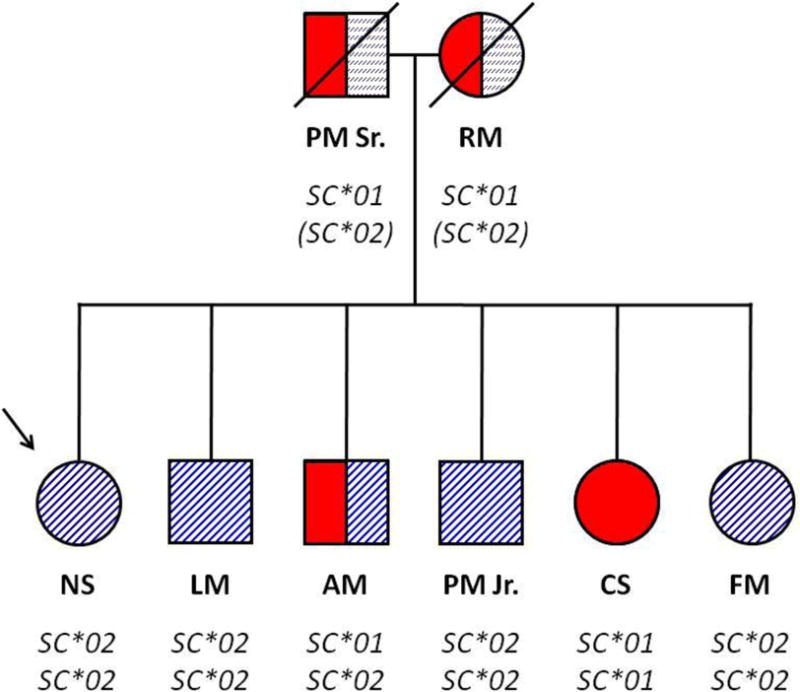
Index family in the characterization of the Sm antigen and demonstration of the antithetical relationship between Sm and Bua antigens. The proband (patient Ms. Scianna) is indicated by the arrow. Solid color represents Sm+ (Sc:1+) antigen test. Striped fill represents Bua+ (Sc:2+) antigen test. Inferred genotype is shown below the symbol for each family member. The proband’s parents were deceased at the time of Bua testing, so are inferred to be heterozygotes, indicated by the interrupted stripes and parentheses in their genotype designation. The combination of Sm and Bua typing confirms that subject AM is a heterozygote, which was suggested by observations of dosage effects in serologic testing. (Redrawn with data from references 2 and 4.)
Concurrent with their suggestion that Sm and Bua were the result of a biallelic polymorphism, the Winnipeg Rh Laboratory also reported an extensive study of a Mennonite population in whom the Bua antigen had a considerably higher frequency of 5 in 348 samples than the 1 in 1,000 prevalence observed in other Caucasian populations.4,5 Although they tested 145 Caucasian families for Bua and rigorously examined 19 based on the presence of Bua, it was not until additional reagent serum was available in 1966 that definitive proof of this relationship was found.
Further examples of anti-Bua were discovered in a group of individuals from Poland and the United Kingdom (particularly the strongest serum Soch.) who had produced anti-Bua after having been artificially immunized with D+ cells to stimulate the production of Rh antibodies.6 Through a careful study of the donors used for these stimulations, three additional subjects who were also stimulated with these cells were found to have created an anti-Bua, although it was much weaker than that in the Soch. serum. The prevalence of the Bua allele was determined as 0.88 percent in a Warsaw and 0.67 percent in a London cohort.
Using this reagent after adsorption to remove the iatrogenically generated anti-D also present in the serum, the Winnipeg Rh Laboratory identified a large, six-generation Mennonite kindred that included two cousin matings of Sm/Bua heterozygotes, which produced 20 offspring.7 Through a methodical review of the serologic data from samples from the kindred, which showed the effects of antibody dosage on antigen expression, this report confirmed and expanded the definition of the new system to exclude all blood groups reported before 1962 (except Diego, Yt, and Auberger) and Doa and Csa. Independence from Yt was established in two British families.8,9 Finally, the currently accepted nomenclature (Table 1) was proposed in 1974 to reflect the surname of the index family in the discovery of anti-Sm, Scianna,10 such that Sm was renamed Sc1 and Bua was renamed Sc2.
Table 1.
Alleles, encoded antigens, and ISBT terminology of the Scianna blood group system
| Antigen | Trivial name | Wild-type
|
Variant
|
||||
|---|---|---|---|---|---|---|---|
| Allele | Genotype | High-prevalence antigen | Allele | Genotype | Low-prevalence antigen | ||
| Sc1 | Sca (Sm) | SC*01 | 169g | 57Gly Sc1 | |||
| Sc2 | Scb (Bua) | SC*02 | 169a | 57Arg Sc2 | |||
| Sc3 | Sc null | SC*03N.01† | 307Δ2 | Frameshift: null | |||
| Sc3 | Sc null | 994c | 332Arg | SC*03N.02† | 994t | 332Stop: null | |
| Sc4 | Rd | 178c | 60Pro Rd− | SC*04 | 178g | 60Ala Rd+ | |
| Sc5 | STAR | 139g | 47Glu STAR+ | SC*05 | 139a | 47Lys STAR− | |
| Sc6 | SCER | 242g | 81Arg SCER+ | SC*06 | 242a | 81Gln SCER− | |
| Sc7 | SCAN | 103g | 35Gly SCAN+ | SC*07 | 103a | 35Ser SCAN− | |
Provisional name suggested by the International Society of Blood Transfusion (ISBT) Working Party on Red Cell Immunogenetics and Blood Group Terminology
The SC Gene Lies on Chromosome 1
The focus then shifted from definition of the system to characterization of the locus responsible for the antigen at a chromosomal level. Much of this seminal work was performed in the Winnipeg Rh Laboratory. So it is not surprising that their detailed investigations of the genetic mapping of chromosome 1 contributed significantly to the progress on SC. Their unique access to informative kindreds certainly hastened this process, and in a series of reports from 1976 to 1978, linkage between RH and SC was established, showing a logarithmic odds ratio score (LOD) of 5.34 at a recombination fraction, θ = 0.10 if paternally segregated,11 and the relative position of SC was determined12–14 and refined.15
A Third Antigen in the System: Sc3 or Scianna Null Alleles
The appearance of “minus-minus” phenotypes, i.e., individuals whose RBCs tested negative for both Sc1 and the antithetical Sc2 antigen (Sc:–1,–2), was first documented in 1973 and demonstrated the existence of apparent SC null alleles.16 However, the term Sc3 was not coined until 1980 when an antibody from an Sc:–1,–2 individual demonstrated no evidence of a separable anti-Sc1 or anti-Sc2.17 The index patient examined in 1973 was a female surgical patient from the Likiep Atoll in the Marshall Islands, who had been transfused 7 months earlier without crossmatching difficulty. Her cells phenotyped as Sc:–1,–2 as did those from one cousin, but both women’s RBCs could still adsorb anti-Sc2. Despite four pregnancies with an Sc:1,–2 husband, the proband’s cousin had a negative antibody screen. Because these RBCs reduced the titer of anti-Sc2, but not anti-Sc1, they may have had some weak Sc2 expression, and a new antigen was not definitively characterized at that time.
However, in 1980, a 67-year-old man in New York being treated with chemoradiotherapy for metastasis to the throat of a carcinoma of unknown origin required a preoperative transfusion, and demonstrated a positive antibody screen.17 He had been transfused four years earlier without event. Unlike the Likiepian Sc:–1,–2 erythrocytes, RBCs from this patient did not reduce the titer of either anti-Sc1 or anti-Sc2. When tested against the Likiepian Sc:–1,–2 cells, the New York serum did not agglutinate the RBCs. No other Sc:–1,–2 cells were found in a family study. No separable anti-Sc1 was present in this patient’s serum, which is somewhat unusual because patients who completely lack all known antigens for a blood group and have been transfused typically make a polyclonal mixture of antibodies directed at the common epitopes of that system.18
The next report of a null phenotype for SC was described in 1986, in another Pacific Islander.19 This patient was a 4-year-old girl from Papua New Guinea with thalassemia major who had been transfused many times. Anti-Sc3 was demonstrated. Erythrocytes from the patient’s mother were compatible, and both mother and daughter were found to have the Sc:–1,–2 phenotype. In this community, the Sc:–1,–2 phenotype was surprisingly common: A survey of 29 family members and apparently unrelated villagers revealed 6 others (2 of whom had no obvious relationship to the patient) who were also Sc:–1,–2. This very high phenotype prevalence (20.6%) makes heterozygosity for a putative recessive Sc3 allele in the patient’s father quite likely (although his RBC phenotype is not reported in the short abstract), as well as explaining the homozygosity for the same allele observed in her mother.
Additional Sc:–1,–2 individuals have been reported (including a patient who experienced an apparent delayed hemolytic transfusion reaction), some of whom have also lacked several other high-prevalence antigens.18 Evidence that each patient in their series produced antibodies with different specificities is discussed in Antibodies section.
Radin: A “New” Low-Prevalence Antigen Is Linked to Scianna (via RH)
At the time of its first description in 1967,20 the relationship between Radin and Scianna was not appreciated. Although the clinical significance of antibodies generated to antigens within the SC system in general is controversial, the analysis of the antibodies that led to the discovery of the Radin antigen is based on its clinical importance, with the initial description encompassing five cases of mild to moderate hemolytic disease of the newborn, one of whom required exchange transfusion and one appearing in the first pregnancy.20 Additional cases and confirmation of the low general frequency (1 in 205) appeared shortly thereafter.21
Once again, it was the unique analysis performed by the Winnipeg Rh Laboratory that elucidated the putative connection between Radin and SC. Linkage analysis of eight propositi in ten nuclear families demonstrated linkage between Rd and RH.22 Similar to the Sc1/Sc2 linkage analysis in the present dataset, the Winnipeg analysis demonstrated heterochiasmy, with the paternal LOD. score exceeding 3 at a recombination fraction of 0.10, whereas the corresponding maternal LOD neither suggests nor refutes linkage (LOD = 0.41 at a recombination fraction of 0.10). This finding was only sufficient to propose the connection between SC and Rd, as heterozygotes at both of these (i.e., double recombinants) had not been found. Incorporation of the gene encoding the Rd antigen into the bigger picture of chromosome 1 mapping placed it so close to SC that it was proposed even at that time that the gene encoding Rd “is either very closely linked to or identical with SC.”23
There were few additional reports on the SC blood group system for more than 20 years until its molecular basis was finally resolved in 2003.1 Since then, molecular analysis has identified the three additional alleles underpinning the three antibodies described by Devine and coworkers in 1988.18
Molecular Basis
The ERMAP Expresses the SC Antigens
The molecular basis of the SC blood group system was identified in 2003 by merging the genetic mapping data with protein chemistry showing that the SC antigens were on a single glycoprotein of approximately 60 to 68 kDa that must be expressed by RBCs.24,25 The SC gene had been mapped to chromosome 1, and based on its linkage to the RH genes, the chromosomal location had been further refined to 1p34 to 1p36. This led to the identification of a strong candidate gene.1
The ERMAP is a 475-amino acid, type 1 single-pass membrane glycoprotein that is a member of the butyrophilin (BTN) family and is encoded by the ERMAP gene.26,27 It consists of a predicted signal sequence of 29 amino acids at the NH3 terminus, an extracellular immunoglobulin V domain (amino acids 50–126), a short transmembrane domain spanning amino acids 157 through 176, and an intracellular carboxyl terminus encompassing a B30.2 domain from amino acids 238 through 395.1
The nomenclature for the transcripts of ERMAP is complicated and has been revised many times. At least two transcript variants have been described, which result from use of an alternative upstream promoter and alternative splicing.28 The longer variant is 3,423 bp and is designated as transcript 1, or as transcript b by GenBank curators29 as it was the second transcript variant identified, and includes an additional upstream exon (GenBank NM_001017922.1). The shorter variant is designated as transcript 2 or as transcript a, is 3,369 bp, excludes this upstream exon (GenBank NM_018538.3), begins transcription 45 bp upstream from the start of exon 2, and was the first variant discovered; thus, early reports describe this gene as consisting of 11 exons. However, the revised current nomenclature for this gene is based on the presence of 12 exons spanning approximately 28 kb. The relative production of these two mRNAs is not known, nor has it been determined whether the use of either promoter is favored in some tissue types or physiologic conditions. Both transcripts share the common ATG start codon in exon 3; thus, there is no predicted difference in the protein product of the two transcripts. They differ in both the transcript initiation site and the DNA stretch of exon 2 that becomes part of the final transcript; consequently, there are two alternative exon 2s, named exon 2a and exon 2b. The longer transcript 1 comprises exon 1, exon 2b, and exons 3 through 12, whereas the shorter transcript 2 starts with exon 2a and also includes exons 3 through 12 (Fig. 2).
Fig. 2.
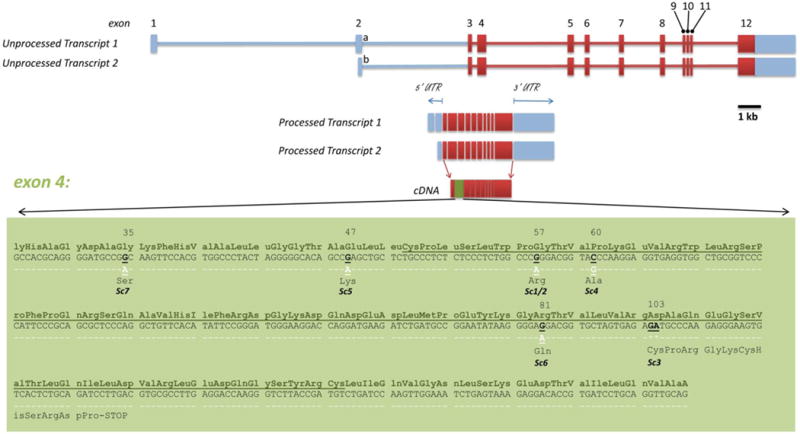
Transcripts, exon structure, and variants of erythroid membrane-associated protein (ERMAP). Codon numbers are indicated above amino acids involved in described variants. The immunoglobulin variable domain is underlined. Exon 4 is expanded to the nucleotide level in the green box to show the locations of most ISBT-recognized Scianna polymorphisms. The wild-type reference sequence is under the corresponding amino acid and is the chromosome 1 reference sequence GenBank NC_000001.10. * = nucleotide deletion.
The Ensembl genome browser curators have delineated five transcripts in ERMAP,30,31 three of which are protein coding and two of which generate a processed transcript only. Two of the Ensembl transcripts correspond to GenBank’s transcripts 1 and 2; however, unique to the Ensembl database is a 3,949-bp transcript (named ERMAP-002),31 which is reported as protein coding and generates a short protein encoding 385 amino acids rather than the usual 475. However, only the two transcripts that encode the 475-amino acid protein are included in the Consensus Coding Sequence Project (CCDS; both with identification number CCDS475), so the biological significance of the 3,949-bp transcript is unclear.
In addition to explaining the Sc1/Sc2 biallelic single nucleotide polymorphism (SNP) at Gly57Arg, a binucleotide GA deletion was identified at nt307 (now known as SC*03N.01, provisional name suggested by the ISBT Working Party on Red Cell Immunogenetics and Blood Group Terminology). This deletion causes a frameshift and premature stop codon which explain the SC null phenotype1 The Rd antigen (Sc4) was defined as the Pro60Ala variation, and two other variants in the presumed leader signal peptide (54C>T and 76C>T) were described in the original study elucidating the molecular basis by Wagner and colleagues1 (Table 2).
Table 2.
Erythroid membrane-associated protein polymorphisms in the coding region and global population frequencies
| Sc name | Descriptive name | dbSNP ID: | cDNA nt | mRNA* | Wild-type (WT) | Variant | Protein effect | Amino acid (aa) codon | WT aa | Variant aa | Exon | Protein domain | Minor allele frequency in blood donors† |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs35757049 | 11 | 281 | c | t | nonsynonymous substitution | 4 | Ala | Val | 3 | Leader | 0.054 (dbSNP) | ||
| rs33950227 | 54 | 324 | c | t | silent | 18 | Leu | Leu | 3 | Leader | 0.28 (German1), 0.1 (dbSNP) | ||
| rs33953680 | 76 | 346 | c | t | nonsynonymous substitution | 26 | His | Tyr | 3 | Leader | 0.33 (German1), 0.25 (Caucasian North American32), 0.1 (dbSNP), 0.05 (African-American32) | ||
| Sc7 | SCAN | 103 | 373 | g | a | nonsynonymous substitution | 35 | Gly | Ser | 4 | NH3-terminus | ||
| Sc5 | STAR | rs56047316 | 139 | 409 | g | a | nonsynonymous substitution | 47 | Glu | Lys | 4 | NH3-terminus | |
| Sc1 vs. Sc2 | rs56025238 | 169 | 439 | g (Sc1) | a (Sc2) | nonsynonymous substitution | 57 | Gly | Arg | 4 | IgV loop | 0.01 (Caucasian North American,33 Brazilian34), 0.008 (German1), 0.005 (Hispanic American33), 0.002 (African-American33), 0 (Asian American33) | |
| Sc4 | Rd | rs56136737 | 178 | 448 | c | g | nonsynonymous substitution | 60 | Pro | Ala | 4 | IgV loop | 0.003 (Slavs33), 0.002 (Caucasian North American,33 Danish,21 German,1 New York Jewish20) |
| rs33954154 | 219 | 489 | c | t | silent | 73 | Arg | Arg | 4 | IgV loop | 0.017 (dbSNP) | ||
| Sc6 | SCER | 242 | 512 | g | a | nonsynonymous substitution | 81 | Arg | Gln | 4 | IgV loop | ||
| Sc null (Δ ga together) | rs55695242 | 307 | 577 | g | del −1 | frameshift | 103 | Asp | frameshift | 4 | IgV loop | ||
| Sc null (Δ ga together) | rs56151267 | 308 | 578 | a | del −1 | frameshift | 103 | Asp | frameshift | 4 | IgV loop | ||
| rs35147822 | 775 | 1045 | t | c | nonsynonymous substitution | 259 | Cys | Arg | 12 | B30.2 domain | 0.022 (dbSNP) | ||
| rs34441268 | 788 | 1058 | g | a | nonsynonymous substitution | 263 | Gly | Glu | 12 | B30.2 domain | 0.011 (dbSNP) | ||
| rs35972628 | 888 | 1158 | g | a | silent | 296 | Glu | Glu | 12 | B30.2 domain | 0.011 (dbSNP) | ||
| Sc null | 994 | 1264 | c | t | nonsense (STOP) | 332 | Arg | STOP | 12 | B30.2 domain | |||
| rs55773259 | 1227 | 1497 | a | g | silent | 409 | Leu | Leu | 12 | C-terminus | |||
| rs55872827 | 1324 | 1594 | t | c | nonsynonymous substitution | 442 | Ser | Pro | 12 | C-terminus | |||
| rs56405033 | 1355 | 1625 | t | c | nonsynonymous substitution | 452 | Leu | Pro | 12 | C-terminus at a 3′ dileucine (LL) phosphorylation motif | |||
| rs55677363 | 1356 | 1626 | g | t | silent | 452 | Leu | Leu | 12 | C-terminus at a 3′ dileucine (LL) phosphorylation motif | 0.054 (dbSNP) |
NCBI Reference Sequence Position: NM_001017922.1
Minor allele frequencies for some populations are determined by reports of serologic phenotype and assuming individuals positive for a rare antigen are heterozygotes.
cDNA = complementary DNA; dbSNP = single nucleotide polymorphism database; IgV = immunoglobulin V; mRNA = messenger RNA; nt = nucleotide.
Understanding the Variants Discovered in the Post-ERMAP Era
The knowledge of the gene responsible for the antigens of this blood group protein opened the floodgates through which variant descriptions of unknown, unresolved serologic cases promptly flow. This was clearly the case for SC, for which three new variants were discovered within just a few years after the characterization of ERMAP as the SC gene. Careful molecular follow-up of the three patients described by Devine and coworkers18 in 1988 revealed three distinct ERMAP/SC variants, demonstrating the importance of molecular testing in the resolution of serologic SC mysteries. The success of these investigations depended not only on the cooperation among an international collaborative, but also on the supportive participation of two patients and a family who kindly released two autologous RBC units. It was appreciated in the initial case reports in 1988 that the SC protein carried multiple high-prevalence antigens other than Sc1/2.18 Sera or eluates from these three Sc:1,–2 patients who had developed high-prevalence antibodies did not react with the Sc:–1,–2 null RBCs, but the samples were not mutually compatible.
Sc5 (STAR)
In 1982, a 65-year-old man with a history of transfusion of three units of crossmatch-compatible whole blood before presentation underwent routine preoperative blood bank testing, demonstrating anti-C and anti-e. He was transfused with another three units of C–e– RBCs, and 1 week later, a new anti-Jkb and an antibody against a high-prevalence antigen were detected. Because his serum reacted with all cells except his own (phenotyped as Sc:1,–2), those of a sibling, and Sc:–1,–2 RBCs, it was suspected that he had developed an antibody to another antigen on the SC protein.35 Blood needs for the patient were met with autologous units. Blood samples from the proband and 16 family members had been frozen. Sequencing of the exons encoding the extracellular and transmembrane domains demonstrated homozygosity at a new nonsynonymous polymorphism at amino acid 47 (glutamic acid to lysine), which is in the N-terminal domain.36 This variation is catalogued as rs56047316 in the SNP database (dbSNP), and it results from the guanine–adenine transition at nucleotide 139 in the complementary DNA (cDNA). Frequencies of this allele in population studies have not been reported, nor have additional cases of this antibody.
Sc6 (SCER) and Sc7 (SCAN)
The remaining two antibody cases described by Devine and colleagues18 were concomitantly resolved using the same approach as for the Sc5 antigen.37 These investigators sequenced all 11 exons of ERMAP known at the time, which encompasses all cDNA. They also solicited others to submit suspected SC variants and the eight other orphan low-prevalence antigens By, Toa, Pta, Rea, Jea, Lia, SARA, and Ska. The proband for case 1 reported by Devine and colleagues18 demonstrated homozygosity for a guanine–adenine transition at cDNA nucleotide 242, predicted to cause an amino acid change at position 81 (from arginine to glutamine), which is in the immunoglobulin V loop.18 The case 2 proband demonstrated a new homozygous, nonsynonymous variant at cDNA nucleotide position 103 (again a guanine–adenine transition), corresponding to a glycine to serine change at amino acid 35 in the NH3 terminus. This specimen also demonstrated two other SNPs (at cDNA nucleotides 54C>T and 76C>T), which had been previously reported, are common in Caucasian populations, and are in tight linkage disequilibrium (LD).1 Because these variants are both in the predicted leader sequence (and the 54C>T is a silent mutation), they are not predicted to directly impact on ERMAP structure. This report also excluded the eight orphan low-prevalence antigens from the SC system because the only variants found were in the leader sequence or in introns.
Molecular Basis of SC Null Phenotypes
Central to the set of specimens originally reported with the discovery of ERMAP as SC was an Sc:–1,–2 sample from a Saudi Arabian pedigree,1 which demonstrated homozygosity at three SNPs: the common 54C>T and 76C>T (described in an earlier section) as well as a 2-bp deletion starting at cDNA nucleotide 307 (307Δga) that is predicted to cause a frameshift mutation and early termination codon after 113 amino acids. The nt54 and nt76 SNPs were genotyped in an additional 111 European blood donors and were found in tight LD. The frameshift would account for a complete lack of an intact ERMAP protein in the cell membrane.
Two Sc:–1,–2 individuals from northern and southern California were then sequenced in a follow-up study,37 and both shared a new SC null allele formed by a nonsense mutation at codon 332. This is the first report of a variant in the B30.2 intracellular domain in a patient immunized to SC. It is not, however, the only variant in the B30.2 domain (Table 2), because three others have been described in dbSNP, two of which are nonsynonymous amino acid changes (C259R and G263E) and one of which is silent (E296E). This result suggests that an early translation termination, even as late as at codon 332 of the expected 475, sufficiently interferes with correct protein trafficking, membrane insertion, or stability so as to render the individual susceptible to alloimmunization at the Sc1/2 site (codon 57).
Other Variants in ERMAP: Using dbSNP and HapMap
Variants in the coding region, particularly those in the extracellular domain of transmembrane proteins, are of particular clinical interest in transfusion medicine, as their location provides an obvious mechanism for putative clinical significance by alloimmunization. However, much variation in the human genome is found outside of coding regions, and ERMAP is no exception. Studies of this variation are a critical tool in the investigation of human disease and basic sciences. Several collaborative projects in human genomics focus on gathering, sharing, and interpreting human genetic variation, and although a full cataloguing of these is beyond the scope of this review, we will present variants in ERMAP that are part of two such projects: dbSNP38 and the International HapMap Project.39 Table 2 integrates some of these variants (identified by their rs numbers) with those described in the transfusion medicine literature. These data allow informative LD studies of ERMAP (Fig. 3). This LD map shows two distinct LD blocks that correspond to the exons encoding the extracellular immunoglobulin-like domains and the intracellular B30.2 domains of ERMAP. The importance of this pattern of LD is that it reveals a possible evolutionary vestige of the modular nature of the butyrophilin-like (BTNL) protein family and is even more pronounced in the Nigerian population. Long stretches of LD such as that illustrated in the extracellular domain of ERMAP, where all of the SC antigens are encoded, raise the possibility that a selective advantage by genetic hitchhiking may be operating at ERMAP.40
Fig. 3.

Pairwise linkage disequilibrium (LD) heat plot of the logarithmic odds ratio (LOD) score for single nucleotide polymorphisms in erythroid membrane-associated protein (ERMAP) in a Nigerian population. Darker shades (red) = areas of high LD, lighter shades (blue) = areas of lower LD. The areas of tightest LD correspond to the extracellular domains of ERMAP, on the left-hand side of the figure. Increased LD is also seen in the intracellular domains, at the right side. This segmental pattern of LD reveals a possible evolutionary vestige of the modular nature of the butyrophilin-like protein family. YRI = Yoruba in Ibadan, Nigeria. (Source: Haploview™ software run at http://hapmap.ncbi.nlm.nih.gov/cgi-perl/gbrowse/hapmap3r2_B36/#search; accessed April 27, 2010.)
Global Variation
Some ethnographic trends have already been historically appreciated with SC variants during the early years of their investigation (Table 2). For instance, Sc4 has been identified most often in Ashkenazi Jews and Slavic populations, and the few Sc:–1,–2 phenotypes have been reported particularly in populations from Oceania. Sc2 appears to be more common in a consanguineous Canadian Mennonite population (derived from a small region in Eastern Europe),5 but it remains very rare or absent from populations of African5,33 or Native American10 descent. However, it is not sufficient to rely on case reports to propose generalizations regarding population frequencies. Fortunately, recent technologies are available to rapidly genotype ERMAP variants using an automated genotype-calling platform, and additional population studies will continue to be reported.33
Biochemistry and Physiology
ERMAP Is a Member of the Butyrophilin-like Family of the Immunoglobulin Superfamily
By virtue of its extracellular immunoglobulin V and the intracellular B30.2 domains, ERMAP is a member of the BTNL protein family, which is a subset of the immunoglobulin superfamily.41 The compact, globular 110–amino acid immunoglobulin domain defines this superfamily and is found in three operational domain subclasses: variable (immunoglobulin V), intermediate (immunoglobulin I), or constant (immunoglobulin C).42,43 These proteins are central to many immunologic processes, especially cell adhesion, costimulation, and signalling.44,45 The immunoglobulin superfamily is one of the largest in all eukaryotic organisms.46 Other members of this family well known to the transfusion medicine community include the Lutheran, LW, OK, JMH, and Indian blood groups, which are found on the B-CAM, ICAM4, CD147, CD108, and CD44 molecules, respectively.47 BTN proteins are within the B7-CD28-like branch of this superfamily48 and have been extensively studied in cows.49 The prototype of the human BTN family of proteins is BTN1A1, which is primarily expressed in the lactating breast, where it makes up 20 percent of the protein in the membrane of milk fat globules. The name derives from the Greek butyros and philos, meaning “having an affinity for butterfat.” Although ERMAP is carried on chromosome 1, the genes for many members of this family are located in the major histocompatibility complex (MHC) on chromosome 6.50
The lactational and immunologic functions of BTNs as a part of the secretory granule–to–plasma membrane zipper complex and as an inhibitor of T-cell activation are only recently described,51,52 and even less is known about the BTNL proteins. There are six known human BTNL proteins, which are defined by their homology to BTN proteins (Fig. 4). The most extensively studied of these is BTNL2, the gene for which is located in the MHC class II cluster at 6p21.3. A truncating splice site mutation in BTNL2 has been associated with sarcoidosis,53 and although this SNP has been investigated in many other infectious and autoimmune diseases, some of these associations have been attributable to LD with the MHC.54 BTNL2 inhibits T-cell activation by inhibiting proliferation in response to the stimulatory T-cell receptor signal.55 This inability for BTNL2 to propagate a cell signal has led some investigators to propose its role as “decoy receptor” because it lacks the B30.2 intracellular domain present in most other BTN and BTNL proteins.54
Fig. 4.
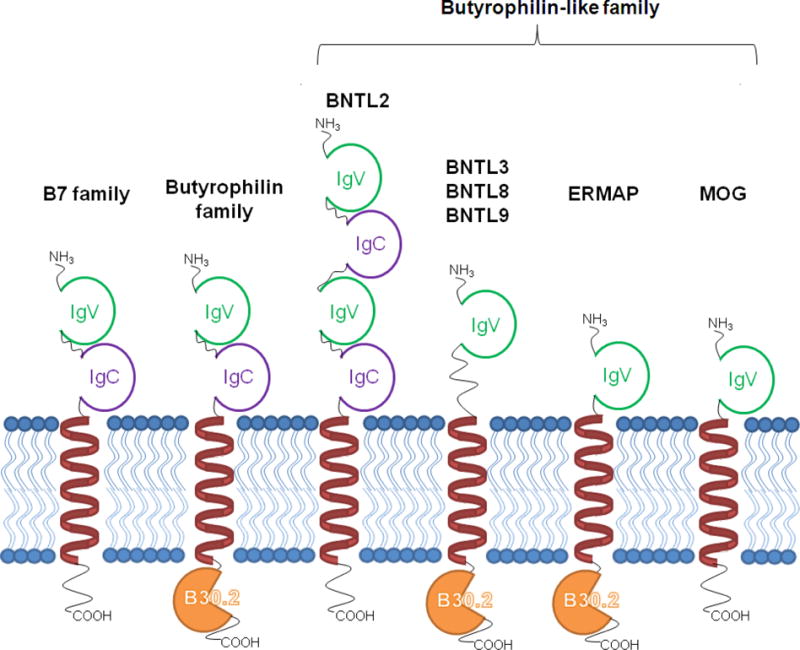
The organization of the butyrophilin-like (BTNL) family in comparison to the butyrophilin (BTN) and B7 families. The B7 branch of the immunoglobulin superfamily is characterized by variable (IgV) and constant (IgC) immunoglobulin domains. The BTN subfamily includes the B30.2 intracellular domain. The BTNL proteins exhibit various combinations of these features. MOG = myelin oligodendrocyte glycoprotein, an autoantigen implicated in multiple sclerosis. (Modified from reference 52.)
The B30.2 Cytoplasmic Domain: A Role in Erythropoiesis?
The B30.2 (or PRYSPRY) domain was described in 1993 with the discovery of an exon in the MHC class I region showing similarity to other mammalian and amphibian proteins, which have since been termed the tripartite motif family.56,57 Such a domain was demonstrated in bovine BTN58 and in human BTN proteins.59 The B30.2 domain is proposed to be a recent evolutionary adaptation in the immune system of mammals as a fusion of two ancient domains (PRY and SPRY), which are found in all eukaryotes.50 The genes for proteins in this family have a modular structure, suggesting that they arose by duplication.56 Mutations in the B30.2 domain in the tripartite motif–branch of the B30.2 proteins have been associated with Opitz syndrome (MID1 protein) and familial Mediterranean fever (pyrin protein). However, human syndromes have not been associated with mutations in the B30.2 domain of the BTN or BTNL families.
The crystal structure of the B30.2 domain of pyrin was recently elucidated. A β-barrel consisting of two antiparallel β-sheets has been described, forming a central cavity.60 The ligand(s) that interact with the B30.2 domain are yet to be described, although binding analyses suggest that it is the site of protein–protein interactions.61 The crystal structure of one other B30.2 domain interacting with a peptide has shown that there is a conformationally rigid peptide binding pocket (consisting of a core β-sandwich around variable loops) binding to a short-sequence motif, which may allow multiple intracellular targets to bind.62 Recently, BTN1A1 has been shown to bind xanthine oxidoreductase via B30.2, apparently stabilizing the milk fat globule membrane in mammary tissue, but it is also hypothesized to function as a novel signaling pathway in nonmammary tissues or perhaps in the innate immune system via generation of reactive oxygen species.63
A role for B30.2 domains in erythropoiesis has been revealed through study of an unlikely model organism: Antarctic icefish. These animals are the only vertebrate taxon that fails to produce RBCs (Fig. 5), and as such have been studied in a hunt for genes important in erythropoiesis and cardiovascular biology.64 Using this approach, a new B30.2-containing protein, called bloodthirsty (bty), was identified in the pronephric kidney of a red-blooded Antarctic rockcod (Notothenia coriiceps), which was present at levels tenfold higher than that in an icefish (Chaenocephalus aceratus).65 Interestingly, disruption of bty synthesis in zebrafish suppressed both erythrocyte production and hemoglobin synthesis, and although interactions between bty and ERMAP have been hypothesized, they have not been empirically demonstrated (H. William Detrich, personal communication, January 20, 2010).
Fig.5.
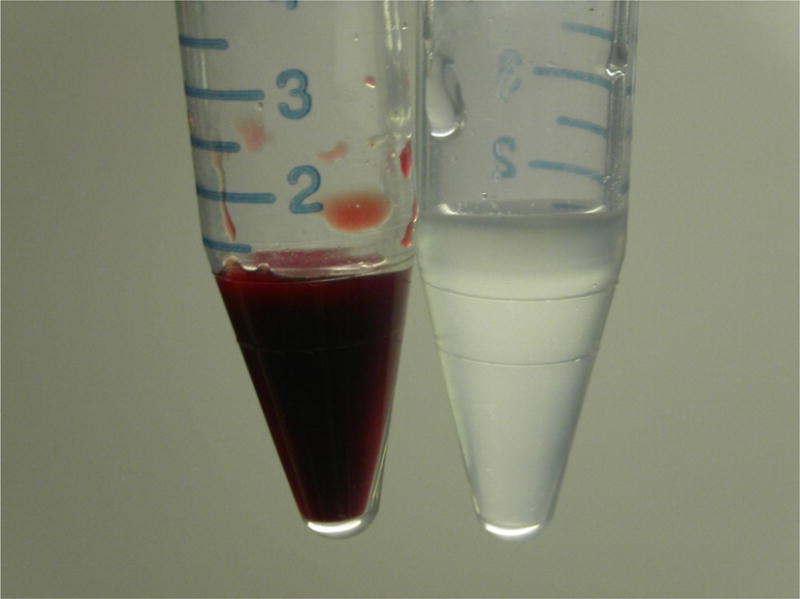
On the left, blood from a fish of the genus Notothenia (with hemoglobin-containing erythrocytes) and on the right is blood from Chaenocephalus aceratus. Icefish transport oxygen strictly in physical solution, without a carrier molecule. The recent discovery that bloodthirsty (bty), a B30.2-domain–containing protein like ERMAP, has dramatically decreased expression in hemoglobinless icefish provides a fascinating link between this protein domain and erythropoiesis. Courtesy of Professor Guillaume Lecointre.
Functional Studies of ERMAP
The function of ERMAP itself, however, is not well understood. The murine homolog, Ermap, was described first and named as such because it was found to be produced exclusively in erythroid cells.66 The human homolog was characterized shortly thereafter, and Northern blots demonstrated high expression in hematopoietic tissues, like fetal liver and bone marrow, with weaker expression in peripheral blood leukocytes, thymus, lymph node, and spleen.26 Sc1 has been demonstrated on phagocytic leukocytes using an antibody absorption technique.67 Recent RNA array expression analysis supports this finding, having detected low levels of ERMAP transcripts in leukocytes, especially monocytes; however, quantitative protein expression remains to be assessed.54 An in silico analysis of nucleotide database searches of human expressed-sequence tags using the ERMAP transcript 1 (GenBank NM_001017922.1) as the probe detected the transcript in cDNA libraries from hematogenous tissues, like bone marrow, a chronic myeloid leukemia cell line, peripheral blood pool, thymus, spleen, and fetal liver, as well as in neural tissues.68 However, it is difficult to definitively ascribe transcript production to the nonerythroid cells in the neural tissues, as contamination from the vasculature cannot be completely eliminated. ERMAP mRNA levels in fetal liver peaks in the 18th through 20th weeks, and is present from the 15th through 32nd weeks in fetal bone marrow, suggesting that ERMAP may be related to the migration of erythroid cells to these sites during hematopoietic development.69
Additional evidence that ERMAP may be involved in erythroid differentiation has been put forward, although only abstracts of these reports are available in the English literature. Using fluorescent quantitative polymerase chain reaction,70 ERMAP expression has been found in the K562 cell line, which is derived from chronic myeloid leukemia and is of undifferentiated granulocytic lineage.71 To test the degree of erythroid-specificity of ERMAP in hematopoiesis, this group used cytarabine (Ara-C) to induce these cells toward erythroid differentiation and 12-O-tetradecanoylphorbol-13-acetate to induce development toward the macrophage lineage, but found an increase in ERMAP mRNA after an Ara-C stimulation only.72 This finding suggests that although ERMAP may be present on cells of the monocyte/macrophage lineage, its functional importance may be limited in these cell types. RNA silencing experiments using an ERMAP shRNA/K562 cell line also reported by this group showed decreased ERMAP expression and morphologic features, including the relative amounts of surface erythrocyte maturation markers, leading the authors to conclude that ERMAP shRNA inhibited Ara-C–induced erythroid differentiation.73 A second model of erythroid differentiation using umbilical cord blood with two naturally occurring hormones (namely stem cell factor and IL-3) and erythropoietin to provoke differentiation also showed a concurrent rise in ERMAP mRNA with erythrocyte maturation.74
Many structural features of ERMAP point toward a putative role in immunity, perhaps by adhering to other cells or pathogens via its extracellular immunoglobulin V domain, or by immunoregulatory mechanisms such as the modulation of cellular activation signals via B30.2 as is observed in its BTN family members.52 Much work is needed in this area to better characterize the proteins that interact with ERMAP, both extracellularly and intracellularly, and to determine whether ERMAP has a central role in erythropoiesis itself.
Antibodies in the System
The discovery of the SC blood group system began with and has relied on the detection and investigation of alloantibodies. The effects of enzymes and chemicals and the in vitro characteristics of SC antibodies were recently reported elsewhere.75 In general, SC antigens are resistant to ficin plus papain, trypsin, α-chymotrypsin, sialidase, and 50 mM dithiothreitol (DTT), and they are sensitive to pronase and 200 mM DTT. The exception to this trend is the variable sensitivity of Sc4 to trypsin and α-chymotrypsin. Enzymatic properties of anti-SCER and anti-SCAN are only described in the initial case report as showing no change in the strength of antibody activity when tested with RBCs that had been treated with ficin, papain, trypsin, ZZAP (mixture of 0.1 M DTT plus 0.1% cysteine-activated papain), or chloroquine diphosphate.18 Anti-STAR demonstrated enhanced reactivity with enzyme-treated RBCs.36 Patients or donors in whom an antibody to an SC antigen has been reported (Table 3) have been thoroughly reviewed recently.76 Thirteen of the 19 reports of alloantibodies to SC system antigens listed in Table 3 have occurred in cases in which they do not appear to manifest a major clinical significance. All six of the reports with clinical significance occurred in cases of HDFN. All six of the reported autoantibodies to SC system antigens in patients were in the context of clinically significant autoimmune hemolytic anemia, although five of these cases were presented as an abstract only, without the full clinical details. Alleles Sc4–7 were not tested in any of these cases. Such an investigation is warranted because heterozygosity for alleles with weak expression in the Rh blood group system has been associated with D autoantibodies.86
Table 3.
Published reports of antibodies to antigens in the Scianna blood group system
| Antibody | Allo or auto? |
Authors | Year | Case identifier |
Scianna phenotype |
Age (years) |
Sex | Ethnicity | Important reactions? |
Clinical synopsis | Transfusion / pregnancy history |
Other antibodies |
Other laboratory data |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| anti-Sc1 | Allo | Schmidt et al.2 | 1962 | Mrs. N.S. | Sc:−1,2 | 25 | F | Caucasian | HDFN | Impossible to distinguish role of anti-Sc1 in presence of anti-D. | Multiparous, after 1st baby with HDFN. | anti-D | |
| Kaye et al.77 | 1990 | Sc:−1,2 | 28 | F | Indian | No HDFN | Neonate with 3+ DAT, eluted anti-Sc1, but Hgb = 13.5 g/dL and no HDFN. Mother B, R1r; infant B, rr; husband Sc:1,−2. | G2, 1st was uncomplicated with negative Ab screen. | Excluded all except E and Lu(a) | IgG3 subclass; steady rise in ADCC despite constant titer throughout pregnancy; 9 weeks postpartum ADCC increased ×4 and titer doubled (16 to 32). | |||
| Auto | Tregellas et al.78 | 1979 | Sc:1, −2 | 49 | Healthy blood donor. | Antibody demonstrated in serum, not in plasma, suggesting autoantibody is only seen when it reacts with a coagulation factor; IgG3; very weak DAT (both IgG and C3b), weak anti-Sc1 eluted. | |||||||
| McDowell et al.79 | 1986 | Case 1 | Sc:1,−2 | Hemolytic anemia | 3+ DAT and anti-Sc1 in IAT. Eluates by heat, ether, and glycine acid were nonreactive, but xylene eluate showed anti-Sc1. | ||||||||
| McDowell et al.79 | 1986 | Case 2 | Sc:1,−2 (1+, but 3+ 2 years later) | Hemolytic anemia | 4 units RBC transfused earlier. | Transient Sc antigen weakening: serum from initial presentation reacted 3+ with patient’s RBCs 2 years later. | |||||||
| Owen et al.80 | 1992 | Sc:1,−2 | 10 months | F | West Indies | Hemolytic anemia | Sudden-onset jaundice, pallor, fever with hepatomegaly, Hgb = 4.1 g/dL. AIHA diagnosed, treated with steroids and IVIG, transfusion, urgent splenectomy. Transfusion-dependent for 4 weeks, then steroids stopped 7 months after splenectomy. | DAT+ (IgG and C3d), elevated unconjugated bilirubin, reduced haptoglobin, hemoglobinuria. DAT negative with follow-up. Serum and eluate demonstrated anti-Sc1. | |||||
| Ramsey and Williams81 | 2010 | Sc:1 | 20 | F | Caucasian | Hemolytic anemia, acute hemolysis | Hodgkin disease in remission, presented with 3 weeks of fatigue, 2-day Hgb decrease (9.1 to 5.7 g/dL). Gross hematuria and Tbili = 5.5 mg/dL during 3 units RBC transfusion. | 15 units RBC 3 years earlier during chemotherapy, previously negative Ab screen. | Ab screen 1−2+ in all cells (anti-Sc1 identified). DAT− (hospital), w+ (ref. lab; negative eluate). Posttransfusion Hgb = 9.2 g/dL. Anti-Sc1 reacted w+ to patient’s cells. Genotyped SC1/SC1. Patient group O−. | ||||
| Auto vs. Allo not resolved | Steane et al.82 | 1982 | Case 2: pt C.D. | 22 | M | Caucasian | Evans-like syndrome with anemia during a lung infection, 2 years of hospitalizations, steroids, splenectomy, immune suppressants. Transfused safely after 16 plasma exchanges. | Not reported. | Reported DAT+, but “presence of an antibody in his plasma which reacted with all erythrocytes tested except his own.” Antibody undetectable after the 9th exchange, but it reappeared, then disappeared after 4 more exchanges. | ||||
| anti-Sc2 | Allo | Anderson et al.3 | 1963 | Mr. Char. | Sc:1,−2 | 50 | M | Caucasian | DSTR vs. DHTR | Carcinoma of the stomach, pretransfusion antibody screen positive. Hemolysis could be the reason for the 2nd blood request, but this is not stated. | 3 units RBC transfused 2 weeks earlier. | Whether the patient was hemolyzing and thus required the pretransfusion testing is not stated; therefore, we cannot distinguish a DSTR from a DHTR as described in this paper. | |
| Seyfried et al.6 | 1966 | Four donors | Healthy donors iatrogenically immunized for anti-D production. | ||||||||||
| DeMarco et al.83 | 1995 | Sc:1,−2 | 29 | F | Caucasian | HDFN | Jaundice in infant (Tbili = 14.3 mg/dL) on DOL2, postphototherapy discharged Hct = 45%, DOL20 Tbili = 6.5 mg/dL, Hct 17.3% with pallor, tachypnea, tachycardia; transfused 45 mL; Hct 4 months later = 36.3% Mother B+; infant B-. | G0, no transfusion history. | Negative maternal Ab screen | Maternal serum 3+ against paternal RBCs (titer of 64) and Sc:−1,2 RBCs; Paternal RBCs Sc:1,2. Neonate cord blood DAT macroscopically + IgG; Neonate peripheral venous blood 2+; Readmission on DOL20 DAT 2+ (polyspec, IgG, and C3); Eluate + on paternal and Sc:1,−2 RBCs. | |||
| anti-Sc3 | Allo | McCreary et al.16 | 1973 | Sc:−1,−2 | F | Likiep Atoll, Marshall Islands | Preoperative testing. | Transfused 7 months earlier without difficulty. | Antibody dropped to below detectable levels shortly after discovery. | ||||
| Nason et al.17 | 1980 | Sc:−1,−2 | 67 | M | Caucasian | DSTR | Preoperative positive antibody screen for metastatic carcinoma surgery. Not transfused owing to deteriorating clinical condition and lack of compatible blood. | Transfused 4 years earlier without incident. | Negative DAT, did not adsorb anti-Sc1 or anti-Sc2. | ||||
| Woodfield et al.19 | 1986 | Sc:−1,−2 | 4 | F | Papua New Guinea | Thalassemic with new IgG Ab, mother used as only available donor (also Sc:−1,−2). Patient underwent splenectomy. | Many transfusions. | Antibody was undetectable after splenectomy and not stimulated by use of XM-compatible blood. | |||||
| Auto | Peloquin et al.84 | 1989 | Case 1 | weak Sc1 and Sc3 | 64 | Hemolytic anemia | Lymphoma patient with severe anemia; 5 units incompatible RBCs transfused without difficulty. | DAT weak with IgG and C3d, eluates negative. Antibody disappeared within 70 days. | |||||
| Peloquin et al.84 | 1989 | Case 2 | weak Sc1 and Sc3 | 54 | Hemolytic anemia | Hodgkin disease, CHF, moderate anemia; 6 units incompatible blood transfused without difficulty. | 4 units RBCs transfused earlier. | DAT 3+ with C3d, negative with IgG, eluate negative. Serum antibody weaker after transfusion; additional follow-up not possible. | |||||
| anti-Sc4 (anti-Rd) | Allo | Rausen et al.20 | 1967 | Family 1 −Rd | Sc:−4 (Rd+) | F | Russian Jewish | HDFN | 2 children Rd+, both had HDFN; 1 child Rd−, did not have HDFN. | “Newborn with hemolytic disease” in 1st and 3rd children. | Presumably very mild HDFN, as case detected by “routine anti-globulin testing of cord blood.” | ||
| Rausen et al.20 | 1967 | Family 2 −Fl | Sc:−4 (Rd+) | F | African American | HDFN severe | 10 children in pedigree: 5 Rd− with no HDFN; 1st 3 Rd+ without HDFN, last 2 Rd+ with HDFN (1st of these requiring exchange transfusion). | “Newborn with hemolytic disease” in 7th and 9th children. | |||||
| Rausen et al.20 | 1967 | Family 3 −Ha | Sc:−4 (Rd+) | F | Northern European | HDFN | 4 children in family: 1 Rd− with no HDFN; 1st 2 Rd+ without HDFN, last 1 Rd+ with HDFN. | “Newborn with hemolytic disease” in 4th child. | In the same pedigree, Rd− grandmother of HDN proband had 2 children, 1st Rd+ and 2nd Rd−, neither had HDFN. | ||||
| Rausen et al.20 | 1967 | Family 4 −We | Sc:−4 (Rd+) | F | Native American | HDFN | 4 children in family: 1 Rd− with no HDFN; 1st 2 Rd+ without HDFN, last 1 Rd+ with HDFN. | ||||||
| Rausen et al.20 | 1967 | Family 5 Gr | Sc:−4 (Rd+) | F | German Jewish or Scotch-Irish | HDFN | 5 children in family: 3 Rd− with no HDFN; 1 stillborn (3rd in birth order) followed by 1 Rd+ with HDFN. | No description of pathology of the stillborn fetus. | |||||
| Lundsgaard and Jensen21 | 1968 | Mrs. J.P. | Sc:−4 (Rd+) | 47 | F | DSTR: No clinical evidence of hemolysis | Moderately strong DAT (polyspecific and IgG-eluate showed anti-Rd), weakened with time. Negative Ab screen preoperatively. | Had a 9-year-old child. 2 units blood transfused during gynecologic operation. | Patient is O+; 2 weeks after transfusion, strongly reactive antibody screen at new hospital. Her child’s RBCs do not react with patient’s posttransfusion plasma. | ||||
| Lundsgaard and Jensen21 | 1968 | Mr. L.C. | Sc:−4 (Rd+) | 55 | M | Preoperative positive antibody screen for atoxic goiter surgery. Considered as a “naturally occurring antibody.” | Never transfused or hospitalized. | ||||||
| Winn et al.85 | 1976 | 19 | M | Caucasian | DSTR: No clinical evidence of hemolysis. | 11 units RBC on 3 dates for gunshot wound surgery (7/5, 7/17, and 8/5). 1 unit found incompatible on crossmatch (RT, 37°C, and AHG) but screening RBCs still negative. | Negative DAT, negative Ab screen before transfusion. | anti-Vw | anti-Vw accounts for incompatible RT crossmatch. | ||||
| anti-Sc5 (anti-STAR) | Allo | Devine et al.18 and Skradski et al.35 | 1988 | Case 3 | Sc:1,−2 | 65 | M | Irish and English | DSTR | 1 week after transfusion of 3 units RBC, Ab screen showed new anti-Jkb and a high-frequency antigen. DAT−, patient eventually diagnosed with lymphoma and transfused with autologous units. | 3 units RBC 3 years earlier. | anti-Canti-eanti-Jkb | Serum reacted with all cells except Sc:−1,−2, autologous, and the patient’s sibling (phenotype Sc:1,−2). Identity of antigen discovered with molecular testing years afterward. |
| anti-Sc6 (anti-SCER) | Allo | Devine et al.18 | 1988 | Case 1 | Sc:1,−2 | 76 | M | German | DSTR | Preoperative positive Ab screen for revision of right total hip arthroplasty, procedure delayed until autologous units could be collected. | 3 units RBC 12 years earlier; 3 units RBC 6 years earlier. | Identity of antigen discovered with molecular testing years afterward. | |
| anti-Sc7 (anti-SCAN) | Allo | Devine et al.18 | 1988 | Case 2 | Sc:1,−2 | 50 | M | German, English, and Native American | DHTR | 12 days after transfusion of 2 units RBC after orthopedic surgery, patient had decrease in hematocrit, pretransfusion Ab screen positive, weakly DAT+ (IgG and C3). | 3 units RBC 8 years earlier; 2 units RBC 3 years earlier. | anti-D | Eluate reacted with all cells except Sc:−1,−2. Antibody no longer detected within 9 months. Identity of antigen discovered with molecular testing years afterward. |
Ab = antibody; ADCC = antibody-dependent cell-mediated cytotoxicity; AHG = antihuman globulin; AIHA = autoimmune hemolytic anemia; CHF = congestive heart failure; DAT = direct antiglobulin test; DHTR = delayed hemolytic transfusion reaction; DOL = day of life; DSTR = delayed serologic transfusion reaction; Hct = hematocrit; HDFN = hemolytic disease of the fetus and newborn; Hgb = hemoglobin; IAT = indirect antiglobulin test; IgG = immunoglobulin G; IVIG = intravenous immunoglobulin; RBCs = red blood cells; RT = room temperature; Tbili = total bilirubin; XM = crossmatch.
Clinically important reactions to an erythrocyte antibody typically result from significant RBC destruction and usually manifest in the patient as a hemolytic anemia or HDFN. In the SC system, significant HDFN defined the blood group in a patient named Ms. Scianna; however, she also had anti-D to account for her severe obstetric complications in addition to the anti-Sc1.2 Anti-Sc2 has been reported in a case of HDFN requiring simple transfusion in a 20-day-old infant.83 Importantly, the search for antibodies to low-prevalence antigens in this patient was only performed because the neonate and the mother were ABO-compatible: had they been ABO-incompatible, the HDFN would likely have been attributed to this, and the anti-Sc2 antibody may not have been detected.
The recent availability of recombinant reagents to assist in the detection of SC antibodies87 may bring this ability into the wider immunohematology arena. This novel technique will allow one to appraise the relevance of SC antibodies in conjunction with other antibodies and tease out their relative clinical importance, which may have been previously underestimated. The ERMAP protein has been expressed in its native form in a eukaryotic system,87 and so although the initial report describes the utility of this reagent for detecting the high-prevalence SC antigens, a screening protein for low-prevalence antigens Sc2 and Sc4 would also be feasible.
Some SC alloantibodies (Table 3) resulted in delayed serologic transfusion reactions without associated hemolysis, including a so-called naturally occurring anti-Sc4 described in a 55-year-old man without a transfusion history.21 Many of these patients were transfused with crossmatch-compatible blood without incident. However, some surgeries were delayed owing to lack of availability of crossmatch-compatible units, and other patients were transfused with autologous products in nonurgent scenarios.18
The remaining reports of important reactions involving SC antibodies are in cases of autoimmune hemolytic anemia. Auto-anti-Sc1 and -Sc3 have been eluted from direct antiglobulin test–positive RBCs of these patients. In many cases, these antibodies were transient and associated with decreased expression of SC antigens.79,84 Several patients underwent splenectomy in addition to treatment with many types of immunosuppressant medications. One patient with erythroid hypoplasia and diagnosed with an Evans-like syndrome even underwent a total of 20 plasma exchanges over the course of 11 weeks.82 After the ninth exchange, the antibody was no longer detectable, although it reappeared 1 week later. He was finally transfused after 16 exchanges, despite the continued presence of the antibody, with a resultant rise in his hematocrit from 14% to 30%.
Clinical Significance
Is It Worth Phenotype or Genotype Matching?
Of the 23 cases of SC alloantibodies catalogued here (Table 3), one case of HDFN83 and one delayed hemolytic transfusion reaction (case 2)18 relate sufficient information in their reports to convincingly attribute clinical relevance to SC antibodies. The HDFN cases described in the initial report of the Radin antigen also speak to the potential importance of antibodies to Sc4 (especially the case that required exchange transfusion); however, these cases are not reported in sufficient detail to definitively implicate anti-Sc4 to the exclusion of all other causes.20 Even the very dramatic presentation of Ms. Scianna’s antibody does not incriminate anti-Sc1 as the cause of her poor obstetric outcomes, as an anti-D of a higher titer was also found.2 The fundamental question of when and whether antibodies to SC system antigens are of clinical importance remains unresolved because these antibodies are not routinely characterized, especially if they are detected along with other, better-understood alloantibodies that can account for a clinical presentation.
On the Detection of SC System Antibodies in Routine Pretransfusion Testing
The most important obstacle to understanding the clinical consequences of transfusing across SC antigens is that serologic reagents to simply define these antigens in both patients and test RBCs have not been widely available. Akin to the Dombrock blood group system,88 molecular approaches help resolve these situations. It has been estimated that approximately 13 percent of the transfused population are immunologic responders capable of creating alloantibodies,89 but unless RBC reagents with SC antigens are included in the routine laboratory workup, these antibodies may go unnoticed. It is also problematic if an antibody to one of the high-prevalence SC antigens is in fact detected because the mere presence of the antibody does not necessarily correspond to clinical importance. Such a qualitative assay is insufficient. The quantitative characterization of the antibody, such as by titer, has only been performed in a few SC antibody cases, and we recommend that this parameter be more routinely evaluated in cases of possible hemolysis caused by antibodies in the SC system and in most other blood group systems. Especially because HDFN has been reported, establishing a better understanding of antibody potency as detected by the titer would help inform practice guidelines.
An economic and rational approach would be the routine use of recombinant SC protein during pretransfusion testing before titration to effectively screen only for antibodies present above a threshold titer.87 In this way, nuisance antibodies (akin to the low-titer cold autoantibodies detected before routine testing at higher temperatures) would fall under the radar as desired and precious laboratory resources would not be spent working up these most likely incidental findings. Only higher titer antibodies would be detected, focusing the laboratory investigation on cases more likely to be of consequence to the patient. If these laboratory techniques do lead to the increased identification of SC system alloantibodies, the medical importance of respecting them in a particular patient’s transfusion recommendation remains debatable.
Should We Genotype for SC Alleles?
The integration of molecular diagnostics to transfusion medicine has been a slow, but steady, process, with applications ranging from individual patient prenatal diagnosis to routine high-throughput donor testing.90,91 Although the rate of implementation of these technologies varies among nations92 and local transfusion services,93 it is expanding.94 The transfusion community should move these procedures from potentially inaccessible research laboratories to standard clinical practice.95 For optimal performance of such an approach toward personalized medicine, ideally all donors and all recipients should be evaluated at a molecular level. Economic barriers to this strategy are falling as high-throughput and multiplexed assays are achieved, because the incremental cost of adding a handful of additional SNPs when designing a DNA microarray is negligible. Consequently, molecular testing strategies are shifting from decisions about which variants to include in a genotyping system that force the prioritization of well-studied variants already known to have medical importance, to finding effective platforms to analyze data derived from genotyping as many known variants as possible, regardless of their allele frequencies or uncertain clinical effects.
The most compelling reason to genotype SC variants in greater depth, both by increasing the number of donors and patients and also by including more variants regardless of their prevalence, is that such data are necessary to clarify the presently ambiguous role of SC in clinical transfusion practice. At the same time, valuable data for research into ERMAP biology are accrued without added costs. Inclusion of the SC*01 to SC*07 alleles in high-throughput assays would be a move in the right direction, and in cases of unresolved serology, a referral to the molecular laboratory for an in-depth investigation may be desirable. By using a high-throughput genetic test to determine the potential for alloimmunization by a variant homozygote, we can collect such genetic data almost for free. The pursuit of alloantibodies in such variant homozygous patients is a feasible strategy to achieve complete resolution of all alloantibodies in a posttransfusion sample submitted as a transfusion reaction investigation. Consequently, knowing these genetic data gives us the opportunity to better define which antibodies are of clinical importance.
Conclusion
The SC story is an excellent example of the essential roles that classic genetics played in the original physical mapping of a blood group system and is an exceptional illustration of transfusion laboratories that were in the right place at the right time to finally identify the responsible locus in the era of whole genome analysis. Parsing out the meaning of global polymorphism frequency distributions in light of founder effects or selection pressures would be a useful adjunct to in vitro studies of transcript utilization and protein expression and function. The continued study of ERMAP homologues, such as the BTNL family of proteins in general, and their physiologic interactions in other species, such as the B30.2-containing bloodthirsty gene in Antarctic icefishes, is a promising avenue to explore fundamental insights into erythropoiesis. Investigators in transfusion medicine are uniquely poised not only to carry out this work but also to lead the way toward a more comprehensive understanding of the “lucky 13th” blood group SC, making us the lucky ones indeed.
Acknowledgments
We gratefully acknowledge Professor Guillaume Lecointre for kindly providing the photograph of blood from the Antarctic icefish (Fig. 5).
References
- 1.Wagner FF, Poole J, Flegel WA. Scianna antigens including Rd are expressed by ERMAP. Blood. 2003;101:752–7. doi: 10.1182/blood-2002-07-2064. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt RP, Griffitts JJ, Northman FF. A new antibody, anti-Sm, reacting with a high incidence antigen. Transfusion. 1962;2:338–40. doi: 10.1111/j.1537-2995.1962.tb00250.x. [DOI] [PubMed] [Google Scholar]
- 3.Anderson C, Hunter J, Zipursky A, Lewis M, Chown B. An antibody defining a new blood group antigen, Bu-a. Transfusion. 1963;3:30–3. doi: 10.1111/j.1537-2995.1963.tb04599.x. [DOI] [PubMed] [Google Scholar]
- 4.Lewis M, Chown B, Schmidt RP, Griffitts JJ. A possible relationship between the blood group antigens Sm and Bu-a. Am J Hum Genet. 1964;16:254–5. [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis M, Chown B, Kaita H, Philipps S. Further observations on the blood group antigen Bu-a. Am J Hum Genet. 1964;16:256–60. [PMC free article] [PubMed] [Google Scholar]
- 6.Seyfried H, Frankowska K, Giles CM. Further examples of anti-bu-a found in immunized donors. Vox Sang. 1966;11:512–16. doi: 10.1111/j.1423-0410.1966.tb04248.x. [DOI] [PubMed] [Google Scholar]
- 7.Lewis M, Chown B, Kaita H. On the blood group antigens Bua and Sm. Transfusion. 1967;7:92–4. doi: 10.1111/j.1537-2995.1967.tb04848.x. [DOI] [PubMed] [Google Scholar]
- 8.Giles CM, Bevan B, Hughes RM. A family showing independent segregation of Bua and Ytb. Vox Sang. 1970;18:265–6. doi: 10.1111/j.1423-0410.1970.tb01458.x. [DOI] [PubMed] [Google Scholar]
- 9.Rowe GP. A second family showing independent segregation of Sc 2(Bua) and Ytb. Vox Sang. 1986;50:191. doi: 10.1111/j.1423-0410.1986.tb04877.x. [DOI] [PubMed] [Google Scholar]
- 10.Lewis M, Kaita H, Chown B. Scianna blood group system. Vox Sang. 1974;27:261–4. doi: 10.1111/j.1423-0410.1974.tb02416.x. [DOI] [PubMed] [Google Scholar]
- 11.Lewis M, Kaita H, Chown B. Genetic linkage between the human blood group loci Rh and Sc (Scianna) Am J Hum Genet. 1976;28:619–20. [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis M, Kaita H, Côté GB, Chown B, Giblett ER, Anderson JA. Chromosome 1: lods on linkage among eight loci: Do, ENO1 Fy, PGM1, Rh, UMPK, Sc, and PGD. Birth Defects Orig Artic Ser. 1976;12:322–5. [PubMed] [Google Scholar]
- 13.Lewis M, Kaita H, Chown B. Relative positions of chromosome 1 loci Fy, PGM1, Sc, UMPK, Rh, PGD and ENO1 in man. Can J Genet Cytol. 1977;19:695–709. doi: 10.1139/g77-076. [DOI] [PubMed] [Google Scholar]
- 14.Lewis M, Kaita H, Giblett ER, Anderson JE. Data on chromosome 1 loci Fy, PGM1, Sc, UMPK, Rh, PGD, and ENO1: two-point lods, R:NR counts, multipoint information, and map. Cytogenet Cell Genet. 1978;22:392–5. doi: 10.1159/000130980. [DOI] [PubMed] [Google Scholar]
- 15.Noades JE, Corney G, Cook PJ, et al. The Scianna blood group lies distal to uridine monophosphate kinase on chromosome 1p. Ann Hum Genet. 1979;43:121–32. doi: 10.1111/j.1469-1809.1979.tb02004.x. [DOI] [PubMed] [Google Scholar]
- 16.McCreary J, Vogler AL, Sabo B, Eckstein EG, Smith TR. Another minus-minus phenotype: Bu(a-)Sm-, two examples in one family [abstract] Transfusion. 1973;13:350. [Google Scholar]
- 17.Nason SG, Vengelen-Tyler V, Cohen N, Best M, Quirk J. A high incidence antibody (anti-Sc3) in the serum of a Sc:-1,-2 patient. Transfusion. 1980;20:531–5. doi: 10.1046/j.1537-2995.1980.20581034505.x. [DOI] [PubMed] [Google Scholar]
- 18.Devine P, Dawson FE, Motschman TL, et al. Serologic evidence that Scianna null (Sc:-1,-2) red cells lack multiple high-frequency antigens. Transfusion. 1988;28:346–9. doi: 10.1046/j.1537-2995.1988.28488265264.x. [DOI] [PubMed] [Google Scholar]
- 19.Woodfield DG, Giles C, Poole J, Oraka R, Tolanu T. A further null phenotype (Sc-1-2) in Papua New Guinea [Abstract]. 21st Congress of the Int Soc Haem and 19th Congress of the ISBT; Sydney, Australia. May 1986; p. 651. [Google Scholar]
- 20.Rausen AR, Rosenfield RE, Alter AA, et al. A “new” infrequent red cell antigen, Rd (radin) Transfusion. 1967;7:336–42. doi: 10.1111/j.1537-2995.1967.tb04864.x. [DOI] [PubMed] [Google Scholar]
- 21.Lundsgaard A, Jensen KG. Two new examples of anti-Rd. A preliminary report on the frequency of the Rd (Radin) antigen in the Danish population. Vox Sang. 1968;14:452–7. doi: 10.1111/j.1423-0410.1968.tb01738.x. [DOI] [PubMed] [Google Scholar]
- 22.Lewis M, Kaita H. Genetic linkage between the Radin and Rh blood group loci. Vox Sang. 1979;37:286–9. doi: 10.1111/j.1423-0410.1979.tb02306.x. [DOI] [PubMed] [Google Scholar]
- 23.Lewis M, Kaita H, Philipps S, et al. The position of the Radin blood group locus in relation to other chromosome l loci. Ann Hum Genet. 1980;44(Pt 2):179–84. doi: 10.1111/j.1469-1809.1980.tb00956.x. [DOI] [PubMed] [Google Scholar]
- 24.Spring FA, Herron R, Rowe G. An erythrocyte glycoprotein of apparent Mr 60,000 expresses the Sc1 and Sc2 antigens. Vox Sang. 1990;58:122–5. doi: 10.1111/j.1423-0410.1990.tb02074.x. [DOI] [PubMed] [Google Scholar]
- 25.Spring FA. Characterization of blood-group-active erythrocyte membrane glycoproteins with human antiseras. Transfus Med. 1993;3:167–78. [Google Scholar]
- 26.Su YY, Gordon CT, Ye TZ, Perkins AC, Chui DH. Human ERMAP: an erythroid adhesion/receptor transmembrane protein. Blood Cells Mol Dis. 2001;27:938–49. doi: 10.1006/bcmd.2001.0465. [DOI] [PubMed] [Google Scholar]
- 27.Xu H, Foltz L, Sha Y, et al. Cloning and characterization of human erythroid membrane-associated protein, human ERMAP. Genomics. 2001;76:2–4. doi: 10.1006/geno.2001.6600. [DOI] [PubMed] [Google Scholar]
- 28.Ensembl entry for ERMAP, number ENSG00000164010 2010. http://www.ensembl.org/Homo_sapiens/Location/View?db=core;r=1:43276635-43326634. Accessed July 14, 2010.
- 29.Entrez Gene entry for ERMAP. http://www.ncbi.nlm.nih.gov/gene/114625. Accessed July 14, 2010.
- 30.Havana entry for ERMAP, transcript numbers OTTHUMT00000020180 through OTTHUMT00000020184 2010. http://vega.sanger.ac.uk/Homo_sapiens/Gene/Summary?g=OTTHUMG00000007619. Accessed July 14, 2010.
- 31.Ensembl Transcript View, ERMAP’s 5 listed transcripts 2010. http://www.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000164010;r=1:43276635-43326634;t=ENST00000470938. Accessed July 14, 2010.
- 32.Fuchisawa A, Lomas-Francis C, Hue-Roye K, Reid ME. The polymorphism nt 76 in exon 2 of SC is more frequent in whites than in blacks. Immunohematology. 2009;25:18–19. [PubMed] [Google Scholar]
- 33.Hashmi G, Shariff T, Zhang Y, et al. Determination of 24 minor red blood cell antigens for more than 2000 blood donors by high-throughput DNA analysis [published correction appears in Transfusion 2007;47:952] Transfusion. 2007;47:736–47. doi: 10.1111/j.1537-2995.2007.01178.x. [DOI] [PubMed] [Google Scholar]
- 34.Ribeiro KR, Guarnieri MH, da Costa DC, Costa FF, Pellegrino J, Jr, Castilho L. DNA array analysis for red blood cell antigens facilitates the transfusion support with antigen-matched blood in patients with sickle cell disease. Vox Sang. 2009;97:147–52. doi: 10.1111/j.1423-0410.2009.01185.x. [DOI] [PubMed] [Google Scholar]
- 35.Skradski KJ, McCreary J, Sabo B, Polesky HF. An antibody against a high frequency antigen absent on red cells of the Scianna:-2,-2 phenotype [abstract] Transfusion. 1982;(22):406. [Google Scholar]
- 36.Hue-Roye K, Chaudhuri A, Velliquette RW, et al. STAR: a novel high-prevalence antigen in the Scianna blood group system. Transfusion. 2005;45:245–7. doi: 10.1111/j.1537-2995.2004.04226.x. [DOI] [PubMed] [Google Scholar]
- 37.Flegel WA, Chen Q, Reid ME, et al. SCER and SCAN: two novel high-prevalence antigens in the Scianna blood group system. Transfusion. 2005;45:1940–4. doi: 10.1111/j.1537-2995.2005.00646.x. [DOI] [PubMed] [Google Scholar]
- 38.Smigielski EM, Sirotkin K, Ward M, Sherry ST. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 2000;28:352–5. doi: 10.1093/nar/28.1.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.The International HapMap Project Homepage. 2010 http://hapmap.ncbi.nlm.nih.gov/. Accessed July 14, 2010.
- 40.Stephan W. Genetic hitchhiking versus background selection: the controversy and its implications. Philos Trans R Soc Lond B Biol Sci. 2010;365:1245–53. doi: 10.1098/rstb.2009.0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams AF, Barclay AN. The immunoglobulin superfamily—domains for cell surface recognition. Annu Rev Immunol. 1988;6:381–405. doi: 10.1146/annurev.iy.06.040188.002121. [DOI] [PubMed] [Google Scholar]
- 42.Cannon JP. Plasticity of the immunoglobulin domain in the evolution of immunity. Integr Comp Biol. 2009;49:187–96. doi: 10.1093/icb/icp018. [DOI] [PubMed] [Google Scholar]
- 43.Harpaz Y, Chothia C. Many of the immunoglobulin superfamily domains in cell adhesion molecules and surface receptors belong to a new structural set which is close to that containing variable domains. J Mol Biol. 1994;238:528–39. doi: 10.1006/jmbi.1994.1312. [DOI] [PubMed] [Google Scholar]
- 44.Aricescu AR, Jones EY. Immunoglobulin superfamily cell adhesion molecules: zippers and signals. Curr Opin Cell Biol. 2007;19:543–50. doi: 10.1016/j.ceb.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 45.Peggs KS, Allison JP. Co-stimulatory pathways in lymphocyte regulation: the immunoglobulin superfamily. Br J Haematol. 2005;130:809–24. doi: 10.1111/j.1365-2141.2005.05627.x. [DOI] [PubMed] [Google Scholar]
- 46.Vogel C, Chothia C. Protein family expansions and biological complexity. PLoS Comput Biol. 2006;2:e48. doi: 10.1371/journal.pcbi.0020048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simon TL, Dzik WH, Snyder EL, Stowell CP, Strauss RG, editors. Rossi’s Principles of Transfusion Medicine. 3rd. Philadelphia, PA: Lippincott Williams & Wilkins; 2002. [Google Scholar]
- 48.Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116–26. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 49.Mather IH, Jack LJ. A review of the molecular and cellular biology of butyrophilin, the major protein of bovine milk fat globule membrane. J Dairy Sci. 1993;76:3832–50. doi: 10.3168/jds.s0022-0302(93)77726-7. [DOI] [PubMed] [Google Scholar]
- 50.Rhodes DA, Stammers M, Malcherek G, Beck S, Trowsdale J. The cluster of BTN genes in the extended major histocompatibility complex. Genomics. 2001;71:351–62. doi: 10.1006/geno.2000.6406. [DOI] [PubMed] [Google Scholar]
- 51.Robenek H, Hofnagel O, Buers I, et al. Butyrophilin controls milk fat globule secretion. Proc Natl Acad Sci U S A. 2006;103:10385–90. doi: 10.1073/pnas.0600795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith IA, Knezevic BR, Ammann JU, et al. BTN1A1, the mammary gland butyrophilin, and BTN2A2 are both inhibitors of T cell activation. J Immunol. 2010;184:3514–25. doi: 10.4049/jimmunol.0900416. [DOI] [PubMed] [Google Scholar]
- 53.Valentonyte R, Hampe J, Huse K, et al. Sarcoidosis is associated with a truncating splice site mutation in BTNL2 [published correction appears in Nat Genet 2005;37:652] Nat Genet. 2005;37:357–64. doi: 10.1038/ng1519. [DOI] [PubMed] [Google Scholar]
- 54.Arnett HA, Escobar SS, Viney JL. Regulation of costimulation in the era of butyrophilins. Cytokine. 2009;46:370–5. doi: 10.1016/j.cyto.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 55.Nguyen T, Liu XK, Zhang Y, Dong C. BTNL2, a butyrophilin-like molecule that functions to inhibit T cell activation. J Immunol. 2006;176:7354–60. doi: 10.4049/jimmunol.176.12.7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meyer M, Gaudieri S, Rhodes DA, Trowsdale J. Cluster of TRIM genes in the human MHC class I region sharing the B30.2 domain. Tissue Antigens. 2003;61:63–71. doi: 10.1034/j.1399-0039.2003.610105.x. [DOI] [PubMed] [Google Scholar]
- 57.Vernet C, Boretto J, Mattei MG, et al. Evolutionary study of multigenic families mapping close to the human MHC class I region. J Mol Evol. 1993;37:600–12. doi: 10.1007/BF00182746. [DOI] [PubMed] [Google Scholar]
- 58.Jack LJ, Mather IH. Cloning and analysis of cDNA encoding bovine butyrophilin, an apical glycoprotein expressed in mammary tissue and secreted in association with the milk-fat globule membrane during lactation. J Biol Chem. 1990;265:14481–6. [PubMed] [Google Scholar]
- 59.Tazi-Ahnini R, Henry J, Offer C, Bouissou-Bouchouata C, Mather IH, Pontarotti P. Cloning, localization, and structure of new members of the butyrophilin gene family in the juxta-telomeric region of the major histocompatibility complex. Immunogenetics. 1997;47:55–63. doi: 10.1007/s002510050326. [DOI] [PubMed] [Google Scholar]
- 60.Weinert C, Grütter C, Roschitzki-Voser H, Mittl PR, Grütter MG. The crystal structure of human pyrin b30.2 domain: implications for mutations associated with familial Mediterranean fever. J Mol Biol. 2009;394:226–36. doi: 10.1016/j.jmb.2009.08.059. [DOI] [PubMed] [Google Scholar]
- 61.Woo JS, Imm JH, Min CK, Kim KJ, Cha SS, Oh BH. Structural and functional insights into the B30.2/SPRY domain. EMBO J. 2006;25:1353–63. doi: 10.1038/sj.emboj.7600994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Woo JS, Suh HY, Park SY, Oh BH. Structural basis for protein recognition by B30.2/SPRY domains. Mol Cell. 2006;24:967–76. doi: 10.1016/j.molcel.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 63.Jeong J, Rao AU, Xu J, et al. The PRY/SPRY/B30.2 domain of butyrophilin 1A1 (BTN1A1) binds to xanthine oxidoreductase: implications for the function of BTN1A1 in the mammary gland and other tissues. J Biol Chem. 2009;284:22444–56. doi: 10.1074/jbc.M109.020446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Garofalo F, Pellegrino D, Amelio D, Tota B. The Antarctic hemoglobinless icefish, fifty five years later: a unique cardiocirculatory interplay of disaptation and phenotypic plasticity. Comp Biochem Physiol A Mol Integr Physiol. 2009;154:10–28. doi: 10.1016/j.cbpa.2009.04.621. [DOI] [PubMed] [Google Scholar]
- 65.Yergeau DA, Cornell CN, Parker SK, Zhou Y, Detrich HW., 3rd bloodthirsty, an RBCC/TRIM gene required for erythropoiesis in zebrafish. Dev Biol. 2005;283:97–112. doi: 10.1016/j.ydbio.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 66.Ye TZ, Gordon CT, Lai YH, et al. Ermap, a gene coding for a novel erythroid specific adhesion/receptor membrane protein. Gene. 2000;242:337–45. doi: 10.1016/s0378-1119(99)00516-8. [DOI] [PubMed] [Google Scholar]
- 67.Kuriyan MA, Oyen RE, Marsh WL. Demonstration of Diego (Dib) and Scianna (Scl) antigens on phagocytic leukocytes of the blood. Transfusion. 1978;18:361–4. doi: 10.1046/j.1537-2995.1978.18378205148.x. [DOI] [PubMed] [Google Scholar]
- 68.Rojewski MT, Schrezenmeier H, Flegel WA. Tissue distribution of blood group membrane proteins beyond red cells: evidence from cDNA libraries. Transfus Apher Sci. 2006;35:71–82. doi: 10.1016/j.transci.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 69.He XR, He YY, Chen Y, Ye TZ. Expression of human ERMAP gene in fetal tissues [in Chinese] Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2006;14:972–5. [PubMed] [Google Scholar]
- 70.Zhang XH, Ye TZ, Hu B, Si WZ. Quantification of human ermap by using real-time FQ-PCR [in Chinese] Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2005;13:154–7. [PubMed] [Google Scholar]
- 71.He YY, Zhang XH, Ye TZ, Wu ZL. Study on the expression of human ERMAP gene in erythropoietic and macrophage differentiation of K562 cells [in Chinese] Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2005;13:553–6. [PubMed] [Google Scholar]
- 72.He YY, Zhang XH, Ye TZ, Wu ZL. Expression of human ERMAP gene in different cell lines [in Chinese] Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2005;13:819–22. [PubMed] [Google Scholar]
- 73.Liang JF, Chen Y, Ye TZ, et al. Effects of human ERMAP-siRNA on erythroid differentiation of K562 cells induced by Ara-C [in Chinese] Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2009;17:49–53. [PubMed] [Google Scholar]
- 74.Lin LD, He XR, Ye TZ, et al. Expression of human ermap gene in umbilical cord blood mononuclear cells during differentiation and development towards erythroid lineage [in Chinese] Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2008;16:328–32. [PubMed] [Google Scholar]
- 75.Reid ME, Lomas-Francis C. The blood group antigen factsbook. 2nd. San Diego, CA: Academic Press; 2004. [Google Scholar]
- 76.Velliquette RW. Review: the Scianna blood group system. Immunohematology. 2005;21:70–6. [PubMed] [Google Scholar]
- 77.Kaye T, Williams EM, Garner SF, Leak MR, Lumley H. Anti-Sc1 in pregnancy. Transfusion. 1990;30:439–40. doi: 10.1046/j.1537-2995.1990.30590296379.x. [DOI] [PubMed] [Google Scholar]
- 78.Tregellas WM, Holub MP, Moulds JJ, Lacey PA. An example of autoanti-Sc1 demonstrable in serum but not in plasma [abstract] Transfusion. 1979;19:650. [Google Scholar]
- 79.McDowell MA, Stocker I, Nance S, Garratty G. Auto anti-Sc1 associated with autoimmune hemolytic anemia [abstract] Transfusion. 1986;26:578. [Google Scholar]
- 80.Owen I, Chowdhury V, Reid ME, Poole J, Marsh JC, Hows JM. Autoimmune hemolytic anemia associated with anti-Sc1. Transfusion. 1992;32:173–6. doi: 10.1046/j.1537-2995.1992.32292180150.x. [DOI] [PubMed] [Google Scholar]
- 81.Ramsey G, Williams LM. Autoimmune hemolytic anemia with auto-anti-Sc1, weakened Sc:1 antigen, and superimposed transfusion-associated acute hemolysis [abstract] Transfusion. 2010;50(2S):156A. [Google Scholar]
- 82.Steane EA, Sheehan RG, Brooks BD, Frenkel EP. Therapeutic plasmapheresis in patients with antibodies to high-frequency red cell antigens. Prog Clin Biol Res. 1982;106:347–53. [PubMed] [Google Scholar]
- 83.DeMarco M, Uhl L, Fields L, Pacini D, Gorlin JB, Kruskall MS. Hemolytic disease of the newborn due to the Scianna antibody, anti-Sc2. Transfusion. 1995;35:58–60. doi: 10.1046/j.1537-2995.1995.35195090664.x. [DOI] [PubMed] [Google Scholar]
- 84.Peloquin P, Moulds M, Keenan J, Kennedy M. Anti-Sc3 as an apparent autoantibody in two patients [abstract] Transfusion. 1989;29(Suppl):49S. [Google Scholar]
- 85.Winn LC, Eska PL, Grindon AJ. Anti-Rd (Radin) following the transfusion of a Radin positive unit of blood. Transfusion. 1976;16:351–2. doi: 10.1046/j.1537-2995.1976.16476247056.x. [DOI] [PubMed] [Google Scholar]
- 86.Wagner FF, Frohmajer A, Ladewig B, et al. Weak D alleles express distinct phenotypes. Blood. 2000;95:2699–708. [PubMed] [Google Scholar]
- 87.Seltsam A, Grueger D, Blasczyk R, Flegel WA. Easy identification of antibodies to high-prevalence Scianna antigens and detection of admixed alloantibodies using soluble recombinant Scianna protein. Transfusion. 2009;49:2090–6. doi: 10.1111/j.1537-2995.2009.02255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Reid ME. Complexities of the Dombrock blood group system revealed. Transfusion. 2005;45(2 Suppl):92S–9S. doi: 10.1111/j.1537-2995.2005.00527.x. [DOI] [PubMed] [Google Scholar]
- 89.Higgins JM, Sloan SR. Stochastic modeling of human RBC alloimmunization: evidence for a distinct population of immunologic responders. Blood. 2008;112:2546–53. doi: 10.1182/blood-2008-03-146415. [DOI] [PubMed] [Google Scholar]
- 90.Reid ME. Transfusion in the age of molecular diagnostics. Hematology Am Soc Hematol Educ Program. 2009:171–7. doi: 10.1182/asheducation-2009.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Veldhuisen B, van der Schoot CE, de Haas M. Blood group genotyping: from patient to high-throughput donor screening. Vox Sang. 2009;97:198–206. doi: 10.1111/j.1423-0410.2009.01209.x. [DOI] [PubMed] [Google Scholar]
- 92.Flegel WA. Blood group genotyping in Germany. Transfusion. 2007;47(1 Suppl):47S–53S. doi: 10.1111/j.1537-2995.2007.01310.x. [DOI] [PubMed] [Google Scholar]
- 93.Hillyer CD, Shaz BH, Winkler AM, Reid M. Integrating molecular technologies for red blood cell typing and compatibility testing into blood centers and transfusion services. Transfus Med Rev. 2008;22:117–32. doi: 10.1016/j.tmrv.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 94.Garraty G, Reid M, Westhoff C, editors. A Workshop on Methods in Molecular Immunohematology. Transfusion. 2010;47:1S–100S. [Google Scholar]
- 95.Denomme GA, Flegel WA. Applying molecular immunohematology discoveries to standards of practice in blood banks: now is the time. Transfusion. 2008;48:2461–75. doi: 10.1111/j.1537-2995.2008.01855.x. [DOI] [PubMed] [Google Scholar]