

Abstract
With the wide-spread emergence of drug resistance, there is an urgent need to search for new antimicrobials, especially those against Gram-negative bacteria. Along this line, the identification of viable targets is a critical first step. SecA is a protein translocase and is commonly believed to be an excellent target for the development of broad-spectrum antimicrobials. In recent years, we have developed three structural classes of SecA inhibitors, which have proven to be very effective against Gram-positive bacteria. However, we have not achieved the same level of success against Gram-negative bacteria, despite the potent inhibition of SecA in enzyme assays by the same inhibitors. In this study, we use representative inhibitors as chemical probes to gain understanding as to why these inhibitors were not effective against Gram-negative bacteria. The results validate our initial postulation that the major difference in effectiveness against Gram-positive and Gram-negative bacteria is in the additional permeability barrier posed by the outer membrane in Gram-negative bacteria. We have also found that expression of efflux pumps, which are responsible for multi-drug resistance, have no effect on the effectiveness of these SecA inhibitors. Identification of an inhibitor-resistant mutant and complementation tests of the plasmids containing secA in a secAts mutant showed that a single secA-azi-9 mutation increased the resistance, providing genetic evidence that SecA indeed is the target of these inhibitors in bacteria. Such results strongly suggest SecA as an excellent target for developing effective antimicrobials against Gram-negative bacteria with the intrinsic ability to overcome MDR. A key future research direction should be the optimization of membrane permeability.
Introduction
Bacterial infection is making a strong comeback as a major threat to human health,[1] especially because of the emergence of newer strains that are resistant to essentially all known antibiotics. Therefore, there is an urgent need to search for new antimicrobials, especially those that are effective against Gram-negative bacteria and bacterial strains that exhibit multi-drug resistance (MDR).[2] SecA is a protein translocase responsible for the secretion of majority of proteins including virulence factors and the integration of some proteins into bacterial membrane, such as efflux pumps. Therefore, SecA is considered an excellent target for the development of broad-spectrum antimicrobials.[3] Prior efforts identified inorganic azide as a SecA inhibitor in the mM range.[2a, 2g, 4] We have recently developed three structural classes of SecA inhibitors (Figure 1) and started examining this issue. Since the publication of our earlier work, there have been additional reports of SecA inhibitors with potencies in the high μM to low mM range.[3a, 5] A recent paper reported a low μM inhibitor against citrus pathogen, Candidatus Liberibacter asiaticus.[6] The three structural classes of small molecule SecA inhibitors (Figure 1) that we developed include Rose Bengal and its Class A analogues,[7] Class B pyrimidine analogues,[8] and Class C triazole-pyrimidine analogues[9] with the best having IC50 and/or MIC values in the high nM range (Tables 1 and 2).[7b]
Figure 1.
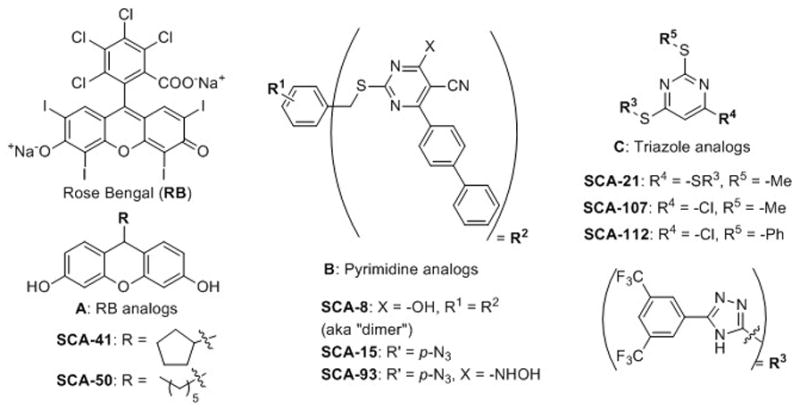
Three structural classes of small molecule SecA inhibitors
Table 1.
Bacteriostatic effects of SecA inhibitors (MIC, μg/mL)
| Strains | Addition | Class A | Class B | Class C | ||||
|---|---|---|---|---|---|---|---|---|
| RB | SCA-50 | SCA-15 | SCA-93 | SCA-107 | SCA-112 | Vancomycin | ||
| MW: 1017 | MW: 298 | MW: 436 | MW: 452 | MW: 472 | MW: 534 | MW: 1446 | ||
| E. coli NR698 | None | 4.5 | 4.7 | 10.9 | 22.7 | 1.8 | 0.8 | ND |
|
| ||||||||
| E. coli MC4100 | None | >1,272* | >373* | >546* | >452* | >590* | >667* | 250 |
| PMBN | 15.9 | 0.9 | 6.8 | 13.6 | 0.7 | 0.8 | 31.3 | |
| NAB | 2.0 | 1.2 | ≤0.3 | ND | 0.9 | ≤0.4 | ND | |
|
| ||||||||
|
Shigella flexneri ATCC12022 |
None | >1,272* | >373* | >546 | >564 | >590* | >667* | >250 |
| PMBN | 7.5 | 1.7 | 10.9 | 11.3 | 2.2 | 2.9 | 62.5 | |
| NAB | 1.6 | 1.2 | 2.0 | 2.1 | 0.3 | 0.3 | 250.0 | |
The permeabilizers used: PMBN: 25 μg/mL; NAB7061: 4 μg/mL. At these concentrations, NAB7061 or PMBN alone had no inhibition on growth.
Highest concentration tested. ND: Not determined.
Table 2.
MIC (μg/mL) of SecA inhibitors for various Gram-N bacteria.
| Addition | Class A | Class B | Antibiotics | ||||
|---|---|---|---|---|---|---|---|
| RB | SCA-50 | SCA-107 | SCA-112 | Vancomycin | Carbenicillin | ||
| MW: 1017 | MW: 298 | MW: 472 | MW: 534 | MW: 1446 | MW: 378 | ||
| E. coli C291-b1 | None* | >1,272* | >373* | >590* | >667* | >250* | >1,000 |
| PMBN | 3.2 | 11.4 | 5.9 | 3.3 | 31 | >1,000 | |
| NAB | >254* | 7.5 | 2.9 | 0.9 | 125 | >1,000 | |
|
| |||||||
| Klebisiella spp. | None* | >1,272* | >373* | >590* | >667* | 1,000 | >1,250 |
| PMBN | >254* | 7.5 | 23.5 | 6.7 | 250 | >1,250 | |
| NAB | >254* | 7.5 | 11.8 | 1.7 | 250 | >1,250 | |
|
| |||||||
| K. pneumoniae BARB 105 | None* | >1,272* | >373* | >590* | >667* | >1000 | >1,000 |
| PMBN | 25.4 | 14.9 | 47.1 | 53.4 | 500 | >1,000 | |
| NAB | >254* | >74.6 | 47.1 | 6.7 | 1000 | >1,000 | |
|
| |||||||
|
P. aeruginosa** T15464 (MDR) |
None* | >1,272* | >373* | >590* | >667* | >1000 | >1,000 |
| PMBN | 0.4 | 1.9 | 0.2 | 0.1 | 31 | >1,000 | |
| NAB | 3.2 | 2.8 | 5.9 | 0.4 | >1000 | >1,000 | |
|
| |||||||
|
A. baumanii ATCC9955 |
None* | >1,272* | >373* | >590* | >667* | 250 | >1,000 |
| PMBN | 80.6 | 7.5 | 1.0 | 1.3 | 250 | >1,000 | |
| NAB | 127.2 | 4.7 | 1.5 | 1.7 | 250 | >1,000 | |
|
| |||||||
| S. typhimurium SL1344 | None* | >1,272* | >373* | >590* | >667* | >250 | >500 |
| PMBN | 17 | 13 | 10 | >53 | ND | >500 | |
| NAB | 254 | 1.6 | 1.0 | 0.4 | >250 | >500 | |
The permeabilizers used: For P. aeruginosa, PMBN at 3 μg/mL; for other strains: PMBN: 25 μg/mL; NAB7061: 4 μg/mL; At these concentrations, NAB7061 or PMBN alone used had no inhibition on growth.
Highest concentration tested
P. aeruginosa T15464 is a clinical isolated strain, and 6 other clinical strains of P. aeruginosa showed similar results. E. coli C291-b1 is an enterotoxigenic strain. ND: Not determined
Our SecA inhibitors have proven to be very effective against Gram-positive bacteria; but success with Gram-negative bacteria has been limited. In earlier studies, SecA inhibitors were found to be ineffective against Gram-negative bacterial strains. The only exception was a strain of mutant E. coli NR698,[10] with compromised outer membrane (OM) and thus possible “leakage.”[7–8] Such results suggest that the existence of an outer membrane in Gram-negative bacteria as the reason for the ineffectiveness of these SecA inhibitors. In this study, we set out to examine this and two others issues by using representative inhibitors from these three structural classes as chemical probes. First, we would like to examine whether outer membrane permeability is a general issue for SecA inhibitors. Second, because SecA is a membrane protein, there are good reasons to believe that the target is more accessible than intracellular soluble protein targets. If this is true, then the effectiveness of these SecA inhibitors should not be attenuated by the presence of MDR efflux pumps. Third, we intend to examine the correlation of the in vitro enzyme inhibition data with antimicrobial efficacy in order to gain assurance that these inhibitors mainly target SecA in achieving antimicrobial effects. Additional experiments include protein pull down, complementation, and examining the effectiveness against mutant strains. All these should help establishing SecA as a viable target in developing antimicrobials against Gram-negative bacteria.
Results and Discussions
The issue of membrane permeability
As discussed in the Introduction section, we suspect that membrane permeability was the key reason that these SecA inhibitors were not effective against Gram-negative bacteria. We set out to examine the role of the outer membrane and mechanistic implications (discussed later) in Gram-negative bacteria using SecA inhibitors as chemical probes through additional experiments.
We thought of using a set of chemical probes that selectively permeabilize the outer membrane of Gram-negative bacteria to see what the effect would be. Specifically, we used two polymyxin derivatives, PMBN and NAB7061 (SFigure 1), which are cationic peptides derived from antibiotic polymyxin B (SFigure 1) and are known to disrupt outer membrane integrity.[11] They are similar to the family of cationic antimicrobial peptides (AMP) that were initially identified as host defense peptides against microbes as part of the innate immunity system.
These polymyxin derivatives were optimized[11a, 11c, 11d] to reduce their effect on mammalian cell membrane and are being explored for human use as antimicrobial agents alone[11] or in combination with other antibiotics against Gram-negative bacteria.[11b–e] We reasoned that if these SecA inhibitors proved to be effective against Gram-negative bacteria in the presence of such membrane permeabilizers, then it lends strong evidence to the notion that the presence of the outer membrane as a permeation barrier is the key reason for the diminished potency of these SecA inhibitors against Gram-negative bacteria. We selected representatives from each of the three classes of SecA inhibitors for this study. We first determined the concentrations of PMBN and NAB7061 that could be used for enhancing the permeability of the inhibitors without affecting the growth of Gram-negative bacteria, and found that PMBN at 25 μg/mL (for P. aeruginosa at 3 μg/mL) and NAB7061 at 4 μg/mL had no effect on bacterial growth, but were capable of rendering SecA inhibitors effective against Gram-negative bacteria with IC50 values similar to or lower than those against mutant E. coli NR698 without a permeabilizers (Table 1). These concentrations of PMBN and NAB7061 were then used throughout. In the presence of either PMBN or NAB7061, E. coli MC4100, which is the parental strain of NR698, and Shigella flexineri were sensitive to all 3 classes of SecA inhibitors in growth inhibition tests (Table 1). We previously showed that SecA inhibitors are not only bacteriostatic against Gram-positive bacteria, but also bactericidal.[7] SCA-107 was selected as an example to examine this aspect in Gram-negative bacteria, and was found to be bactericidal (Figure 2). Specifically, at 25 μM, SCA-107 showed reduction of 6 log units of bacterial colonies against MC4100 in the presence of PMBN and about 4 log units against NR698 without a membrane permeabilizer. Against E. coli 4100 and Shigella flexineri, all three classes of SecA inhibitors showed significantly improved potency in the presence of a membrane permeabilizer (Table 1). For example, in the presence of PMBN or NAB, the MIC value for all three classes in SecA inhibitors ranged from 0.3–15.9 μg/mL, while in the absence of a permeabilizer, the MIC ranged from 373 to least 1,272 μg/mL. The situation with Shigella flexineri is essentially the same as with E. coli.
Figure 2. Bactericidal effects of SCA-107.

E. coli NR698 and MC4100 with or without PMBN (25 μg/mL) were treated with SCA-107 at the concentration indicated at 37 °C for 2 hours. Then CFU were determined.
We extended the synergistic studies with SecA inhibitors against other Gram-negative bacteria using two classes of inhibitors: SCA-50 as a representative of Class A, and SCA-107 and/or SCA-112 as representatives of Class C. Combination treatments were shown to be very effective in inhibiting the growth of other Gram-negative bacteria including ETEC E. coli, S. typhimurium, P. aeruginosa, Acinetobacter baumannii, and Klebsiella pneumonia with MIC in the range of mid μg/mL to high ng/mL for the SecA inhibitors (Table 2) with the exception of RB. Several of these compounds are better than many antibiotics in clinical use in the MDR pathogens, including carbenicillin and vancomycin, which is generally considered as a last resort option (Table 2). Our results showed that SecA inhibitors have both bacteriostatic and bactericidal effects against Gram negative bacteria once they overcome out membrane barrier.
We have noted with interest that PMBN and NAB7061 have differential potentiating effects with our lead inhibitors, SCA-107 and SCA-112 on different species of bacteria (Table 2). Such results are understandable because the outer membrane compositions and molecular scaffolds are different for different bacterial strains and species.
Overall, the results clearly demonstrate that the outer membrane in Gram-negative bacteria was the reason for the diminished potency of the SecA inhibitors. On first glance, the results are not surprising because many antibiotics are more effective against Gram-positive bacteria than Gram-negative bacteria, precisely because of the outer membrane in Gram-negative bacteria, which posses a permeation barrier. However, in light of the results that the potency of SecA inhibitors against Gram-positive bacteria is not affected by efflux pumps, there are additional mechanistic issues that one should consider. Specifically, protein secretion machineries in Gram-negative bacteria clearly need to span the range of inner membrane, periplasmic space, and outer membrane. The potency difference between Gram-negative and positive bacteria clearly indicates that accessing SecA is different from accessing the secretion machineries in achieving inhibition. It is generally believed that the “motor” component of SecA and SecA-driven protein secretion machineries are located on the inner membrane. Thus for Gram-positive bacteria, the lack of effect of efflux pumps on the potency of SecA inhibitors indicates that the SecA inhibitors does not have to accumulate intracellularly in order to achieve the needed potency. However, the diminished potency of the same SecA inhibitors in Gram-negative bacteria combined with the restored potency in the presence of membrane permeabilizers clearly suggests that the inhibitors need to accumulate in the periplasmic space. Such results further indicate that accessing the portion of the secretion machineries that extend beyond the inner membrane would not allow for the SecA inhibitors to reach its binding site, which most likely is on the inner membrane. With these results in mind, then there is the question as to whether SecA would be able to overcome the effect of efflux pumps in Gram-negative bacteria. Thus we next examined this issue.
SecA inhibitors’ permeation through the outer membrane and inhibition of cell growth of MDR strains
A major challenge in discovering inhibitors of Gram-negative bacteria is not only to overcome the outer membrane barrier, but also to resolve the issue of multidrug resistance (MDR).[3b, 12] The seriousness of the MDR problem of Gram-negative bacteria is exemplified by the thought of reviving the cytotoxic polymyxin B as the last resort antibiotic to treat such pathogens.[3b, 12a] Among the Gram-negative bacteria we tested in the synergistic use of PMBN or NAB7061 and SecA inhibitors (Table 2), many are MDR pathogens. In some tests, RB was used a reference compound, which has been extensively tested [7b] and is the lead compound of Class A RB analogues,[7a, 13] with sub-μM potency in inhibiting SecA ATPase and other functional activities.[7a, 13] Many other inhibitors (Table 2) showed potency in the low μg/mL to high ng/mL MIC range. All 3 classes of inhibitors exhibited bactericidal effects to different degrees. In side-by-side comparisons, the potency of some SecA inhibitors, e.g., SCA-107 or SCA-112, surpasses that of commonly used antibiotics such as carbenicillin and vancomycin (Table 2). For example, for clinical MDR P. aeruginosa strain T15464, the MIC100 values for SCA-112 are 0.1–2 μg/mL, as compared to over 1,000 μg/mL for carbenicillin, erythromycin and vancomycin.
The lack of effect of SecA inhibitors on MIC for various strains including wild-type, null, and over-expression of efflux pumps
Virtually the majority of surface proteins,[14] virulence factors,[14–15] and efflux pumps[2f] in Gram-negative bacteria possess signal peptides, most of which are SecA-dependent (from genome annotations, Signal IP program predicts 39 out of 45 proteins in P. aeruginosa relating to efflux possess signal peptides);[2d, 16] thus inhibition of SecA would also inhibit the secretion and functions of many of such components important to efflux and pathogenesis. It is well-known that the efflux pumps possess broad substrate specificity. Thus it is very difficult to design inhibitors to simultaneously act on various efflux pumps.[2c–f] We reason that inhibition of SecA should inhibit the assemblies of most bacterial efflux pumps, which are SecA-dependent. Thus the SecA inhibitors could render many SecA-dependent efflux pumps ineffective, in addition to inhibiting the secretion of virulence factors, and cell growth. The results strongly support our hypothesis that SecA inhibitors can overcome the effects of efflux and thus may not be subjected to MDR problems. Validating this point would represent a major advance in combating MDR problem.
We evaluated the ability of SecA inhibitors against a range of MDR strains with or without efflux pump expressions. MDR of P. aeruginosa is particularly problematic because of its pathogenicity.[17] Because SecA functions in the membrane as a protein-conducting channel,[18] it is possible that SecA is accessible from outside of the cytoplasmic membrane as is the case for Gram-positive bacteria. Five clinical P. aeruginosa MDR strains tested[19] were shown to be sensitive to SecA inhibitors (Table 2, and data not shown).
Most efflux pumps in E. coli (including AcrA and TolC)[20] and P. aeruginosa depend on outer membrane proteins that have signal peptides and are SecA-dependent.[16] Again, virtually all surface proteins,[14] virulence factors and efflux pumps [2f] in Gram-negative bacteria possess signal peptides, most of which are SecA-dependent; thus inhibition of SecA could also inhibit the functions of many of such components important to efflux and pathogenesis. As a result, SecA inhibitors may intrinsically be insensitive to the effect of efflux pumps. We tested the hypothesis by examining the antimicrobial effects of Class C SecA inhibitors SCA-107 and SCA-112 with several strains possessing different levels of activity of efflux pumps (Table 3). These included strains with the major efflux pumps in E. coli AcrAB/TolC efflux pump[21] from R. Misra,[20, 22] and P. aeruginosa PAO1 with wild-type, deletion and over-expressed pumps MexJK/OprM-OprH efflux pumps from H. Schweizer.[3a, 23] Our data showed that in the presence of NAB7061, deletion or over-production of MexJK/OprM pump in P. aeruginosa had no effect on the MIC of SCA-112 (Table 3). Furthermore, the deletion or overproduction of the major AcrAB pump in E. coli also has no effect on the MIC of SCA-107 or SCA-112 (Table 4). The E. coli strains with a plasmid carrying P. aureus pumps MexCD/OprJ or MexCD also had no effect on the MIC of SCA-107 or SCA-112. These results showed that the different levels of the efflux pumps in these strains have no effect on the MIC of the SecA inhibitors, validating the ability for SecA inhibitors to overcome the effect of efflux pumps in both Gram-positive and Gram-negative bacteria.
Table 3.
Effects of SCA-112 on P. aeruginosa strains with various efflux pumps, MIC (μg/mL)
| Addition | PAO1 Wild-type | PAO397 Δefflux pumps | MexJK-OprM in PAO397 | MexJK-OprH in PAO397 | MexJK-OprM-OmpH in PAO397 |
|---|---|---|---|---|---|
| None | >667 | >667 | >667 | >667 | >667 |
| PMBN | 0.9 | 0.2 | 0.2 | 0.3 | 0.1 |
| NAB7061 | 0.2 | 0.2 | 0.1 | 0.2 | 0.2 |
PMBN 5 μg/mL NAB7061. 10 μg/mL. PAO397 (PAO1 Δ(mexAB-oprM) nfxB Δ(mexCD-oprJ) Δ(mexJKL) Δ(mexXY) Δ(mexEF-oprN) OpmH+). The other specific efflux complexes are in PAO0397.
Table 4.
Effects of SecA inhibitors on levels of efflux pumps in E. coli. MIC (μg/mL)
| Inhibitors | Addition | W4573 Ram1292 | AcrAB− Ram1340 | AcrAB++ in Ram1340 | MexCD-OprJ+ in Ram1340 | MexCD+ in Ram1340 |
|---|---|---|---|---|---|---|
| SCA-112 | None | >667* | >667* | >667* | >667* | >667* |
| PMBN | 0.3 | 0.2 | 0.3 | 0.4 | 0.4 | |
| NAB7061 | 0.2 | 0.1 | 0.2 | 0.1 | ≤0.1 | |
| SCA-107 | None | >590* | >590* | >590* | >590* | >590* |
| PMBN | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | |
| NAB7061 | 0.7 | 0.7 | 0.7 | 0.4 | ≤0.05 |
PMBN used: 12.5 μg/mL; NAB7061 used : 2 μg/mL. E. coli parental strain W4573(Ram1292) and strains Ram1340(AcrAB−) contain P. aeruginosa efflux pump MexCD+ or MexCD-OprJ+.
Next we were interested in correlating the antimicrobial results with SecA inhibition outcomes in order to establish that the antimicrobial effects are indeed largely from the inhibition of SecA.
SecA inhibition kinetics
We tested the effects of these small molecules inhibitors on SecA functions in the liposomes or membranes.[7–8] Some of these compounds have been evaluated for their ability to inhibit in vitro functions of SecAs from Gram-positive bacteria including enzymatic ATPase activity, protein translocation, and protein-conducting channel activity, and to inhibit in vivo viability of Gram-positive bacteria.[7–8] Even though the original screenings were done by assaying the intrinsic ATPase of a truncated SecA without a regulatory C-terminus,[7b, 8b] it is worth emphasizing that SecA functions mainly in the lipid environment and that these inhibitors may exhibit varying affinities for different forms of SecA. Thus inhibition studies do need to employ various assays in order to achieve a thorough understanding of the ability for these inhibitors to interfere SecA functions. Our recent publications detail the discussion of the various assays available and their utilities.[3a, 7b, 9] In particular, we have developed two very sensitive assays to evaluate the functions of SecA in the lipid-environments.[18a, 24] Both are reconstituted SecA-only liposomes systems specific for SecA functions in lipid environment for ion channel activity and protein translocation activity [SFig. 2]. Employing these assays, we previously discovered several SecA inhibitors that are effective at low micromolar concentrations.[7–8, 25]
We determined the functional activities of SecA purified from Gram-negative E. coli (EcSecA), P. aeruginosa (PaSecA) and S. typhymurium (StSecA) in the presence of these Class C inhibitors in comparison with published RB[26] or SCA-50.[13] Both the channel activity and protein translocation in the SecA-only liposomes systems were strongly inhibited by these compounds with IC50 values in the range of 1.0–3.5 μM for SCA-107 and SCA112 (Table 5A, B). Even though RB is the most effective in the in vitro assays in inhibiting SecA functions, it affects other ATPases such as H+-ATPase (Table 5C) and is known to have cytotoxicity.[13] Its smaller analogues such as SCA-50 are more selective.[7a, 13] Kinetic studies using the channel activity of SecA-only liposomes assayed in the oocytes (Figure 3) showed that all 3 classes of inhibitors Class A SCA-50, Class B SCA-15, and Class C SCA-107 ([27] used here for comparison) are non-competitive against ATP for EcSecA, PaSecA or StSecA (Figure 3). Thus major inhibitory effects in the membrane may not be on the high-affinity ATPase active site directly (Table 5C); rather, the inhibition is probably allosteric in a membrane lipid environment (SFig. 3). This is significant because by not targeting the high affinity ATP site, it allows for an improved chance of minimizing toxicity issues since there are many ATP-binding proteins in the body. The lack of effects of these SecA inhibitors on other ATPases, such as H+-ATPase and SpuB, indicated the selectivity of the inhibitors for SecA. Furthermore, SecA alone may function as a channel in the membrane;[18a] the inhibitors may not need to enter into the cytoplasmic to exert their effect. The results with Gram-negative bacteria in the presence and absence of membrane permeabilizers also indicate that the binding site in the segment embedded in the inner membrane, not beyond. The finding that our inhibitors are non-competitive inhibitors for ATP may explain the earlier unsuccessful attempts to develop inhibitors that focused on the inhibition of intrinsic ATPase activity of SecA. These inhibitors are 1,000 fold more active than the well-known SecA inhibitor, azide,[2g] and the IC50 and MIC are in the range of 2–5 μM.
Table 5.
Effects of SecA inhibitors in channel activity, protein translocation, and ATPase IC50 (μM).
| Assays | Protein | RB | SCA-50 | SCA-107 | SCA-112 |
|---|---|---|---|---|---|
| A. Channel activity | EcSecA | 0.4 | 2.3 | 1.6 | 1.3 |
| PaSecA | 0.3 | 3.0 | 1.3 | 1.1 | |
|
| |||||
| B. Protein translocation | EcSecA | 1.0 | ND | 3.5 | ND |
| PaSecA | 1.0 | ND | 3.0 | ND | |
|
| |||||
| C. ATPase activity | H+-ATPase | 60.0 | >200* | >200* | >200* |
| SpuB ATPase | >200* | >200* | >200* | >200* | |
(A). Inhibition on ion-channel activity of SecAs in oocytes. SecA of E. coli (EcSecA), and P. aeruginosa (PaSecA), were reconstituted with liposomes, and then were assayed for ion-channel activity in oocytes with various inhibitors for 3 hrs. The recordings were for 1 min. n =30. (B). Inhibition on SecA-liposomes protein translocation activity, (C) Effects against other ATPase. ND: not determined.
Highest concentration tested.
Figure 3. Non-competitive Inhibition of channel activity of SecA-liposomes in oocytes.

(A–C) All 3 classes of SecA inhibitors with EcSecA show non-competitive inhibition with ATP; the data of SCA-107 is from ref. [9], presented here for comparisons with 3 classes of inhibitors, and with SecA homologs (D) PaSecA and (E) StSecA
Molecular docking studies using methods as described previously[13, 23a] show that SCA-107 lies outside the ATP pocket of chain A (SFig. 3A). This is different from the binding site of SCA-50 which blocks the ATP entrance in chain B.[13] Also, docking studies indicate that EcSecA-L515, (a point SecA mutation which results in resistance to SCA-107, see below) is located away (SFig. 3B) from the SCA-107 binding pockets (and possibly SCA-50’s), suggesting potential allosteric effects. The 3 classes of SecA inhibitors are structurally different (Figure 1), and molecular modeling work suggests that they bind to different pockets of the SecA dimer. Earlier, we have noticed that these classes do not bind directly to or inside the ATP containing pockets in chains A and B, but lie close to and outside the ATP pocket, (SCA-50, and SCA-15),[13, 23a] whereas SCA-107 binds on the interface of chains A and B.[9] Such observations are consistent with the kinetic data obtained showing non-competitive inhibition with ATP (Fig. 3).
Probing Protein Binding
In addition to the specific inhibition by SecA-only liposomes systems, we also use SecA inhibitors as chemical probes to examine direct interactions with SecA in whole cell lysates. Specifically, we employed two assays commonly used to identify drug-binding targets.[13] These were pull-down assays with conjugated inhibitors (SFig. 4) and Altered Trypsin Sensitivity (DARTS) study that have been used successfully in identifying binding targets [28] (Figures 5A, B). Thus we synthesized conjugates of SCA-104, SCA-105 and SCA-113 with biotin or beads (SFig. 4), which were successfully used to pull down SecA in E. coli lysates (Figure 4). By adding conjugated inhibitor SCA-104, a major protein of the size of SecA was detected (Figure 4A). All three conjugated inhibitors pulled down SecA as detected by immunoblot with antibodies (Figure 4B), validating direct interaction of the inhibitors with SecA in whole cell lysates. Moreover, addition of SCA-15 and SCA-21 (an analogue of SCA-107) to E. coli lysates containing thousands of proteins altered the sensitivity of SecAs to trypsin digest (altered sensitivity indicated by arrows, Figure 5B). Such results are consistent with inhibitor binding to SecA, leading to altered accessibility of its hydrolytic site to trypsin. Both methods supported the idea that SecA is a target of the inhibitors [see also [7a]]. These experiments lend evidence to support that SecA is a target in bacteria in achieving the observed antimicrobial effects. In order to further assess whether SecA inhibition is the key mechanism through which antimicrobial effect was achieved, we conducted additional experiments using SecA mutants.
Figure 5. Altered trypsin sensitivity tests by SecA inhibitors.

(A) E. coli MC4100 cell lysates (200 μg) were incubated with or without 2.5 mM SCA-15 on ice for 2 hrs. The mixtures were digested with trypsin (50 μg) at 25 °C for 20 min. The digestion was stopped by adding SDS sample buffer and boiling for 20 min. and run on 10–20% gradient SDS-PAGE gel. (B) Immunoblot by EcSecA antibodies. E. coli MC4100 whole cell lysates with or without addition of purified EcSecA protein were similarly incubated with or without SCA-15 or SCA-21, and treated with trypsin as in (A). Arrows indicate the bands altered by treatment with the inhibitors.
Figure 4. Target pulled-down of SecA from E. coli lysates by Inhibitors conjugates.

E. coli MC4100 whole cell lysates (33 μg) were treated with SecA inhibitor conjugates as indicated, and the pull- down bead samples were washed, run on SDS gels, and analyzed: (A) Coomassie Blue staining. Arrow indicates SecA band. (B) Immunoblots by EcSecA antibodies.
Specific SecA mutation azi-9 (resulting in L515F) is SCA-107 resistant
In addition to the biochemical evidences above, perhaps the most critical evidence is our discovery of an E. coli azide-resistant mutant (a single azi-9 mutation resulting in EcSecA-L515F) that is resistant to SCA-107 (Table 6A). This mutant is among many azide-resistant strains we have tested for SecA mutants and PrlA-PrlD suppressors of D. Oliver, and T. Silhavy.[2g, 2h, 25, 29] It is noted that azide (at mM) inhibits only translocation ATPase, not intrinsic or membrane ATPase, and many suppressors mutants of prlD/secA (Table 6A), prlA/secY and prlG/secE are also azide resistant, but are sensitive to our SecA inhibitors at single digit μM concentrations (Table 6A, and data not shown). We have found that an azide-resistant strain DO318 carrying a single seca-azi-9 mutation [2g] is resistant to SCA-107 with an increased MIC of about 10–20 folds, providing important genetic evidence of SecA as a key target for SCA-107 (Table 6A) in achieving its antimicrobial effect. Interesting, the mutant is also partially resistant to Class A SCA-50 (data not shown). We have cloned this mutated ecsecA gene, and identified the single mutation resulting in SecAL515F. We expressed and purified the SecA L515F protein and carried out the liposomes assays to verify that this mutated SecA-L515F is indeed resistant to SCA-107. The results showed that the EcSecA-L515F were about 3 folds more resistant to SCA-107 and about 2–3 fold to SCA-50 in both channel activity and protein translocation activity assays (Table 7). Though the in vitro resistance is modest, it is similar to the azide-resistant mutants for in vitro activity [2g]. Moreover, we have also cloned the corresponding azi-9 in B. subtilis SecA, and purified the BsSecA-Azi9. Similarly, IC50 value increases of 2–3 fold were observed in channel activity and protein translocation activity assays (data not shown).
Table 6.
Effects of mutation azi-9 on MIC SecA inhibitors resistance in E. coli
| Assays | E. coli strains | PMBN | SCA-107 |
|---|---|---|---|
| A. Inhibitor sensitivity at 37°C, MIC (μg/mL). | NR698 | − | 2.2 |
| MC4100 | − | >590* | |
| + | 2.6 | ||
| DO318 (azi-9) | + | >37.8 | |
| Other prlD* | + | 0.7–3.0 | |
|
| |||
| B. Complementation at 42°C, MIC (μg/mL). | MC4100 | − | >566* |
| + | 2.0 | ||
| DO318 | − | >566* | |
| + | >50 | ||
| BA13/secA | − | >357* | |
| + | 1.8 | ||
| BA13/secA-azi-9 | − | >566 | |
| + | 11.0 | ||
MIC (μg/mL) of E. coli azide resistant strain DO318 (azi-9, EcSecA L515F) at 37°C. PMBN when used was at 25 μg/mL. *Other E. coli prlD mutants are all azide resistant or supersensitive strains, including az-R mutation in prlD5(A373V), prlD2(A488V), prlD22(Y134C), prlD43(H484Q), prlD4(T111N), azi-4 (L645Q), azi-630(A630V), azi-656(R656C), azi-7(N179Y), azi-6, SecA(Δ519–547) and az-SS mutation prlD20(A507V), and prlD43(H484O).
MIC (μg/mL) of complementation test of azi-9 mutation. Complementation of azi-9 mutation (EcSecA L515F) in vivo drug resistance, E. coli secAam, supFts mutant BA 13 containing plasmid ecsecA wild-type or ecsecA-azi-9 mutant were subjected to MIC test at 42C with or without 25 μg/mL PMBN.
Table 7.
Effects of azi-9 mutation of EcSecA on SecA inhibitors
| IC50: μM | EcSecA | Ion channel Activity | Protein Translocation | ||
|---|---|---|---|---|---|
| SCA-50 | SCA-107 | SCA-50 | SCA107 | ||
| SecA-Liposomes | Wild-type | 3.1 | 1.8 | ND | 3.5 |
| L515F | 9.3 | 3.8 | ND | 11.4 | |
|
| |||||
| Membranes | Wild-type | 2.4 | 1.1 | 3.9 | 2.8 |
| L515F | 4.7 | 4.1 | 11.2 | 8.2 | |
Effects of azi-9 mutation (L515F) of EcSecA on SecA inhibitors, IC50 (μM). Liposomes and SecA-depleted BA13 membranes were used for ion channel activity, while urea-treated 773 membrane was used for protein translocation. ND: Not determined
Complementation test of azi-9 mutation
To further verify that the azi-9 mutation alone confers resistance to SecA-107, we determined its ability to complement the drug-resistance of a secA ts mutant at 42 °C. We used the plasmid with cloned secA-azi-9 gene to compare to that with wild-type secA in the same strain. We tested the cloned azi-9 for its effectiveness in complementing the drug-resistance in vivo in a ts-secA mutant BA13.[11d, 30] As expected, the strain with the plasmid carrying secA-azi-9, like wild-type secA, complemented the growth of secA ts mutant BA13 at 42 °C. While the ecsecA wild-type strain remained sensitive to SCA-107 with a similar MIC of about 1.8 μg/mL, the complementation of secA-azi-9 occurred with a six-fold increase of MIC 11.0 μg/mL of SCA-107 (Table 6B). Thus, the plasmid carrying the azi-9 mutation conferred the in vivo resistance to SCA-107, further verifying that SecA is the target of SCA-107 and the EcSecA-L515F confers the resistance. Moreover, the complementation test confirmed the resistance by this single mutation on SecA for the drug resistance (Tables 6 and 7), thus providing the most convincing genetic evidences that SecA is the target for SCA-107. It should be noted that the original DO318 strain is much more resistant to SCA-107 (10–20 fold), but the complementation test for azi-9 mutation in a different genetic background showed significant but less resistance (6 fold, below). It is possible that the complementation is intrinsically not as efficient, but it is also possible that different genetic background accounts for the in vivo difference in resistance.
Conclusions
We have demonstrated that the potency difference for SecA inhibitors between Gram-positive and Gram-negative bacteria is indeed in the presence of the outer membrane in Gram-negative bacteria, which posses a permeation barrier. Using outer membrane permeabilizers as probes, we found that SecA inhibitors have potent effects on Gram-negative pathogens as well. The potency levels in antimicrobial activity and enzyme inhibition activity are consistent with SecA being the key target. In addition, the effectiveness of these SecA inhibitors is not affected by MDR strains with elevated levels of efflux pumps, indicating the intrinsic ability of our inhibitors to overcome/bypass the MDR issues. Because developing effective treatment against drug resistant Gram-negative bacteria is a significant challenge, this finding is very important. Enzyme kinetics results reveal non-competitive inhibition, indicating that the binding sites of these inhibitors are not the high affinity ATP site in SecA. This is important because there are many ATP-binding proteins in the body. By not targeting the ATP site, it positions these classes of inhibitors for the possibility of not having many off-target effects. Additional experiments in protein pull down, complementation, and using mutant SecA are consistent with SecA being the key target for the antimicrobial effects.
Because these SecA inhibitors are non-competitive against ATP, they are also well-positioned for further optimization effort without having to be concerned of off-target effects on other ATP-binding proteins. In addition, because of the role that SecA plays in the secretion of virulence factors, its inhibitors should also have the ability to attenuate bacterial pathogenicity. With the demonstrated outer membrane permeability problems, future research in targeting Gram-negative bacteria should focus on optimizing membrane permeability.
Materials and Methods
Strains and Plasmids
Escherichia coli MC4100 derived from a K12 strain, was obtained from J. Beckwith [31]; E. coli strains BA13 and BL21.19 [32] were obtained from D. Oliver; E. coli NR698 with an outer membrane mutation from T. Silhavy [10]; E. coli enterogenic strain from J. Scott [33]; mutants with azi and suppressors from D. Oliver [2g] and T. Silhavy [29a]; E. coli acr strains from R. Misra [34]; Pseudomonas aeruginosa MDR strains from H. Schweitzer [35] and CD Lu [19]; and Salmonella typhimurium SL1344 from A. Gewirtz [36]. Others strains are from ATCC in GSU Biology Department stocks.
Growth, inhibition and complementation
The growth conditions have been reported previously [13]. Briefly, 0.5 mL culture of bacterial cells (exponential phase, OD600 ≈ 0.5) was mixed with 4 mL of Luria-Bertani (LB) with 0.2% of glucose, with shaking at 37 °C. All cultures where appropriate were grown in the presence of 2% DMSO which had no effect on bacterial growth and was final concentration used with SecA inhibitors.
Bacteriostatic effect
Bacteriostatic effects were tested according to the guidelines of the Clinical and Laboratory Standards Institute (http://clsi.org/). This assay was performed in a 96-well microtiter plate in triplicates in three separate experiments at 37 °C, unless indicated otherwise, with shaking at 250 rpm for 24 hrs as described previously [13, 37]. MIC is the lowest concentration of inhibitor at which cells were not able to grow.
Bactericidal effect
Bactericidal effect was determined as described previously [7a]. Log-phase cells (OD 600 ≈ 0.5) were mixed with different concentrations of inhibitors, and incubated in an Eppendorf Thermomixer R (Brinkman Instruments) at 37 °C with shaking (1,000 rpm) for 2 hr. Cultures were serially diluted with LB broth and spread on LB plates (1.5% agar), and incubated at 37 °C overnight to determine colony forming units (CFU). Bactericidal effect was determined by the reduction of CFU as described previously [13, 37].
Complementation of secA-azi-9 mutation
The complementation of SecA function in vivo was carried out in BA13 secAts strain carrying the pT7 plasmid with wild-type secA or secA with azi-9 mutation (resulting in L515F) at 42 °C as described [30, 38]. The MIC was determined with various SCA-107 and SCA-50 concentrations as above.
Inner membrane vesicles preparation
Wild-type MC4100 membranes, and OmpA-deleted 773 membranes used for OmpA translocation, were prepared and washed with 8 M urea solution to inactivate SecA as described [39]. SecA-depleted BA-13 membranes were prepared from secAts BA13 mutant. Briefly, BA13 cells were grown at 30 °C to the mid-log phase, and then shifted to 42 °C until growth ceased due to SecA depletion. The cells were collected and the BA13 SecA-depleted membranes were prepared as described [39].
Liposomes preparation
Liposomes of E. coli total lipids (Avanti Lipids) were prepared as described previously [18a]. Briefly, the lipids were dried by spin vacuum, re-suspended in 150 mM KCl solution and sonicated 3–5 mins until the solution was clear. Liposome preparations were stored in small aliquots at −80°C and thawed only once for use.
Cloning and protein purification
EcSecA was cloned, over-expressed from BL21 (λDE3)/pET7a-SecA in E. coli BL21 (λDE3) and were purified as described [18a]. SecA from P. aerus (PasecA), B. subtilis (BsSecA), S. typhimurium (StSecA) were cloned in pET7a and expressed in E. coli BL21.19 after depletion of E.coli SecA at 42°C as described [7b, 18a, 40]. The ecsecA with azi-9 mutation was amplified from DO318 chromosome DNA, cloned, and a single mutation is identified by DNA sequencing resulting in SecA L515F. EcSecA-azi-9 cloned in pET7a was purified as above. A corresponding azi-9 mutation in bssecA was similarly constructed and the BsSecA-Azi-9 similarly purified. Precursor pOmpA was prepared as previously described [18a, 39]. Protein amounts were estimated from A280/A260 ratios, and confirmed by Bradford assay as described [18a, 24].
In vitro ATPase activity assay
ATPase activity for SecA, SpuB and H+-ATPase were assayed with minor modifications as described previously [7b]. Briefly, 50 μL reaction mixtures contained 1.5 μg of protein, 20 μg ovalbumin, 1.2 mM ATP, 50 mM Tris-HCl (pH 7.6), 20 mM KCl, 20 mM NH4Cl, 2 mM Mg(OAc)2, 1 mM DTT at 40°C for 30 mins. The ATPase activity was determined by the release of inorganic phosphate detected by a photometric method [7b] by measuring absorption at 660 nm (SmartSpec Plus, Bio-Rad Laboratories, Inc.). All assays were performed at least in triplicate.
In vitro protein translocation
Translocation of pOmpA was conducted as described previously [18a]. (See Supplement SFig 2). Unless otherwise indicated, the translocation mixtures in 0.1 mL contained 120 μg of liposomes or 4.5 μg of OmpA-depleted 773 membranes, 1 μg SecA, 0.1 μg SecB and 150 ng substrates pOmpA and ATP energy sources [18a]. The mixtures were incubated at 37 °C for 30 min. The translocation mixtures were treated with 400 μg/mL proteinase K in ice water for 30 min to remove untranslocated pOmpA. Liposomes or membranes were collected by centrifugation, and translocated OmpA proteins were detected by immunoblots as described previously [1a, 18a].
Xenopus oocyte injection and whole cell recording
Oocytes were collected, prepared and injected with sample mixtures as described previously [13, 41]. Briefly, the 50 nl sample mixture containing 120 ng liposomes, 120 ng SecA, 14 ng protein pOmpA, 2 mM ATP, and 1 mM Mg2+ was injected into oocytes. For kinetic studies, ATP concentrations were varied as indicated. All experiments were carried out in the presence of 4 mM puromycin to remove oocytes’ endogenous precursors. The effective concentration of the reagents in 50 nl injected mixtures was estimated based on an average oocytes volume of 500 nl. A voltage clamp was used to measure the opening of protein-conducting channels in the oocytes as described previously [3c, 18a, 41], (schematically, see SFig. 2). After 3 hours of incubation at 23 °C, the current of the oocytes was recorded continuously for 1 min. The inward current, which is minimal, and outward current were recorded with the two-electrode voltage clamp technique using KCl as the bath solution to measure the net currents [3c, 41].
SecA inhibitors, antibiotics and other reagents
The synthesis of the SecA inhibitors and their antimicrobial evaluation against Gram-positive bacteria have been described previously.[3a, 7a, 13, 23a, 27] All SecA inhibitors were dissolved in DMSO with a final DMSO concentration of 2% in all cultures or in all biochemical assays, which has no effect on cell growth or assays. Polymyxin derivative NAB7061 and PMBN were from Northern Antibiotics Ltd., Helsinki, Finland and Sigma-Aldrich, respectively, and used as received. Other antibiotics and chemicals were of reagent grade and obtained from Sigma-Aldrich.
Supplementary Material
Acknowledgments
We thank D. Oliver, T. Silhavy, CD Lu, R. Misra, and H. Schweizer for strains and plasmids. We thank Ping Jiang for DNA sequencing in the Biology Core facilities in the Center of Biotechnology and Drug Design, which were supported in parts by Georgia Research Alliance. This work was support by a NIH grant AI104168, and Fellowships of Molecular Basis of Diseases Program at GSU (JSJ, YHH, and ASC).
This article is dedicated to the memory of Bernard Davis for his work on antibiotics and protein secretion, and his influence on PCT in this study.
References
- 1.a) Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]; b) Bonomo RA, Szabo D. Clin Infect Dis. 2006;43(Suppl 2):S49–56. doi: 10.1086/504477. [DOI] [PubMed] [Google Scholar]; c) Chopra I, Schofield C, Everett M, O’Neill A, Miller K, Wilcox M, Frere JM, Dawson M, Czaplewski L, Urleb U, Courvalin P. Lancet Infect Dis. 2008;8:133–139. doi: 10.1016/S1473-3099(08)70018-5. [DOI] [PubMed] [Google Scholar]; d) Cornaglia G, Rossolini GM. Clin Microbiol Infect. 2010;16:99–101. doi: 10.1111/j.1469-0691.2009.03114.x. [DOI] [PubMed] [Google Scholar]; e Livermore DM. J Antimicrob Chemother. 2009;64(Suppl 1):i29–36. doi: 10.1093/jac/dkp255. [DOI] [PubMed] [Google Scholar]
- 2.a) Nakane A, Takamatsu H, Oguro A, Sadaie Y, Nakamura K, Yamane K. Microbiology. 1995;141(Pt 1):113–121. doi: 10.1099/00221287-141-1-113. [DOI] [PubMed] [Google Scholar]; b) Nathan C. Nature. 2004;431:899–902. doi: 10.1038/431899a. [DOI] [PubMed] [Google Scholar]; c) Nikaido H. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Nikaido H. Annu Rev Biochem. 2009;78:119–146. doi: 10.1146/annurev.biochem.78.082907.145923. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Nikaido H, Pages JM. FEMS Microbiol Rev. 2012;36:340–363. doi: 10.1111/j.1574-6976.2011.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Nikaido H, Zgurskaya HI. Curr Opin Infect Dis. 1999;12:529–536. doi: 10.1097/00001432-199912000-00001. [DOI] [PubMed] [Google Scholar]; g) Oliver DB, Cabelli RJ, Dolan KM, Jarosik GP. Proc Natl Acad Sci USA. 1990;87:8227–8231. doi: 10.1073/pnas.87.21.8227. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Osborne RS, Silhavy TJ. EMBO J. 1993;12:3391–3398. doi: 10.1002/j.1460-2075.1993.tb06013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Chaudhary AS, Chen W, Jin J, Tai PC, Wang B. Future Med Chem. 2015;7:989–1007. doi: 10.4155/fmc.15.42. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Evans ME, Feola DJ, Rapp RP. Ann Pharmacother. 1999;33:960–967. doi: 10.1345/aph.18426. [DOI] [PubMed] [Google Scholar]; c) Lin BR, Hsieh YH, Jiang C, Tai PC. The Journal of membrane biology. 2012;245:747–757. doi: 10.1007/s00232-012-9477-8. [DOI] [PubMed] [Google Scholar]; d) Rao S, De Waelheyns CVE, Economou A, Anné J. Biochimi Biophys Acta (BBA) - Mol Cell Res. 2014;1843:1762–1783. doi: 10.1016/j.bbamcr.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 4.a) Knott TG, Robinson C. J Biol Chem. 1994;269:7843–7846. [PubMed] [Google Scholar]; b) Sugie Y, Inagaki S, Kato Y, Nishida H, Pang CH, Saito T, Sakemi S, Dib-Hajj F, Mueller JP, Sutcliffe J, Kojima Y. J Antibiot. 2002;55:25–29. doi: 10.7164/antibiotics.55.25. [DOI] [PubMed] [Google Scholar]
- 5.Jang MY, De Jonghe S, Segers K, Anne J, Herdewijn P. Bioorg Med Chem. 2011;19:702–714. doi: 10.1016/j.bmc.2010.10.027. [DOI] [PubMed] [Google Scholar]
- 6.Nagaraju A, Zheng H, Han FQ, Wang N. Bioorg Med Chem Lett. 2011;21:4183–4188. doi: 10.1016/j.bmcl.2011.05.086. [DOI] [PubMed] [Google Scholar]
- 7.a) Cui JM, Jin JS, Shieh YH, Yang H, Ke B, Damera K, Tai PC, Wang B. ChemMedChem. 2013;8:1384–1393. doi: 10.1002/cmdc.201300216. [DOI] [PubMed] [Google Scholar]; b) Huang YJ, Wang H, Gao FB, Li M, Yang H, Wang B, Tai PC. ChemMedChem. 2012;7:571–577. doi: 10.1002/cmdc.201100594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Chen W, Huang YJ, Gundala SR, Yang H, Li M, Tai PC, Wang B. Bioorg Med Chem. 2010;18:1617–1625. doi: 10.1016/j.bmc.2009.12.074. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li M, Huang YJ, Tai PC, Wang B. Biochem Biophys Res Commun. 2008;368:839–845. doi: 10.1016/j.bbrc.2008.01.135. [DOI] [PubMed] [Google Scholar]
- 9.Cui J, Jin J, Chaudhary AS, Hsieh Y-h, Zhang H, Dai C, Damera K, Chen W, Tai PC, Wang B. ChemMedChem. 2016;11(1):43–56. doi: 10.1002/cmdc.201500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruiz N, Falcone B, Kahne D, Silhavy TJ. Cell. 2005;121:307–317. doi: 10.1016/j.cell.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 11.a) Ali FE, Cao G, Poudyal A, Vaara T, Nation RL, Vaara M, Li J. J Antimicrob Chemother. 2009;64:1067–1070. doi: 10.1093/jac/dkp331. [DOI] [PubMed] [Google Scholar]; b) Vaara M. Microbiol Rev. 1992;56:395–411. doi: 10.1128/mr.56.3.395-411.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Vaara M, Fox J, Loidl G, Siikanen O, Apajalahti J, Hansen F, Frimodt-Moller N, Nagai J, Takano M, Vaara T. Antimicrob Agents Chemother. 2008;52:3229–3236. doi: 10.1128/AAC.00405-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Vaara M, Siikanen O, Apajalahti J, Fox J, Frimodt-Moller N, He H, Poudyal A, Li J, Nation RL, Vaara T. Antimicrob Agents Chemother. 2010;54:3341–3346. doi: 10.1128/AAC.01439-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Vaara M, Viljanen P. Antimicrob Agents Chemother. 1985;27:548–554. doi: 10.1128/aac.27.4.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Falagas ME, Kasiakou SK. Clin Infect Dis. 2005;40:1333–1341. doi: 10.1086/429323. [DOI] [PubMed] [Google Scholar]; b) Mogi T, Kita K. Cell Mol Life Sci. 2009;66:3821–3826. doi: 10.1007/s00018-009-0129-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsieh YH, Zou J, Jin JS, Yang H, Chen Y, Jiang C, Yang J, Tai PC. Analytical biochemistry. 2015;480:58–66. doi: 10.1016/j.ab.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 14.Kline KA, Falker S, Dahlberg S, Normark S, Henriques-Normark B. Cell Host Microbe. 2009;5:580–592. doi: 10.1016/j.chom.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 15.Mudrak B, Kuehn MJ. J Bacteriol. 2010;192:1900–11. doi: 10.1128/JB.01542-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewenza S, Gardy JL, Brinkman FS, Hancock RE. Genome Res. 2005;15:321–329. doi: 10.1101/gr.3257305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breidenstein EB, de la Fuente-Nunez C, Hancock RE. Trends Microbiol. 2011;19:419–426. doi: 10.1016/j.tim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 18.a) Hsieh YH, Zhang H, Lin BR, Cui N, Na B, Yang H, Jiang C, Sui SF, Tai PC. J Biol Chem. 2011;286:44702–44709. doi: 10.1074/jbc.M111.300111. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang HW, Chen Y, Yang H, Chen X, Duan MX, Tai PC, Sui SF. Proc Natl Acad Sci USA. 2003;100:4221–4226. doi: 10.1073/pnas.0737415100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Kwon DH, Lu CD. Antimicrob Agents Chemother. 2006;50:1615–1622. doi: 10.1128/AAC.50.5.1615-1622.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kwon DH, Lu CD. Antimicrob Agents Chemother. 2007;51:2070–2077. doi: 10.1128/AAC.01472-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Hackett J, Misra R, Reeves P. FEBS Lett. 1983;156:307–310. doi: 10.1016/0014-5793(83)80518-3. [DOI] [PubMed] [Google Scholar]; b) Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. J Bacteriol. 1993;175:6299–6313. doi: 10.1128/jb.175.19.6299-6313.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a) Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. Nature. 2006;443:173–179. doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]; b) Yu EW, McDermott G, Zgurskaya HI, Nikaido H, Koshland DE., Jr Science. 2003;300:976–980. doi: 10.1126/science.1083137. [DOI] [PubMed] [Google Scholar]
- 22.Sulavik MC, Houseweart C, Cramer C, Jiwani N, Murgolo N, Greene J, DiDomenico B, Shaw KJ, Miller GH, Hare R, Shimer G. Antimicrob Agents Chemother. 2001;45:1126–1136. doi: 10.1128/AAC.45.4.1126-1136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Chaudhary AS, Jin J, Chen W, Tai PC, Wang B. Bioorg Med Chem. 2015;23:105–117. doi: 10.1016/j.bmc.2014.11.017. [DOI] [PubMed] [Google Scholar]; b) Srikumar R, Kon T, Gotoh N, Poole K. Antimicrob Agents Chemother. 1998;42:65–71. doi: 10.1128/aac.42.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsieh Y, Zhang H, Yang H, Jiang C, Sui S-F, Tai Phang C. Biochem Biophys Res Commun. 2013;431:388–392. doi: 10.1016/j.bbrc.2013.01.042. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt MG, Rollo EE, Grodberg J, Oliver DB. J Bacteriol. 1988;170:3404–3414. doi: 10.1128/jb.170.8.3404-3414.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsieh YH, Huang YJ, Jin JS, Yu L, Yang H, Jiang C, Wang B, Tai PC. Biochem Biophys Res Commun. 2014;454:308–312. doi: 10.1016/j.bbrc.2014.10.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cui J, Jin J, Chaudhary AS, Hsieh YH, Zhang H, Dai C, Damera K, Chen W, Tai PC, Wang B. ChemMedChem. 2016;11(1):43–56. doi: 10.1002/cmdc.201500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S, Wang J, Wu RP, Gomez F, Loo JA, Wohlschlegel JA, Vondriska TM, Pelletier J, Herschman HR, Clardy J, Clarke CF, Huang J. Proc Natl Acad Sci USA. 2009;106:21984–21989. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.a) Huie JL, Silhavy TJ. J Bacteriol. 1995;177:3518–3526. doi: 10.1128/jb.177.12.3518-3526.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fikes JD, Bassford PJ., Jr J Bacteriol. 1989;171:402–409. doi: 10.1128/jb.171.1.402-409.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a) De Waelheyns E, Segers K, Sardis MF, Anne J, Nicolaes GA, Economou A. J Antibiot (Tokyo) 2015 doi: 10.1038/ja.2015.53. [DOI] [PubMed] [Google Scholar]; b Floyd JH, You Z, Hsieh YH, Ma Y, Yang H, Tai PC. Biochem Biophys Res Commun. 2014;453:138–142. doi: 10.1016/j.bbrc.2014.09.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silhavy TJ, Beckwith J. Methods Enzymol. 1983;97:11–40. doi: 10.1016/0076-6879(83)97115-x. [DOI] [PubMed] [Google Scholar]
- 32.Cabelli RJ, Chen LL, Tai PC, Oliver DB. Cell. 1988;55:683–692. doi: 10.1016/0092-8674(88)90227-9. [DOI] [PubMed] [Google Scholar]
- 33.Froehlich B, Parkhill J, Sanders M, Quail MA, Scott JR. J Bacteriol. 2005;187:6509–6516. doi: 10.1128/JB.187.18.6509-6516.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soparkar K, Kinana AD, Weeks JW, Morrison KD, Nikaido H, Misra R. J Bacteriol. 2015 doi: 10.1128/JB.00547-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.a Chuanchuen R, Murata T, Gotoh N, Schweizer HP. Antimicro Agents Chemother. 2005;49:2133–2136. doi: 10.1128/AAC.49.5.2133-2136.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chuanchuen R, Narasaki CT, Schweizer HP. J Bacteriol. 2002;184:5036–5044. doi: 10.1128/JB.184.18.5036-5044.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanders CJ, Franchi L, Yarovinsky F, Uematsu S, Akira S, Nunez G, Gewirtz AT. Eur J Immunol. 2009;39:359–371. doi: 10.1002/eji.200838804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das S, Stivison E, Folta-Stogniew E, Oliver D. J Bacteriol. 2008;190:7302–7307. doi: 10.1128/JB.00593-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang H, Na B, Yang H, Tai PC. J Bacteriol. 2008;190:1413–1418. doi: 10.1128/JB.01633-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tai PC, Tian G, Xu H, Lian JP, Yu JN. Methods Cell Biol. 1991;34:167–187. [PubMed] [Google Scholar]
- 40.Yu L, Yang H, Ho Q, Tai PC. Protein Expr Purif. 2006;50:179–184. doi: 10.1016/j.pep.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 41.Lin BR, Gierasch LM, Jiang C, Tai PC. The Journal of membrane biology. 2006;214:103–113. doi: 10.1007/s00232-006-0079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.