

Abstract
The first example of a biocatalytic [2,3]-sigmatropic rearrangement reaction involving allylic sulfides and diazo reagents (Doyle-Kirmse reaction) is reported. Engineered variants of sperm whale myoglobin catalyze this synthetically valuable C–C bond forming transformation with high efficiency and product conversions across a variety of sulfide substrates (i.e., aryl-, benzyl-, and alkyl-substituted allylic sulfides) and α-diazo esters. Moreover, the scope of this myoglobin-mediated transformation could be extended to the conversion of propargylic sulfides to give substituted allenes. Active site mutations proved effective toward enhancing the catalytic efficiency of the hemoprotein in these reactions as well as modulating its enantioselectivity, resulting in the identification of a myoglobin variant, Mb(L29S, H64V, V68F), capable of mediating asymmetric Doyle-Kirmse reactions with an enantiomeric excess up to 71%. This work extends the toolbox of currently available biocatalytic strategies for realizing the asymmetric formation of carbon–carbon bonds.
Biocatalytic Doyle-Kirmse reaction
Engineered variants of sperm whale myoglobin catalyze the tandem sulphur ylide formation / [2,3]-sigmatropic rearrangement of allylic and propargylic sulfide in the presence of α-diazo esters. This transformation could be applied to a variety of aryl, benzylic, and alkyl sulfide substrates resulting in high product conversions and enantioselectivity up to 71% ee.
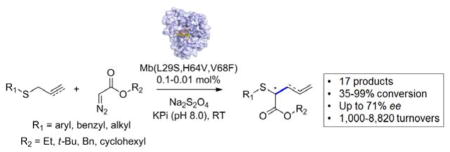
Biocatalytic transformations can provide key opportunities toward the development of economical and sustainable processes for the synthesis and manufacturing of fine chemicals and pharmaceuticals.[1] Enzyme-catalyzed carbon-carbon bond forming reactions have traditionally involved the use of aldolases, thiamine diphosphate-dependent enzymes, hydroxynitrile lyases, and terpene cyclases, with protein engineering providing a means to expand the scope of these enzymes to non-native substrates.[2] More recently, engineered and artificial metalloenzymes have made possible other valuable C–C bond forming transformations, including olefin cyclopropanation,[3] Suzuki coupling,[4], Diels–Alder reactions,[5] Friedel-Crafts indole alkylation[6], Wittig olefination,[7], and olefin metathesis[8]. Despite this progress, the toolbox of biocatalytic systems useful for the construction of C–C bonds remains limited when compared to synthetic methods.
Our laboratory and the Arnold group have recently reported the ability of heme-containing proteins such as myoglobin[3d, 7, 9] and P450[3a–c], respectively, to engage α-diazo ester reagents in carbene transfer reactions. In particular, we found that engineered variants of sperm whale myoglobin can provide highly active and selective biocatalysts for carbene-mediated transformations such as olefin cyclopropanation,[3d], Y–H carbene insertion (Y = N, S),[9] and aldehyde olefination[7]. The transition metal-catalyzed reaction between allyl sulfides and a diazo reagent (a.k.a. Doyle-Kirmse reaction)[10] represents a powerful method for the creation of new C–C bonds, which has found application in the synthesis of various biologically active molecules.[11] This process involves a reaction between the allyl sulfide and a metallo-carbenoid species, leading to formation of a sulfur ylide which undergoes a [2,3]-sigmatropic rearrangement.[10c, 11] While several organometallic catalysts, including rhodium,[10b, 12] copper,[12a–c, 13] and cobalt[14] complexes, have proven useful for promoting this transformation,[15] the development of catalytically efficient and enantioselective variants of this reaction has proven very challenging.[11b] Here, we report the development of myoglobin-based biocatalysts capable of promoting asymmetric Doyle-Kirmse reactions with high catalytic efficiency across a broad panel of allylic and propargylic sulfide substrates and different α-diazoesters.
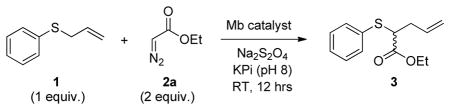
Initially, we investigated the activity of sperm whale myoglobin (Mb) in catalyzing the [2,3]-sigmatropic rearrangement of allyl phenyl sulfide 1 in the presence of ethyl α-diazo acetate (EDA, 2a). Gratifyingly, this reaction resulted in the formation of the desired [2,3]-sigmatropic rearrangement product 3 with 44% conversion (445 TON) under optimized conditions (Table 1, Entry 1). No product formation was observed under aerobic conditions (Table 1, Entry 2), indicating that oxygen, i.e. Mb native ligand, inhibits this reactivity. Reactions performed in the presence and absence of reductant (Na2S2O4) showed that ferrous Mb is catalytically more efficient than the ferric counterpart (445 vs. 195 TON, Table 1), although the latter remains a viable catalyst for this reaction as supported by these results and additional experiments (SI Table S1 and Figure S2). Varying the pH between 6 and 9 had negligible effect on Mb-dependent catalytic activity, whereas improved conversions were obtained with a two-fold excess of EDA (55% vs. 19% with one equiv. EDA). As observed for hemin, the Mb-catalyzed formation of 3 show no enantiomeric excess (<1% e.e.) indicating that the native Mb scaffold is unable to exert any asymmetric induction during the reaction.
Table 1.
Myoglobin-catalyzed tandem sulfur ylide formation and [2,3]-sigmatropic rearrangement of phenyl allyl sulfide with EDA.[a]
| ||||
|---|---|---|---|---|
| Entry | Catalyst | Deviation from s.r.c. | % conv.[b] | TON[c] |
| 1 | WT Mb | - | 44% | 445 |
| 2 | WT Mb | aerobic | 0% | 0 |
| 3 | WT Mb | no Na2S2O4 | 19% | 195 |
| 4 | WT Mb | 1 equiv. 2a | 19% | 185 |
| 5 | WT Mb | 0.5 equiv. 2a | 11% | 115 |
| 6 | Mb(L29S,H64V,V68F) | - | >99% | >995 |
| 7 | Mb(L29S,H64V,V68F) | 0.025 mol% | 87% | 3,500 |
| 8 | Mb(L29S,H64V,V68F) | 0.01 mol% | 63% | 6,270 |
| 9 | Mb(L29S,H64V,V68F) | 30 min | >99% | >995 |
Standard reaction conditions (s.r.c.) = 10 mM 1, 20 mM 2a, 10 μM Mb catalyst (0.1 mol%), 10 mM Na2S2O4 in oxygen-free potassium phosphate buffer (50 mM, pH 8) at room temperature.
As determined by gas chromatography.
= nmol product / nmol catalyst. Errors are within ± 10%.
In order to identify more efficient and selective Mb-based biocatalysts for this reaction, we evaluated a panel of engineered Mb variants containing one to three amino acid substitutions at the level of the five residues defining the distal cavity of the hemoprotein (Leu29, Phe43, His64, Val68, Ile107; Figure S1). Previously, we found that mutations at these positions can dramatically alter the activity and selectivity of Mb variants as carbene[3d, 7, 9, 16] and nitrene[17] transfer catalysts. Upon testing in the reaction with 1 and EDA, a number of Mb variants with significantly improved catalytic activity compared to the wild-type protein were identified (Figure S3). Among them, Mb(L29S,H64V,V68F) emerged as the most active biocatalyst for this reaction, leading to the quantitative conversion of 1 into 3 at a catalyst loading of only 0.1 mol%. Notably, a product conversion of 87% (3,500 TON) and 63% (6,270 TON) were obtained with even lower catalyst loadings of 0.025 and 0.01 mol%, respectively (Table 1, Entries 6–8). These results are notable considering that similar yields in related Doyle-Kirmse reactions were achieved with catalyst loadings of 1–5 mol% for Rh-based complexes[12a, 12d, 12f] and of 5–20 mol% for synthetic catalysts based on non-precious metals.[12c, 13b, 13c, 14] In addition, in contrast to the need for slow addition of the diazo reagent in Rh-[12a, 12d] and Cu-catalyzed reactions[13a], the Mb(L29S,H64V,V68F)-catalyzed reaction proceeds with excellent chemoselectivity, i.e. without carbene dimerization, even upon direct mixing of the sulfide and diazo reactants. Time course experiments further showed that the biocatalytic formation of 3 occurs with an initial rate of 167 turnovers per minute (SI Table S2) and reaches completion within 30 min (Table 1, Entry 9). These kinetics also compare very favourably with those of organometallic catalysts, and in particular those involving Cu and Co complexes, for which reaction times of 10–36 hours have been reported.[13b, 13c, 14]
Controlling the enantioselectivity of Doyle-Kirmse reactions has proven challenging (typically, <10–50% ee), a phenomenon that has been attributed to the difficulty of discriminating, using chiral catalysts, between the heterotopic lone pairs of the sulfide during attack to the metal-carbenoid species to yield a chiral sulfur ylide,[12a, 13a, 13c, 14b] whose stereochemical information is then readily transferred to the carbon atom during the bond rearrangement process.[18] While the native Mb scaffold produces 3 in racemic form, moderate to good levels of enantioselectivity were obtained with some of the engineered Mb variants (Figure S3). Importantly, the highly active Mb(L29S,H64V,V68F) variant also showed the highest degree of stereocontrol, yielding 3 with an enantiomeric excess (ee) of 71% (Figure S4). Notably, Mb(H64V,V68A) favors formation of the opposite enantiomeric product with 46% ee (49% conv., 490 TON).
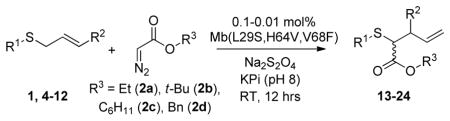
To examine the substrate scope of Mb(L29S,H64V,V68F), variously substituted α-diazo esters and allyl sulfides were tested. As shown in Table 2, quantitative or nearly quantitative conversions to the desired products 13–15 (94–99%) were achieved starting from 1 and diazo reagents such as tert-butyl (2b), cyclohexyl (2c), or benzyl α-diazo-acetate (2d). Good to excellent conversions (57–99%) were also obtained for reactions involving allyl phenyl sulfides with substituted phenyl rings (4–6) to give products 16–18. Next, the Mb(L29S,H64V,V68F)-catalyzed transformation of benzyl- (8–9) and alkyl-substituted allyl sulfides (10–12) in the presence of EDA was examined. The high yields measured for 20 and 21 (78–93%) indicated that benzyl-substituted allyl sulfides are also efficiently processed by the biocatalyst. Except for the poorly water-soluble octyl allyl sulfide (11), moderate to high product conversions (35–86%) were achieved for the reactions with other alkyl-substituted allyl sulfides (22, 24), further supporting the broad substrate scope of Mb(L29S,H64V,V68F). Finally, the successful synthesis of 19 from phenyl but-2-enyl sulfide (7) and EDA (>99% conv.) showed that substitutions at the level of the allyl group are also tolerated by the Mb variant. Under catalyst-limited conditions (i.e., using 0.01 mol%), Mb(L29S,H64V,V68F) were found to support thousands of catalytic turnovers (1,000–8,800) for all of the tested substrates except 11 (Table 2).
Table 2.
Substrate scope of Mb(L29S,H64V,V68F).[a]
| |||||
|---|---|---|---|---|---|
| Sulfide | Diazo reag. | Product | % conv. (TON) | TTN[b] | ee [%][c] |
| 1 | 2b |
 13 |
>99% (>1,000) | 8,170 | 6% |
| 1 | 2c |
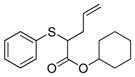 14 |
>99% (>1,000) | 8,820 | 9% |
| 1 | 2d |
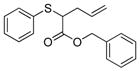 15 |
94% (940) | 3,570 | 47% |
| 4 | 2a |
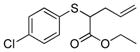 16 |
>99% (>1,000) | 5,050 | 20% −60%[d] |
| 5 | 2a |
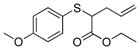 17 |
>99% (>1,000) | 5,960 | 40% |
| 6 | 2a |
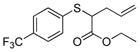 18 |
57% (570) | 1,000 | 18% 58%[d] |
| 7 | 2a |
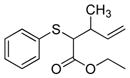 19 |
>99% (>1,000) | 8,120 | 57/59% 1:1 d.r. |
| 8 | 2a |
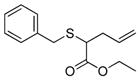 20 |
78% (780) | 7,040 | 10% |
| 9 | 2a |
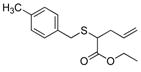 21 |
93% (930) | 5,470 | 43% |
| 10 | 2a |
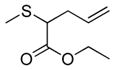 22 |
35% (350) | 3,570 | 38% |
| 11 | 2a |
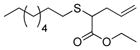 23 |
8% (80) | 125 | n.d.[e] |
| 12 | 2a |
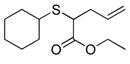 24 |
86% (860) | 4,190 | 19% |
Under standard reactions conditions as described in Table 1.
Using 1 μM Mb catalyst instead of 10 μM.
As determined by chiral GC or Supercritical Fluid Chromatography (SFC).
Using Mb(H64V,V68A).
Enantiomers could not be resolved.
The [2,3]-sigmatropic rearrangement of propargylic sulfides offers a convenient route to generate allenes, which are valuable intermediates for a host of synthetic transformations.[19] To assess the scope of the Mb(L29S,H64V,V68F) catalyst in the context of this reaction, variously substituted phenyl propargylic sulfides in combination with ethyl or benzyl α-diazo-acetate as carbene precursors were tested (Scheme 1). Notably, the corresponding allenyl-substituted sulfide products (28–31) were obtained in high yields (71–83%) in most cases, thereby demonstrating the functionality of the Mb-based catalyst in promoting the [2,3]-sigmatropic rearrangement of propargylic sulfide substrates.
Scheme 1.

Mb(L29S,H64V,V68F)-catalyzed [2,3]-sigmatropic rearrangement of propargylic sulfides.
Chiral GC/SFC analyses showed that Mb(L29S,H64V,V68F) exhibits moderate to good enantioselectivity (20–60% ee) in the transformation of several of the allylic sulfide substrates of Table 2. In comparison, the biocatalytic transformation of the propargylic sulfides occurred with significantly reduced enantiocontrol (<15% ee; Scheme 1). These experiments also revealed that the degree of asymmetric induction can be influenced by the structure of the diazo reagent (e.g., 47–71% ee for 3 and 15 vs. 4–9% ee for 13 and 14). Finally, a larger scale reaction with 15 mg of phenyl allyl sulfide (1), 2 equiv. of EDA (2a), and 0.1 mol% Mb(L29S,H64V,V68F) enabled the isolation of 19.4 mg of 3 in 84% yield, thus demonstrating the scalability of the biocatalytic process.
Scheme 2 depicts a plausible mechanism for the Mb-catalyzed reaction reported here. We envisage the initial formation of an iron-porphyrin bound carbenoid species (II), which has electrophilic character[3d, 20] and can react with nucleophiles.[7, 9] Accordingly, nucleophilic attack by action of the allylic sulfide on this intermediate is envisioned to give rise to a sulfonium ylide (III), followed by a rapid [2,3]-sigmatropic rearrangement to yield the final product. The proposed role of the sulfide substrate as nucleophile is consistent with the experimentally observed higher reactivity of electronrich allyl sulfides versus isosteric, electrodeficient counterparts (17 vs. 18; Table 2). This trend was reproduced with other Mb variants other than Mb(L29S,H64V,V68F) (i.e., 83–95% conversion for 17 vs. 13–30% for 18 with Mb(H64V,V68A) and Mb(F43V,V68F)), suggesting that the differential reactivity is largely dictated by the electronic properties of the substrate rather than the biocatalyst. The enantioselectivity-determining step in asymmetric Doyle-Kirmse reactions is generally assumed to be associated with formation of the (chiral) sulfonium ylide,[12a, 13a, 13c, 14b] an assumption based on the high degree of stereoretention observed during the rearrangement of in situ prepared, optically active sulfonium ylides.[18, 21] Within this mechanistic framework, we envision two alternative, albeit not mutually exclusive scenarios by which enantiocontrol is exerted by the engineered Mb catalysts, namely (i) by influencing the pre-attack orientation of the sulfide so that approach to the heme-carbene intermediate through either of the lone pairs on the sulfur atom is preferred, and/or (ii) by dictating which face of the heme-bound carbenoid group is exposed to attack by the sulfide nucleophile, in analogy to our proposed stereochemical model for Mb-catalyzed olefin cyclopropanation.[3d]
Scheme 2.

Proposed mechanism and catalytic steps for the myoglobin-catalyzed Doyle-Kirmse reaction.
While further studies are warranted to discriminate among these scenarios, experiments were performed to clarify the beneficial role of the active site mutations in Mb(L29S,H64V,V68F). To this end, we characterized and compared the catalytic activity and selectivity of a set of single and double reversion mutants in the reaction with phenyl allyl sulfide (1) and EDA (SI Table S2). In line with our previous observations,[3d, 9a, 16] mutation of the distal His residue (H64V) increases both the TTN (2,230 vs. 1,615 for Mb) and product formation rate (121 vs. 79 min−1), possibly through facilitating access of the substrate to the heme cavity (SI Figure S1). The L29S mutation further enhances the catalytic competency of the hemoprotein, as suggested by the increased total turnovers (3,085 TTN) supported by Mb(L29S,H64V). The V68F mutation has no beneficial effect when alone or in combination with H64V, but it contributes synergistically with L29S to improving both TTN (6,280 for Mb(L29S,H64V,V68F) vs. 3,085 for Mb(L29S,H64V)) and rate (167 vs. 118 min−1, respectively). Interestingly, none of the single-site or double-site variants showed significantly improved enantioselectivity compared to wild-type Mb (<5% ee; SI Table S2). These results indicate that the enhanced enantioselectivity of Mb(L29S,H64V,V68F) stems from a synergistic effect of all three active site mutations, which is likely to be facilitated by the close proximity of these residues within the heme pocket (~7Å for C(β)···C(β) distance; SI Figure S1)
In summary, this study demonstrates that engineered Mb variants can serve as efficient biocatalysts for mediating asymmetric Doyle-Kirmse reactions. Using the optimized variant Mb(L29S,H64V,V68F), good to excellent product conversions as well as high numbers of catalytic turnovers (up to 8,820) could be achieved across a variety of allylic and propargylic sulfides in the presence of α-diazo ester-based carbene precursors. Importantly, the enantioselectivity of the Mb catalyst could be tuned and optimized via mutations within the distal pocket of the protein. This work expands the toolbox of biocatalytic strategies useful for mediating sigmatropic rearrangements[22] and the asymmetric formation of carbon-carbon bonds.[2a]
Supplementary Material
Acknowledgments
This work was supported by the U.S. National Institute of Health grant GM098628. We are grateful to Prof. Daniel Weix (U. Rochester) for providing access to the SFC instrumentation. LC-MS instrumentation was supported by the U.S. NSF grant CHE-0946653.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K. Nature. 2012;485:185–194. doi: 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]; b) Turner NJ. Nature Chem Biol. 2009;5:568–574. doi: 10.1038/nchembio.203. [DOI] [PubMed] [Google Scholar]; c) Bommarius AS, Blum JK, Abrahamson MJ. Curr Opin Chem Biol. 2011;15:194–200. doi: 10.1016/j.cbpa.2010.11.011. [DOI] [PubMed] [Google Scholar]; d) Reetz MT. J Am Chem Soc. 2013;135:12480–12496. doi: 10.1021/ja405051f. [DOI] [PubMed] [Google Scholar]
- 2.a) Schmidt NG, Eger E, Kroutil W. ACS Catal. 2016;6:4286–4311. doi: 10.1021/acscatal.6b00758. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Faber K, Fessner WD, Turner NJ. Science of Synthesis. 1–3. Georg Thieme; Stuttgart: 2015. Biocatalysis in Organic Synthesis. [Google Scholar]; c) Fesko K, Gruber-Khadjawi M. Chemcatchem. 2013;5:1248–1272. [Google Scholar]; d) Hammer SC, Syren PO, Seitz M, Nestl BM, Hauer B. Curr Opin Chem Biol. 2013;17:293–300. doi: 10.1016/j.cbpa.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 3.a) Coelho PS, Brustad EM, Kannan A, Arnold FH. Science. 2013;339:307–310. doi: 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]; b) Coelho PS, Wang ZJ, Ener ME, Baril SA, Kannan A, Arnold FH, Brustad EM. Nat Chem Biol. 2013;9:485–487. doi: 10.1038/nchembio.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang ZJ, Renata H, Peck NE, Farwell CC, Coelho PS, Arnold FH. Angew Chem Int Ed. 2014;53:6810–6813. doi: 10.1002/anie.201402809. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bordeaux M, Tyagi V, Fasan R. Angew Chem Int Ed. 2015;54:1744–1748. doi: 10.1002/anie.201409928. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Srivastava P, Yang H, Ellis-Guardiola K, Lewis JC. Nat Commun. 2015;6:7789. doi: 10.1038/ncomms8789. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Gober JG, Rydeen AE, Gibson-O’Grady EJ, Leuthaeuser JB, Fetrow JS, Brustad EM. Chembiochem. 2016;17:394–397. doi: 10.1002/cbic.201500624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Abe S, Niemeyer J, Abe M, Takezawa Y, Ueno T, Hikage T, Erker G, Watanabe Y. J Am Chem Soc. 2008;130:10512–10514. doi: 10.1021/ja802463a. [DOI] [PubMed] [Google Scholar]; b) Chatterjee A, Mallin H, Klehr J, Vallapurackal J, Finke AD, Vera L, Marsh M, Ward TR. Chem Sci. 2016;7:673–677. doi: 10.1039/c5sc03116h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Coquiere D, Bos J, Beld J, Roelfes G. Angew Chem Int Ed. 2009;48:5159–5162. doi: 10.1002/anie.200901134. [DOI] [PubMed] [Google Scholar]; b) Podtetenieff J, Taglieber A, Bill E, Reijerse EJ, Reetz MT. Angew Chem Int Ed. 2010;49:5151–5155. doi: 10.1002/anie.201002106. [DOI] [PubMed] [Google Scholar]
- 6.Bos J, Browne WR, Driessen AJ, Roelfes G. J Am Chem Soc. 2015;137:9796–9799. doi: 10.1021/jacs.5b05790. [DOI] [PubMed] [Google Scholar]
- 7.Tyagi V, Fasan R. Angew Chem Int Ed. 2016;55:2512–2516. doi: 10.1002/anie.201508817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Mayer C, Gillingham DG, Ward TR, Hilvert D. Chem Commun. 2011;47:12068–12070. doi: 10.1039/c1cc15005g. [DOI] [PubMed] [Google Scholar]; b) Philippart F, Arlt M, Gotzen S, Tenne SJ, Bocola M, Chen HH, Zhu L, Schwaneberg U, Okuda J. Chemistry. 2013;19:13865–13871. doi: 10.1002/chem.201301515. [DOI] [PubMed] [Google Scholar]; c) Basauri-Molina M, Verhoeven DGA, van Schaik AJ, Kleijn H, Gebbink RJMK. Chemistry. 2015;21:15676–15685. doi: 10.1002/chem.201502381. [DOI] [PubMed] [Google Scholar]
- 9.a) Sreenilayam G, Fasan R. Chem Commun. 2015;51:1532–1534. doi: 10.1039/c4cc08753d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tyagi V, Bonn RB, Fasan R. Chem Sci. 2015;6:2488–2494. doi: 10.1039/c5sc00080g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Kirmse W, Kapps M. Chem Ber. 1968;101:994–996. [Google Scholar]; b) Doyle MP, Griffin JH, Chinn MS, Vanleusen D. J Org Chem. 1984;49:1917–1925. [Google Scholar]; c) Doyle MP, Forbes DC. Chem Rev. 1998;98:911–936. doi: 10.1021/cr940066a. [DOI] [PubMed] [Google Scholar]
- 11.a) Sweeney JB. Chem Soc Rev. 2009;38:1027–1038. doi: 10.1039/b604828p. [DOI] [PubMed] [Google Scholar]; b) West TH, Spoehrle SSM, Kasten K, Taylor JE, Smith AD. ACS Catal. 2015;5:7446–7479. [Google Scholar]
- 12.a) Nishibayashi Y, Ohe K, Uemura S. J Chem Soc Chem Comm. 1995:1245–1246. [Google Scholar]; b) Aggarwal VK, Ferrara M, Hainz R, Spey SE. Tetrahedron Lett. 1999;40:8923–8927. [Google Scholar]; c) Zhang XM, Ma M, Wang JB. Tetrahedron Asymmetr. 2003;14:891–895. [Google Scholar]; d) Wee AGH, Shi Q, Wang ZY, Hatton K. Tetrahedron Asymmetr. 2003;14:897–909. [Google Scholar]; e) Muller P, Grass S, Shahi SP, Bernardinelli G. Tetrahedron. 2004;60:4755–4763. [Google Scholar]; f) Liao M, Wang B. Green Chem. 2007;9:184–188. [Google Scholar]; g) Zhang H, Wang B, Yi H, Zhang Y, Wang JB. Org Lett. 2015;17:3322–3325. doi: 10.1021/acs.orglett.5b01542. [DOI] [PubMed] [Google Scholar]
- 13.a) McMillen DW, Varga N, Reed BA, King C. J Org Chem. 2000;65:2532–2536. doi: 10.1021/jo991842n. [DOI] [PubMed] [Google Scholar]; b) Zhang XM, Qu ZH, Ma ZH, Shi WF, Jin XL, Wang JB. J Org Chem. 2002;67:5621–5625. doi: 10.1021/jo025687f. [DOI] [PubMed] [Google Scholar]; c) Ma M, Peng LL, Li CK, Zhang X, Wang JB. J Am Chem Soc. 2005;127:15016–15017. doi: 10.1021/ja055021d. [DOI] [PubMed] [Google Scholar]
- 14.a) Fukuda T, Katsuki T. Tetrahedron Lett. 1997;38:3435–3438. [Google Scholar]; b) Fukuda T, Irie R, Katsuki T. Tetrahedron. 1999;55:649–664. [Google Scholar]
- 15.a) Zhou CY, Yu WY, Chan PWH, Che CM. J Org Chem. 2004;69:7072–7082. doi: 10.1021/jo049540v. [DOI] [PubMed] [Google Scholar]; b) Davies PW, Albrecht SJC, Assanelli G. Org Biomol Chem. 2009;7:1276–1279. doi: 10.1039/b822584b. [DOI] [PubMed] [Google Scholar]; c) Carter DS, Van Vranken DL. Org Lett. 2000;2:1303–1305. doi: 10.1021/ol005740r. [DOI] [PubMed] [Google Scholar]; d) Holzwarth MS, Alt I, Plietker B. Angew Chem Int Ed. 2012;51:5351–5354. doi: 10.1002/anie.201201409. [DOI] [PubMed] [Google Scholar]
- 16.Giovani S, Singh R, Fasan R. Chem Sci. 2016;7:234–239. doi: 10.1039/c5sc02857d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bordeaux M, Singh R, Fasan R. Bioorg Med Chem. 2014;22:5697–5704. doi: 10.1016/j.bmc.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trost BM, Hammen RF. J Am Chem Soc. 1973;95:962–964. [Google Scholar]
- 19.Krause N, Hashmi ASK, editors. Modern Allene Chemistry. Wiley-VCH; Weinheim, Germany: 2004. [Google Scholar]
- 20.a) Khade RL, Fan WC, Ling Y, Yang L, Oldfield E, Zhang Y. Angew Chem Int Ed. 2014;53:7574–7578. doi: 10.1002/anie.201402472. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wolf JR, Hamaker CG, Djukic JP, Kodadek T, Woo LK. J Am Chem Soc. 1995;117:9194–9199. [Google Scholar]
- 21.Trost BM, Biddlecom WG. J Org Chem. 1973;38:3438–3439. [Google Scholar]
- 22.a) Hilvert D, Carpenter SH, Nared KD, Auditor MTM. P Natl Acad Sci USA. 1988;85:4953–4955. doi: 10.1073/pnas.85.14.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jackson DY, Jacobs JW, Sugasawara R, Reich SH, Bartlett PA, Schultz PG. J Am Chem Soc. 1988;110:4841–4842. [Google Scholar]; c) Haynes MR, Stura EA, Hilvert D, Wilson IA. Science. 1994;263:646–652. doi: 10.1126/science.8303271. [DOI] [PubMed] [Google Scholar]; d) Yoon SS, Oei Y, Sweet E, Schultz PG. J Am Chem Soc. 1996;118:11686–11687. [Google Scholar]; e) Ulrich HD, Mundroff E, Santarsiero BD, Driggers EM, Stevens RC, Schultz PG. Nature. 1997;389:271–275. doi: 10.1038/38470. [DOI] [PubMed] [Google Scholar]; f) Zhou ZHS, Flohr A, Hilvert D. J Org Chem. 1999;64:8334–8341. doi: 10.1021/jo991299a. [DOI] [PubMed] [Google Scholar]; g) Prier CK, Hyster TK, Farwell CC, Huang A, Arnold FH. Angew Chem Int Ed. 2016;55:4711–4715. doi: 10.1002/anie.201601056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.