

Summary
Aims
Hypoxic‐ischemia alters mitochondrial membrane potential (Δψm), respiratory‐related enzymes, and mitochondrial DNA (mtDNA). Drugs acting on mitochondria, such as cyclosporine A (CsA), may reveal novel mitochondria‐based cell death signaling targets for stroke. Our previous studies showed that Parkinson's disease‐associated protein DJ‐1 participates in the acute endogenous neuroprotection after stroke via mitochondrial pathway. DJ‐1 was detected immediately after stroke and efficiently translocated into the mitochondria offering a new venue for developing treatment strategies against stroke. Here, we examined a molecular interaction between CsA and mitochondrial integrity in the in vitro acute stroke model of oxygen glucose deprivation/reperfusion (OGD/R) injury with emphasis on DJ‐1.
Methods
Primary rat neuronal cells (PRNCs) were exposed to OGD/R injury and processed for immunocytochemistry, ELISA, and mitochondria‐based molecular assays to reveal the role of DJ‐1 in CsA modulation of mitochondrial integrity.
Results
Administration of CsA before stroke onset (24 h pre‐OGD/R) afforded significantly much more robust neuroprotective effects than when CsA was initiated after stroke (2 h post‐OGD/R), revealing that CsA exerted neuroprotection in the early phase of ischemic stroke. CsA prevented the mitochondria‐dependent cell death signaling pathway involved in cytochrome c (Cyt c)‐induced intrinsic apoptotic process. CsA preserved cellular ATP content, but not hexokinase activity under hypoxic conditions. CsA prevented both mtDNA decrement and Δψm degradation after reperfusion, and enhanced secretion of DJ‐1 in the mitochondria, coupled with reduced oxidative stress.
Conclusion
These observations provided evidence that CsA maintained mitochondrial integrity likely via DJ‐1 upregulation, supporting the concept that mitochondria‐based treatments targeting the early phase of disease progression may prove beneficial in stroke.
Keywords: Cytochrome c, Mitochondrial DNA, Mitochondrial membrane potential (Δψm), Mitochondrial permeability transition pore, Oxygen glucose deprivation/reperfusion injury
Introduction
Stroke is characterized by neural tissue death due to deprivation of oxygen, glucose, and other nutrients that results from a reduction in blood flow to the brain. Disease progression with stroke primarily involves a primary insult characterized by an infarcted core, and subsequently the formation of an ischemic penumbra, which over a subacute period remains as salvageable neural tissue, thereby amenable to therapeutic intervention 1, 2, 3. Secondary cell death processes, including oxidative stress, can further exacerbate cell death in the penumbra limiting neurorestoration 1, 2, 3. Oxidative stress has been implicated in the pathogenesis of many central nervous system (CNS) disorders, including Alzheimer's disease and Parkinson's disease (PD) 4, 5, 6, 7.
The mitochondria are the energy factories of the cells and play essential roles in energy metabolism, including electron transport and adenosine triphosphate (ATP) synthesis, and an important source of reactive oxygen species (ROS) which at abnormally high levels can cause cellular damage 8. Mitochondrial biogenesis is a tightly regulated metabolic process in healthy cells controlled by the mitochondrial DNA (mtDNA). Damage to the energetic integrity of the mitochondria accompanies adult ischemia, with aberrant opening of the mitochondrial permeability transition pore (MPTP), a core mediator of ischemic cell death 9, 10, 11, 12, 13. Ca2+ accumulation contributes to normal neuronal function; however, excessive mitochondrial Ca2+ overload causes a severe reduction in mitochondrial membrane potential (Δψm), and triggers the aberrant opening of the MPTP and membrane permeability leading to the release of apoptotic proteins, such as cytochrome c (Cyt c) and apoptosis‐inducing factor (AIF), from the mitochondrial inner membrane space 14, 15. To this end, inhibition of MPTP may afford neuroprotective effects. Cyclosporine A (CsA) has been shown to be a potent inhibitor of MPTP opening in animal models of CNS disorders, including stroke, traumatic brain injury, and Parkinson's disease (PD) 10, 11, 12, 13, 24, 25, 26, 27, 28. CsA acts through binding with cyclophilin–calcineurin (CN) complex to suppress cytokine gene expression and block of T lymphocyte action. CsA is the treatment of choice for immunosuppression in organ and neural transplantation 10, 17, 20. The inhibition of CN can result in the inhibition of nitric oxide synthase activation, free radical formation 10, 21, or the mitochondria‐induced cell death 10, 22, 23. However, to date, the exact mechanism of CsA in exerting neuroprotection remains not fully understood.
Parkinson's disease‐associated protein DJ‐1 is a multifunctional redox‐sensitive protein that mediates neuroprotection by dampening mitochondrial oxidative stress 6, 7, 24, 25, 26, 27, 28, molecular chaperoning of PD‐aggregating protein a‐synuclein 4, stimulating antiapoptotic and antioxidative gene expression 29 and facilitating the prosurvival Akt while suppressing apoptosis signal‐regulating kinase (ASK1) pathways 30. DJ‐1 is localized both in the cytoplasm and nucleus, translocates to mitochondria of a variety of mammalian cells by oxidative stress or mitogen stimulation 31, and is secreted into the serum under pathologic conditions such as breast cancer and melanoma 32. Our previous studies demonstrated a novel observation of DJ‐1 translocation into the mitochondria after oxygen glucose deprivation/reperfusion (OGD/R) injury in human neural progenitor cells (hNPCs) and primary rat neuronal cells (PRNCs), which opens new avenues of research and therapeutic development targeting DJ‐1 for rescuing stroke and other neurological disorders characterized by rampant mitochondrial deficits 25, 26, 27. Accumulating evidence has implicated the role of mitochondria in abrogating free radical generation 5 which served as impetus for us to determine whether DJ‐1 translocated into the mitochondria might attenuate mitochondrial injury or reduce the mitochondrial ROS production in neurological disorders 25, 26, 27.
We hypothesized that in addition to DJ‐1 acting as an intracellular therapeutic molecule against oxidative stress, the protein also functions as an extracellular signaling molecule, thereby allowing coordination between neighboring neuronal cells via paracrine and/or autocrine cues. Here, we tested whether CsA stimulated DJ‐1 secretion in neuronal cells. The aims of this study were to explore the neuroprotective mechanism of CsA in PRNCs under hypoxic‐ischemia condition using an in vitro acute stroke model of OGD/R injury, in particular focusing on the role of neuronal mitochondria in cell death signaling and as a therapeutic target for stroke.
Materials and Methods
Cell Culture and OGD/R
PRNCs were obtained from BrainBits (E18 rat cortex; Springfield, IL, USA). According to the protocol, cells (4 × 104 cells/well) were suspended in 200 μL Neural Medium (NbActive 4; BrainBit) containing 2 mm L‐glutamine and 2% B27 in the absence of antibiotics and grown in Poly‐L‐Lysine‐coated 96‐well plates (354516; BD Biosciences, Franklin Lakes, NJ, USA) at 37°C in humidified atmosphere containing 5% carbon dioxide in 40% of the neuron and 60% astrocyte cell population (determined immunocytochemically using vesicular glutamate trransporter‐1). After 5‐day culturing (approximately cell confluence of 70%), PRNCs were exposed to OGD/R as described previously with few modifications 26, 33. The cells were initially exposed to OGD/R medium (glucose‐free Dulbecco's Modified Eagle Medium, Gibco, Life Technologies, Waltham, MA, USA), then placed in an anaerobic chamber (Plas‐Labs, Inc., Lansing, MI, USA) containing 95% nitrogen and 5% carbon dioxide for 15 min at 37°C, and finally, the chamber was sealed and incubated for 90 min at 37°C (hypoxic‐ischemic condition). Control cells were incubated in same buffer containing 5 mm glucose at 37°C in a regular CO2 (5%) incubator (normoxic condition). OGD/R was terminated by adding 5 mm glucose to medium, and cell cultures re‐introduced to the regular CO2 incubator (normoxic condition) at 37°C for 2 h, of which period represented a model of “reperfusion.”
Administration of CsA
For pre‐stroke administration of CsA (Paddock Laboratories, Inc., Minneapolis, MN, USA), cells were pre‐treated with CsA 24 h before stroke onset (24 h pre‐OGD/R), while for post‐stroke regimen, cells were exposed to CsA at 2 h after stroke (2 h post‐OGD/R). PRNCs were subjected to OGD/R for 90 min, followed by a 2‐h reperfusion period under normoxic condition.
Measurement of Cell Viability: Calcein‐AM Fluorescence Dye
Measurement of cell viability was performed by both fluorescent live/dead cell assay and trypan blue exclusion method 34, 35. This dye is ferrous iron sensitive 36. A two‐color fluorescence cell viability assay was performed by Calcein‐AM (L3224; Invitrogen, Waltham, MA, USA) to be retained within live cells, including an intense uniform green fluorescence and ethidium homodimer (EthD‐1) to bind the nuclei of damaged cells (bright red fluorescence). After 2‐h reperfusion, the PRNCs were incubated with 2 μ m Calcein‐AM and 4 μ m EthD‐1 for 45 min at room temperature in darkness according to the manufacturer's instructions. Green fluorescence of the live cells was measured by the Gemini EX florescence plate reader (Molecular Device, Sunnyvale, CA, USA), excitation at 485 nm and emission at 538 nm. In addition, trypan blue (15250‐061, Gibco, Life Technologies) exclusion method was conducted and mean viable cell counts were calculated in four randomly selected areas (1 mm2, n = 10) to reveal the cell viability. To precisely calibrate the cell viability, the values were standardized form florescence intensity and trypan blue data 26, 35.
Measurement of Oxidative Stress: Glutathione (GSH) Activity
As glutathione has been validated as an antioxidant component of oxidative defense system in eukaryotic cell 25, 26, 37 and that increased total intracellular glutathione level provides a measure of toxicological response precluding cell death 25, 26, 37, we performed glutathione assay using manufacturer's protocol for GHS‐Glo™ Glutathione Assay Kit (V6911; Promega, Madison, WI, USA). The optical density of solubilized purple formazan was measured at 570 nm on a Synergy HT plate reader (Bio‐Tex, Inc., Houston, TX, USA). The value of EC 50, concentration of CsA that gives half‐maximal response, was calculated from the equation, y = A + [(B−A)/(1 + (x/EC50)h)], where y is the observed value, A is minimal value, B is maximal value, x is the concentration of CsA, and the Hill coefficient (h, 1.0) gives the largest absolute value of the slope of the curve 38.
Measurement of mtDNA Stability: Picogreen Fluorescence Intensity
Effect of CsA with mtDNA stability measured by Picogreen fluorescence (P11496; Invitrogen) intensity 39. To quantitating, the degree of mtDNA depletion within living cells was performed by Picogreen according to the manufacturer's instructions. After 2‐h reperfusion, the PRNCs were incubated with Quant‐iT Picogreen dsDNA reagent for 5 min at room temperature in darkness. The green fluorescence of the live cells was measured by the Gemini EX florescence plate reader (Molecular Device), excitation at 480 nm and emission at 520 nm.
Measurement of Δψm: Tetramethylrhodamine Methyl Ester (TMRM)
For the measurement of Δψm, PRNCs were incubated with 25 nm tetramethylrhodamine methyl ester (TMRM) (88065; Sigma‐Aldrich, Fremont, CA, USA) for 45 min before completed reperfusion 39, 40, 41 and the fluorescence was measured by the Gemini EX florescence plate reader (Molecular Device), excitation at 549 nm and emission at 573 nm.
Measurement of ATP Levels
The measurement of ATP content was analyzed using ATP bioluminescence assay kit (11699709001; Roche Life Science, Indianapolis, IN, USA) according to the manufacturer's instructions. After 2‐h reperfusion, the PRNCs were incubated with cell lysis reagent (Sigma, Fremont, CA, USA) and protease inhibitor cocktail (Sigma) to the samples for 5 min at room temperature in darkness. Luciferase reagent was added to the samples, and the luminescence of the live cells was measured by the Gemini EX florescence plate reader (Molecular Device).
Measurement of Hexokinase Activity: G6P
The measurement of hexokinase activity was measured by glucose 6 phosphate dehydrogenase assay kit (ab102529; Abcam, Cambridge, MA, USA) according to the manufacturer's instructions. After 2‐h reperfusion, the PRNCs were incubated with reaction mix to the samples for 30 min at room temperature in darkness. Absorbance from each sample was measured in duplicate using a Synergy HT plate reader (Bio‐Tex, Inc.) at wavelengths of 450 nm.
Measurement of Extracellular DJ‐1 Concentration
The quantitative measurement of DJ‐1/PARK‐7 in PRNCs supernatant was analyzed using a CircuLex DJ‐1/PARK‐7 ELISA Kit (CY‐9050; MBL International Corporation, Woburn, MA, USA) according to the manufacturer's instructions 42. Under baseline conditions (without OGD/R conditions), dose range of 50 nm – 10 μ m of concentration of CsA cell supernatant was measured with extracellular DJ‐1 levels. Absorbance from each sample was measured in duplicate using a Synergy HT plate reader (Bio‐Tex Inc.) at dual wavelengths of 450/540 nm.
Immunocytochemistry Analysis
PRNCs (8 × 104 cell/well) in 400 μL Neural medium containing 2 mm L‐glutamine and 2% B27 in the absence of antibiotics and grown in Poly‐L‐Lysine 8 chamber (354632; BD Biosciences) were fixed in 4% paraformaldehyde for 20 min at room temperature after OGD/R or non‐OGD/R treatment 26. For Cyt c, mitochondria staining and for DJ‐1, mitochondria staining, cells were blocked for 60 min at room temperature with 5% normal goat serum (50‐062Z; Invitrogen, Camarillo, CA, USA) in PBS containing 0.1% Tween‐20 (PBST) (Sigma‐Aldrich). After blocking reaction, the cells were incubated overnight at 4°C with rabbit monoclonal anti‐cytochrome c (1:250, ab76237; Abcam), mouse monoclonal anti‐ATP synthase β‐chain (Mitochondria) (1:200, 05‐709; Cell Signaling Technology, Danvers, MA, USA), and rabbit monoclonal anti‐DJ‐1 (1:100, ab76008; Abcam) with 5% normal horse serum. The cells were washed five times for 10 min in PBST and then soaked in 5% normal goat serum in PBST containing corresponding secondary antibodies, goat anti‐rabbit IgG‐Alexa 488 (green; 1:1000, A11034; Invitrogen) and goat anti‐mouse IgG‐Alexa 594 (red; 1:1000, A11032; Invitrogen), for 90 min. Finally, cells were washed five times for 10 min in PBST and three times for 5 min in PBS, and subsequently embedded with mounting medium. Immunofluorescent images were visualized using Zeiss Axio Imager Z1 (Zeiss, Thornwood, NY, USA). Control experiments were performed with the omission of the primary antibodies yielding negative results.
Statistics
The data were evaluated using ANOVA followed by post hoc Bonferroni's test. Statistical significance was preset at P < 0.05. Data are presented as mean ± SE from quintuplicates of each treatment condition.
Results
Pretreatment of CsA Enhances Neuroprotection Dose‐Dependently
Administration of 10 μ m CsA before 24 h pre‐OGD/R afforded significantly much more robust neuroprotective effects compared to OGD/R‐treated cells without CsA (P < 0.01) (Figure 1A). On the other hand, CsA was initiated after 2 h post‐OGD/R has no inductive effect on cell survival (P > 0.05) (Figure 1A), suggesting that CsA facilitates the delayed intracellular neuroprotective signaling, but not acute signal transduction. We evaluated the dose–response of CsA efficiency (500 nm ~ 10 μ m) during 24‐h treatment, as half‐life time of CsA is approximately within 24 h in rodents 43. PRNCs viability significantly increased in administration of between 500 nm, 1 μ m, and 10 μ m (P < 0.05, P < 0.05, P < 0.01, respectively) (Figure 1B). These results indicate that effect of CsA occurs predominantly within the confined of concentrations and the time window.
Figure 1.
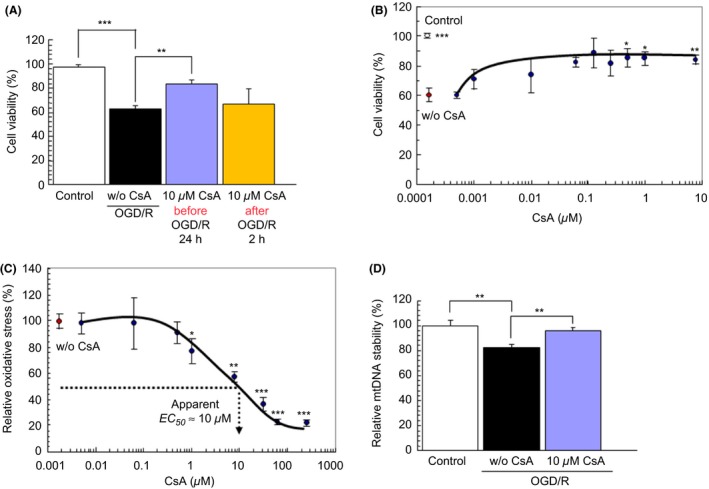
CsA possesses neuroprotective effects and attenuates the mtDNA degradation under hypoxic‐ischemic condition. Administration of CsA before stroke onset (24 h pre‐OGD/R) and CsA was initiated after stroke (2 h post‐OGD/R). PRNCs were subjected to OGD/R for 90 min, followed by a 2‐h reperfusion period under normoxic condition. Under hypoxic‐ischemic condition, cell viability tested by Calcein‐AM fluorescence dye (A). Cell viability dose‐response curve (B). Effect of CsA with oxidative stress (C). Relative oxidative stress percentage (%) is calculated from the equation described in materials and methods. Effect of CsA with mtDNA stability measured by Picogreen fluorescence intensity (D). *P < 0.05, **P < 0.01, and ***P < 0.001 versus w/o CsA.
Effects of CsA on Mitochondrial Activity of PRNCs after OGD/R
A cascade of hypoxic‐ischemic cell death events may principally arise from a dysfunctional mitochondrial complex I or NADH dehydrogenase, culminating with aberrant accumulation of ROS, a hallmark biochemical feature of oxidative stress. The EC 50 value of CsA on reducing the oxidative stress after OGD/R was approximately 10 μ m (P < 0.01) (Figure 1C). Therefore, the following experiments were performed with treatment of 10 μ m CsA during 24 h prior to OGD/R. Ten μ m CsA significantly protected mtDNA degradation (P < 0.01) (Figure 1D), Δψm reduction (P < 0.05) (Figure 2A), and ATP decrement (P < 0.05) (Figure 2B) after OGD/R compared to without CsA. These data prompted us to examine whether CsA influenced ATP synthesis. ATP content was significantly different between CsA treatment at 24 h and nontreatment controls (P < 0.05) (Figure 2B). A hexokinase is an important enzyme which regulates phosphorylation of a hexose. G6P controls intercellular metabolic processes, such as glycolysis and glycogen synthesis, and directly regulates thioredoxin‐interacting protein expression 44. Accordingly, we next investigated whether CsA increased the glucose transport activity. G6P activity was not significantly different between CsA treatment with 24 h and non‐treatment controls (39.3 ± 1.00 pmoles/min/104 cells versus 41.8 ± 2.03 pmoles/min/104 cells, respectively, P value 0.324). Because ATP synthesis depends on the mitochondrial membrane potential, we subsequently tested whether CsA modified the membrane potential, and showed that CsA significantly protected the degradation of Δψm (P < 0.05) (Figure 2A). These data indicate that CsA did not influence glycolysis, but suppressed the abnormal accumulation of mitochondrial ROS in preventing damages to both complexes I and III.
Figure 2.
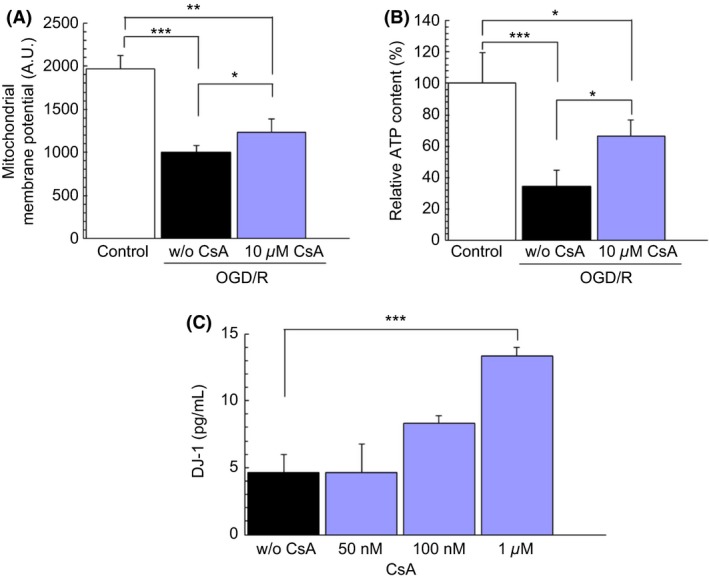
CsA enhances the secretion of DJ‐1 and prevents the decline of both Δψm and cellular ATP. Administration of CsA before stroke onset (24 h pre‐OGD/R). PRNCs were subjected to OGD/R for 90 min, followed by a 2‐h reperfusion period under normoxic condition. Under hypoxic‐ischemic condition, Δψm was measured by TMRM (A) and ATP levels were measured by ATP bioluminescence assay (B). Under baseline conditions (without OGD/R conditions), DJ‐1 expression levels were detected by the CircuLex DJ‐1/PARK ELISA kit (C). *P < 0.05 **P < 0.01, and ***P < 0.001.
CsA Stimulates the Secretion of DJ‐1
The observed therapeutic benefits of CsA coincided with increased levels of DJ‐1. Results revealed that there was a significant upregulation of DJ‐1 at 1 μ m of CsA compared to the baseline levels (without OGD/R conditions) (P < 0.001) (Figure 2C).
CsA Decreases the Release of Cyt c from Mitochondria
To establish an interaction between CsA and mitochondria in the in vitro acute stroke model of OGD/R injury with emphasis on DJ‐1, we examined CsA and DJ‐1 in the mitochondria at the level of immunocytochemical assays (Figure 3). At normoxic condition, Cyt c was retained within the mitochondria (Figure 3A–C). However, under hypoxic condition, the Cyt c was released outside the mitochondria (Figure 3D–F). Under hypoxic condition, treatment with 10 μ m of CsA demonstrated that Cyt c was maintained within the mitochondria (Figure 3G–I).
Figure 3.
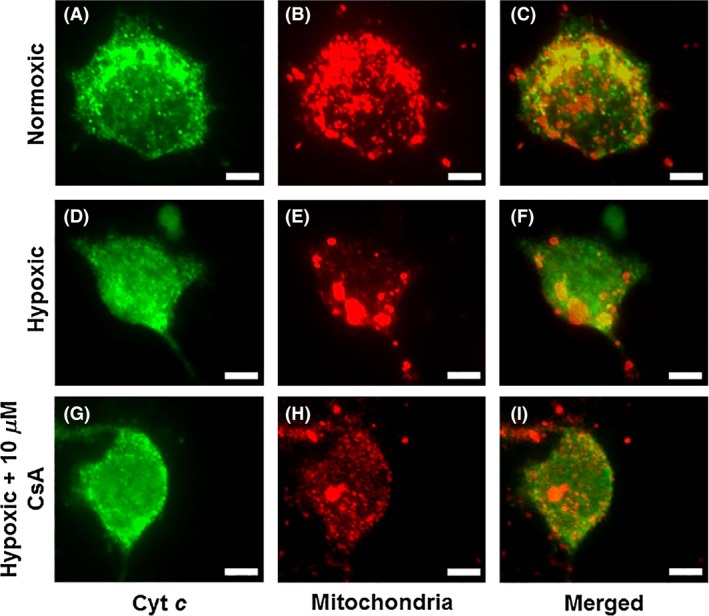
CsA decreases the releasing Cyt c from mitochondria, which turns on the caspases‐induced apoptotic signal pathway. Cyt c (A, D, G) and Mitochondria (B, E, H) staining revealed an apparent maintained within the mitochondria under hypoxic condition when treated with 10 μ m of CsA (G–I) compared to without CsA. Cyt c and Mitochondria double‐positive cells are shown in panels: C, F, and I. Green: Cyt c, Red: Mitochondria. Scale bars = 5 μm.
CsA Enhances DJ‐1 Translocation into the Mitochondria
To relate to this to the DJ‐1 expression, we also analyzed the localization within the mitochondria (Figure 4). We found similar result, at normoxic condition, Cyt c was retained within the mitochondria (Figure 4A–C). However, under hypoxic condition, the Cyt c was released outside the mitochondria (Figure 4D–F). DJ‐1 was co‐localized with the mitochondria under hypoxic condition when treated with 10 μ m of CsA (Figure 4G–I). These results suggest a direct interaction between DJ‐1 and mitochondria in the observed CsA neuroprotection.
Figure 4.
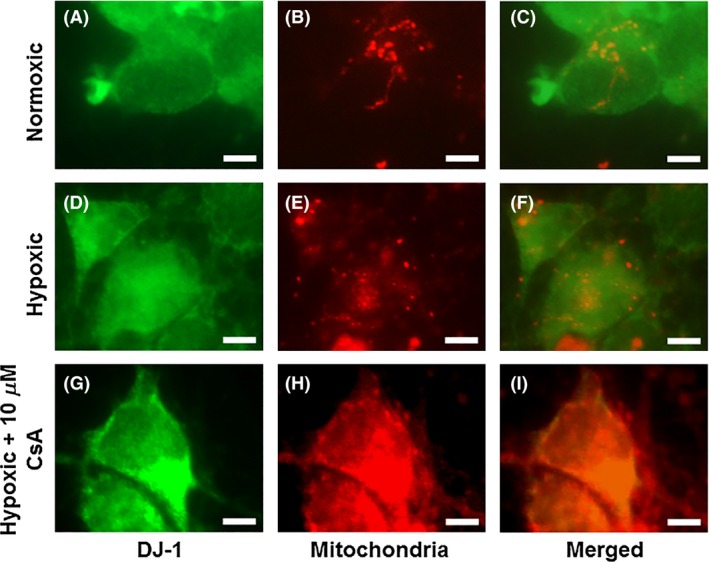
CsA enhances DJ‐1 translocation into the mitochondria. DJ‐1 (A, D, G) and Mitochondria (B, E, H) staining revealed an apparent maintained within the mitochondria under hypoxic condition when treated with 10 μ m of CsA (G–I) compared to without CsA. DJ‐1 and Mitochondria double‐positive cells are shown in panels: C, F, and I. Green: DJ‐1, Red: Mitochondria. Scale bars = 5 μm.
Discussion
This study reports that under hypoxic‐ischemia condition, the Δψm, respiratory‐related enzymes, and mitochondrial DNA deteriorate resulting in the aberrant accumulation of free radicals and ROS. CsA, traditionally considered a robust immunosuppressant drug, has been shown as a potent protective agent against neuronal cell death 10, 11, 12, 13, 25, 26, 27, 28. However, the molecular mechanism by which CsA interacts with mitochondrial membrane‐associated proteins remains not fully understood. Using the in vitro acute stroke model of OGD/R injury, we examined here this interaction between CsA and the Parkinson's disease‐associated protein DJ‐1, which has been recently implicated in the regulation of mitochondrial integrity 6, 7, 25, 26, 27, 28. Administration of CsA before stroke onset (24 h pre‐OGD/R), but not after stroke (2 h post‐OGD/R), afforded significant neuroprotective effects compared to OGD/R‐treated cells without CsA, characterized by the following cellular and molecular events: (1) CsA prevented the mitochondria‐dependent apoptotic cell death associated with Cyt c release; (2) CsA protected cellular ATP decline (2‐fold higher) without altering the hexokinase activity and the Δψm; (3) blocked mtDNA degradation; and, interestingly, (4) enhanced secretion of DJ‐1 in the mitochondria (Figure 5). This neuroprotection was achieved with the effective dose range of 500 nm – 10 μ m of CsA.
Figure 5.
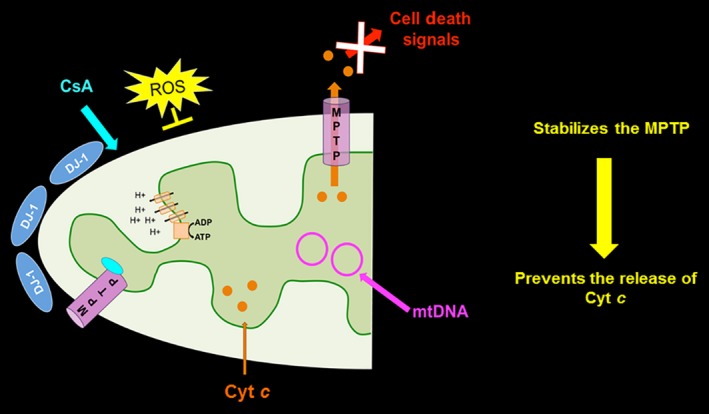
Schematic diagram of proposed neuroprotective mechanism of CsA and DJ‐1 in mitochondria in stroke. This schematic diagram represents a speculative reparative mechanism underlying the both CsA and DJ‐1 neuroprotective for mitochondria in stroke. Treatment of CsA upregulates DJ‐1, stabilizes the MPTP and prevents the release of Cyt c from the mitochondria. The MPTP is thought to open in response to hypoxic‐ischemia to the neuron cell. In combination, there is a reduction in mtDNA degradation and prevention of cellular ATP reduction.
CsA Protects the Opening of MPTP
Mitochondrial dysfunction is an important contributor to neurodegeneration 45, 46, including stroke 47. Hypoxic‐ischemic cell death events may consist of mitochondria complex I spontaneously releasing ROS, a hallmark biochemical feature of oxidative stress 48. A cell death mechanism characterized by the collapse of Δψm, which in turn triggers the aberrant disruption of the impermeability of the inner mitochondrial membrane, has been shown to accompany oxidative stress 25, 26, 27, 49, 50. Initial stroke‐induced ROS acts upon neighboring mitochondria, precipitating MPTP opening, and thereafter generating additional ROS 51. The MPTP is thought to open in response to hypoxic‐ischemia to the neuron cell 52. Intracellular calcium and oxidative stress trigger a conformational change in the adenine nucleotide translocase (ANT) when associated with voltage‐dependent anion channel (VDAC). This is facilitated by cyclophilin D (CypD), and blocked by CsA, which specifically binds to CypD. Of note, the initial accumulation of stroke‐induced ROS may act upon neighboring mitochondria, thereafter precipitating the MPTP opening and generation of additional ROS. Once MPTP is breached, Cyt c is released from mitochondria to cytosol and induces the activation of apoptosis cascade events. Employing the anti‐ATP synthase β‐chain antibody, of which antigen is localized in the mitochondrial inner membrane and Cyt c antibody, the present results revealed that CsA prevents Cyt c releasing from mitochondria.
CsA Stimulates the Secretion of DJ‐1 and Translocates into the Mitochondria
A major finding in the present study is the demonstration that neural cells secreted DJ‐1, with significant upregulation of DJ‐1 generated at 1 μ m of CsA. This observation parallels similar reports documenting that breast cancer and melanoma tumor cells release DJ‐1 to the serum in vitro and in vivo 53, 54, 55. The elevated levels of extracellular DJ‐1 following treatment with CsA, and the antibody sequestration of this secreted protein suggests an intimate involvement of DJ‐1 in the initial endogenous neuroprotective process in response to stroke. Moreover, the translocation of DJ‐1 into the mitochondria coinciding with reduced oxidative stress implies that DJ‐1 may facilitate the molecular link between mitochondria and oxidative stress in establishing a potent neuroprotective pathway to halt the progression of secondary cell death inherent in stroke 25, 26, 27.
CsA Reduces mtDNA Degradation and Prevents of Cellular ATP Reduction
Maintenance of mitochondrial integrity has been suggested to be an important mechanism of extending lifespan, as decreased mitochondrial integrity, impaired ATP generation, and increased ROS levels have been implicated in aging 56. To this end, understanding the role of CsA in mitochondrial integrity may reveal its neuroprotective action. CsA is a hydrophobic peptide drug, with its immunosuppressive activity closely associated with its binding to specific proteins of immune cells, such as CypD. Because of its hydrophobicity, CsA may interact with biological membranes, which may mediate its therapeutic effect 57. DJ‐1 protein stability and dimerization have been shown to be disrupted across the entire dimer interface, characterized by extended hydrophobic surfaces involved in dimer formation 58, 59, 60. This significant increase in DJ‐1 hydrophobic surface area 59 may contribute to its translocation into the mitochondria 25, 26, 27. Our results suggest that CsA and DJ‐1 may specifically interact within mitochondrial membranes, in that CsA facilitates DJ‐1 translocation into the mitochondria allowing maintenance of mitochondrial integrity against ischemic cell death.
CsA Role in Apoptosis Associated with Cyt c Release
Cyt c induction during apoptosis involves (1) the binding of Cyt c to cardiolipin (CL) in the inner membrane of mitochondria (IM); (2) release of Cyt c upon complex‐I‐dependent oxidation of CL; (3) pro–caspase‐8 (pro‐8) binding to the outer membrane of mitochondria (OM), which leads oligomerization, and subsequently undergoes autocatalytic processing in a CL‐dependent manner; (4) pro‐8 cleaving Bid and forming truncated Bid (t‐Bid), in turn facilitating the activation of Bax/Bak; and (5) phospholipid scramblase 3 (PLS3) allows export of CL to OM, mediating normal mitochondrial integrity 52. In the present study, pretreatment of CsA upregulates DJ‐1, stabilizes MPTP and prevents the release of Cyt c from the mitochondria. In tandem with this CsA stabilization of DJ‐1 in the mitochondria, we found that there is a reduction in mtDNA degradation and at the same time prevention of cellular ATP reduction. Preserving the integrity of the mitochondria via regulation of ATP production and Cyt c release is key to abrogating oxidative stress. CsA, via DJ‐1 upregulation, preserves mitochondrial integrity and exerts antioxidative stress, demonstrating a novel neuroprotective mechanism for treatment of stroke and possibly other neurological disorders. These novel observations indicate that CsA‐ and DJ‐1‐neuroprotection amplify the maintenance of mitochondrial integrity and that mitochondria‐based treatments targeting the early phase of disease progression may prove beneficial in stroke. Future studies will be required to use a DJ‐1 knockdown or DJ‐1 knockout cells into which CsA is administered. To translate this in vitro finding into an in vivo model, use a DJ‐1 knockdown or knockout rodent model to further investigate the CsA‐DJ‐1 neuroprotective pathway in stroke.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
CVB is supported by NIH NINDS 1R01NS071956‐01, James and Esther King Foundation for Biomedical Research Program, SanBio Inc., Celgene Cellular Therapeutics, KMPHC, and NeuralStem Inc. NT is supported by Byrd Institute Small Grants Program (BRD 220). The funders have no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Ms. Sussannah Kaelber for her technical assistance in journal formatting of this manuscript.
References
- 1. Heiss WD. Ischemic penumbra: Evidence from functional imaging in man. J Cereb Blood Flow Metab 2000;20:1276–1293. [DOI] [PubMed] [Google Scholar]
- 2. Warach S. Measurement of the ischemic penumbra with MRI: It's about time. Stroke 2003;34:2533–2534. [DOI] [PubMed] [Google Scholar]
- 3. Lo EH. A new penumbra: Transitioning from injury into repair after stroke. Nat Med 2008;14:497–500. [DOI] [PubMed] [Google Scholar]
- 4. Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in parkinson's disease. Science 2003;302:819–822. [DOI] [PubMed] [Google Scholar]
- 5. Nakamura T, Lipton SA. Redox regulation of mitochondrial fission, protein misfolding, synaptic damage, and neuronal cell death: Potential implications for alzheimer's and parkinson's diseases. Apoptosis 2010;15:1354–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ariga H, Takahashi‐Niki K, Kato I, Maita H, Niki T, Iguchi‐Ariga SM. Neuroprotective function of dj‐1 in parkinson's disease. Oxid Med Cell Longev 2013;2013:683920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takahashi‐Niki K, Inafune A, Michitani N, et al. Dj‐1‐dependent protective activity of dj‐1‐binding compound no. 23 against neuronal cell death in mptp‐treated mouse model of parkinson's disease. J Pharmacol Sci 2015;127:305–310. [DOI] [PubMed] [Google Scholar]
- 8. Achanta G, Sasaki R, Feng L, et al. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA pol gamma. EMBO J 2005;24:3482–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J 1999;341(Pt 2):233–249. [PMC free article] [PubMed] [Google Scholar]
- 10. Khaspekov L, Friberg H, Halestrap A, Viktorov I, Wieloch T. Cyclosporin a and its nonimmunosuppressive analogue n‐me‐val‐4‐cyclosporin a mitigate glucose/oxygen deprivation‐induced damage to rat cultured hippocampal neurons. Eur J Neurosci 1999;11:3194–3198. [DOI] [PubMed] [Google Scholar]
- 11. Guzman JN, Sanchez‐Padilla J, Wokosin D, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by dj‐1. Nature 2010;468:696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Giaime E, Yamaguchi H, Gautier CA, Kitada T, Shen J. Loss of dj‐1 does not affect mitochondrial respiration but increases ros production and mitochondrial permeability transition pore opening. PLoS One 2012;7:e40501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dongworth RK, Mukherjee UA, Hall AR, et al. Dj‐1 protects against cell death following acute cardiac ischemia‐reperfusion injury. Cell Death Dis 2014;5:e1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 2009;46:821–831. [DOI] [PubMed] [Google Scholar]
- 15. Osman MM, Lulic D, Glover L, et al. Cyclosporine‐a as a neuroprotective agent against stroke: Its translation from laboratory research to clinical application. Neuropeptides 2011;45:359–368. [DOI] [PubMed] [Google Scholar]
- 16. Hatton J, Rosbolt B, Empey P, Kryscio R, Young B. Dosing and safety of cyclosporine in patients with severe brain injury. J Neurosurg 2008;109:699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Borlongan CV, Emerich DF, Hoffer BJ, Bartus RT. Bradykinin receptor agonist facilitates low‐dose cyclosporine‐a protection against 6‐hydroxydopamine neurotoxicity. Brain Res 2002;956:211–220. [DOI] [PubMed] [Google Scholar]
- 18. Borlongan CV, Yu G, Matsukawa N, et al. Acute functional effects of cyclosporine‐a and methylprednisolone treatment in adult rats exposed to transient ischemic stroke. Life Sci 2005;76:1503–1512. [DOI] [PubMed] [Google Scholar]
- 19. Lulic D, Burns J, Bae EC, van Loveren H, Borlongan CV. A review of laboratory and clinical data supporting the safety and efficacy of cyclosporin a in traumatic brain injury. Neurosurgery 2011;68:1172–1185; discussion 1185‐1176 [DOI] [PubMed] [Google Scholar]
- 20. Borlongan CV, Stahl CE, Keep MF, Elmer E, Watanabe S. Cyclosporine‐a enhances choline acetyltransferase immunoreactivity in the septal region of adult rats. Neurosci Lett 2000;279:73–76. [DOI] [PubMed] [Google Scholar]
- 21. Butcher SP, Henshall DC, Teramura Y, Iwasaki K, Sharkey J. Neuroprotective actions of fk506 in experimental stroke: In vivo evidence against an antiexcitotoxic mechanism. J Neurosci 1997;17:6939–6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Friberg H, Ferrand‐Drake M, Bengtsson F, Halestrap AP, Wieloch T. Cyclosporin a, but not fk 506, protects mitochondria and neurons against hypoglycemic damage and implicates the mitochondrial permeability transition in cell death. J Neurosci 1998;18:5151–5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Uchino H, Elmer E, Uchino K, et al. Amelioration by cyclosporin a of brain damage in transient forebrain ischemia in the rat. Brain Res 1998;812:216–226. [DOI] [PubMed] [Google Scholar]
- 24. Bandopadhyay R, Kingsbury AE, Cookson MR, et al. The expression of dj‐1 (park7) in normal human cns and idiopathic parkinson's disease. Brain 2004;127:420–430. [DOI] [PubMed] [Google Scholar]
- 25. Kaneko Y, Shojo H, Burns J, Staples M, Tajiri N, Borlongan CV. Dj‐1 ameliorates ischemic cell death in vitro possibly via mitochondrial pathway. Neurobiol Dis 2014;62:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kaneko Y, Tajiri N, Shojo H, Borlongan CV. Oxygen‐glucose‐deprived rat primary neural cells exhibit dj‐1 translocation into healthy mitochondria: A potent stroke therapeutic target. CNS Neurosci Ther 2014;20:275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pantcheva P, Elias M, Duncan K, Borlongan CV, Tajiri N, Kaneko Y. The role of dj‐1 in the oxidative stress cell death cascade after stroke. Neural Regen Res 2014;9:1430–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yasuda T, Kaji Y, Agatsuma T, et al. Dj‐1 cooperates with pycr1 in cell protection against oxidative stress. Biochem Biophys Res Commun 2013;436:289–294. [DOI] [PubMed] [Google Scholar]
- 29. Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP. Dj‐1, a cancer‐ and parkinson's disease‐associated protein, stabilizes the antioxidant transcriptional master regulator nrf2. Proc Natl Acad Sci USA 2006;103:15091–15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Junn E, Taniguchi H, Jeong BS, Zhao X, Ichijo H, Mouradian MM. Interaction of dj‐1 with daxx inhibits apoptosis signal‐regulating kinase 1 activity and cell death. Proc Natl Acad Sci USA 2005;102:9691–9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Canet‐Aviles RM, Wilson MA, Miller DW, et al. The parkinson's disease protein dj‐1 is neuroprotective due to cysteine‐sulfinic acid‐driven mitochondrial localization. Proc Natl Acad Sci USA 2004;101:9103–9108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tsuboi Y, Munemoto H, Ishikawa S, Matsumoto K, Iguchi‐Ariga SM, Ariga H. Dj‐1, a causative gene product of a familial form of parkinson's disease, is secreted through microdomains. FEBS Lett 2008;582:2643–2649. [DOI] [PubMed] [Google Scholar]
- 33. Matsukawa N, Yasuhara T, Hara K, et al. Therapeutic targets and limits of minocycline neuroprotection in experimental ischemic stroke. BMC Neurosci 2009;10:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bell E, Cao X, Moibi JA, et al. Rapamycin has a deleterious effect on min‐6 cells and rat and human islets. Diabetes 2003;52:2731–2739. [DOI] [PubMed] [Google Scholar]
- 35. Kaneko Y, Sullivan R, Dailey T, Vale FL, Tajiri N, Borlongan CV. Kainic acid‐induced golgi complex fragmentation/dispersal shifts the proteolysis of reelin in primary rat neuronal cells: An in vitro model of early stage epilepsy. Mol Neurobiol 2016;53:1874–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kon K, Kim JS, Uchiyama A, Jaeschke H, Lemasters JJ. Lysosomal iron mobilization and induction of the mitochondrial permeability transition in acetaminophen‐induced toxicity to mouse hepatocytes. Toxicol Sci 2010;117:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu SC. Regulation of glutathione synthesis. Mol Aspects Med 2009;30:42–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Neubig RR, Spedding M, Kenakin T, Christopoulos A, International Union of Pharmacology Committee on Receptor N, Drug C . International union of pharmacology committee on receptor nomenclature and drug classification. Xxxviii. Update on terms and symbols in quantitative pharmacology. Pharmacol Rev 2003;55:597–606. [DOI] [PubMed] [Google Scholar]
- 39. Ashley N, Harris D, Poulton J. Detection of mitochondrial DNA depletion in living human cells using picogreen staining. Exp Cell Res 2005;303:432–446. [DOI] [PubMed] [Google Scholar]
- 40. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008;183:795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tanaka A. Parkin‐mediated selective mitochondrial autophagy, mitophagy: Parkin purges damaged organelles from the vital mitochondrial network. FEBS Lett 2010;584:1386–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bande MF, Santiago M, Blanco MJ, et al. Serum dj‐1/park 7 is a potential biomarker of choroidal nevi transformation. Invest Ophthalmol Vis Sci 2012;53:62–67. [DOI] [PubMed] [Google Scholar]
- 43. Vachon P, Beaudry F, Marier JF, Ste‐Marie L, Montgomery J. Cyclosporin a in blood and brain tissue following intra‐carotid injections in normal and stroke‐induced rats. Brain Res 2002;943:1–8. [DOI] [PubMed] [Google Scholar]
- 44. Stoltzman CA, Peterson CW, Breen KT, Muoio DM, Billin AN, Ayer DE. Glucose sensing by mondoa: Mlx complexes: A role for hexokinases and direct regulation of thioredoxin‐interacting protein expression. Proc Natl Acad Sci USA 2008;105:6912–6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Parone PA, Martinou JC. Mitochondrial fission and apoptosis: An ongoing trial. Biochim Biophys Acta 2006;1763:522–530. [DOI] [PubMed] [Google Scholar]
- 46. Yang Y, Candelario‐Jalil E, Thompson JF, et al. Increased intranuclear matrix metalloproteinase activity in neurons interferes with oxidative DNA repair in focal cerebral ischemia. J Neurochem 2010;112:134–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci 2008;31:151–173. [DOI] [PubMed] [Google Scholar]
- 48. Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov 2010;9:447–464. [DOI] [PubMed] [Google Scholar]
- 49. Reyes RC, Parpura V. Mitochondria modulate ca2 + ‐dependent glutamate release from rat cortical astrocytes. J Neurosci 2008;28:9682–9691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Krebiehl G, Ruckerbauer S, Burbulla LF, et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of parkinson's disease‐associated protein dj‐1. PLoS One 2010;5:e9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ros)‐induced ros release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 2000;192:1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol 2011;192:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yan H, Pu XP. Expression of the parkinson's disease‐related protein dj‐1 during neural stem cell proliferation. Biol Pharm Bull 2010;33:18–21. [DOI] [PubMed] [Google Scholar]
- 54. Rabinowitz JD, White E. Autophagy and metabolism. Science 2010;330:1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Barsoum MJ, Yuan H, Gerencser AA, et al. Nitric oxide‐induced mitochondrial fission is regulated by dynamin‐related gtpases in neurons. EMBO J 2006;25:3900–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sahin E, DePinho RA. Axis of ageing: Telomeres, p53 and mitochondria. Nat Rev Mol Cell Biol 2012;13:397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Azouzi S, Morandat S, El Kirat K. The potent antimalarial peptide cyclosporin a induces the aggregation and permeabilization of sphingomyelin‐rich membranes. Langmuir 2011;27:9465–9472. [DOI] [PubMed] [Google Scholar]
- 58. Premkumar L, Dobaczewska MK, Riedl SJ. Identification of an artificial peptide motif that binds and stabilizes reduced human dj‐1. J Struct Biol 2011;176:414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Anderson PC, Daggett V. Molecular basis for the structural instability of human dj‐1 induced by the l166p mutation associated with parkinson's disease. Biochemistry 2008;47:9380–9393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Quigley PM, Korotkov K, Baneyx F, Hol WG. The 1.6‐a crystal structure of the class of chaperones represented by escherichia coli hsp31 reveals a putative catalytic triad. Proc Natl Acad Sci USA 2003;100:3137–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]